0022-538X/92/020615-08$02.00/0
Copyright C) 1992, AmericanSocietyforMicrobiology
Defects in Moloney Murine Leukemia
Virus
Replication Caused
by
a
Reverse
Transcriptase Mutation
Modeled on the
Structure
of
Escherichia coli RNase H
ALICE TELESNITSKY, STACY W. BLAIN, AND STEPHEN P. GOFF* Department of BiochemistryandMolecularBiophysics, Columbia University College
of
Physicians
andSurgeons, New
York,
New York 10032Received 19 July1991/Accepted 17 October 1991
We have studied a mutant Moloney murine leukemia virus with a deletion in reverse transcriptase (RT) which ispredictedto makeits RNase H domain resemble structurally that of human immunodeficiency virus RT. This deletion was based onimproved RNase H homologyalignmentsmade possible by the recently solved three-dimensionalstructure forEscherichiacoli RNase H. Thismutant Moloney murine leukemia virus RT was fullyactive in theoligo(dT)-poly(rA) DNApolymerase assay and retained nearly all of wild-type RT's RNase Hactivity in an in situ RNase H gel assay. However, provirusesreconstructed to include this deletion were noninfectious. Minus-strand strong-stop DNA was made by the deletion mutant, but the amount of minus-strandtranslocation wasintermediateto thevery low level measured with RNase H-null virions and the high levelseenwithwild-type RT. The average length oftranslocatedminus-strand DNA was shorter for the deletion mutantthan for wild type, suggesting that mutations in the RNase H domain of RT also affect DNApolymerase activity.
Retroviral replication requires virally encoded reverse transcriptase (RT) to synthesize a double-stranded DNA copy ofthe genomic RNA (1, 11, 46). All retroviral RTs contain botha DNApolymerase activity, capable of synthe-sis on either RNA or DNA templates, and a nuclease activity, called RNase H, which specifically degrades RNA when the RNA is present in RNA:DNA hybrid form (27). Both of these activities arebelieved to be required for the
completion
ofthe complex series of steps involved in the RT-catalyzed biosynthesis ofa double-stranded DNA copy of the retroviral RNA genome (10). RNase H has been implicated in several of these steps, and mutant viruses which lack RNase H activity are noninfectious (30, 34, 43, 47). Minus-strand strong-stop DNA, the first DNA interme-diatetoappearduringreversetranscription, is maintainedin RNA:DNA hybrid form among the abortive products of reverse transcription by virions which lack RNase H (43). This finding suggests that one of the first critical steps mediated by retroviral RNase H is the degradation ofthe RNA strandof thishybrid in order that minus-strand trans-location and its subsequent elongation may occur.Sequence comparisons
reveal that RT is the most highly conserved protein among retroviruses, and sequence con-servation exists between retroviralRTandproteins of other retroelements (9,20, 25). Even relatedenzymes from cellu-lar sourcessuchasEscherichiacoli andyeast RNases H can be aligned with the carboxy termini of retroviral RTs (51). However,the RTsof various retroviruses differstructurally intermsof subunitcomposition
andorganization.
Inthecase of theMoloneymurine leukemia virus (M-MuLV) enzyme, the DNA polymerase and RNase H activities reside in physically separable domains ofasingle
monomericprotein
(41). The human
immunodeficiency
virus(HIV)enzyme isa heterodimerwhose subunits contain identical amino-termi-nal DNApolymerase regions,butonlyonesubunit includes*Correspondingauthor.
the carboxy-terminal RNase H region (8, 14, 23, 39). RT fromtheaviansarcoma-leukosis virusgroup,includingRous sarcoma virus RT, is also aheterodimer, but both subunits retainthe DNApolymerase andRNase Hdomains and differ bytheinclusion ofthe integraseregionattheC terminus of oneofthe subunits.
The three-dimensional structure ofE. coli RNase H was determined recently (21,51). On thebasis of this structure, improved homologyalignments, which seek topreserve the secondary structure features and putative catalytic core geometryofE. coliRNase H, havebeenmade betweenthe E.colienzyme and the RNase Hdomains of retroviral RTs. The validity ofthe resulting predicted retroviral RNase H structures has been partially confirmed by the recent eluci-dation ofthe structure of an HIV RT-derived polypeptide containing the region of RNase H homology (6). What emergesfromthese studies is astriking structuralsimilarity among RNases H from evolutionarily distant sources. Al-though many RNases Hdifferinminorways, such asin the numberofresidues between someof theirsecondary struc-ture features, a more
significant
difference exists between HIV RNase H and thepredicted
structurefortheM-MuLV RNase Hdomain.Tertiarystructurepredictionssuggest that the RNase Hdomains ofM-MuLV RTandE.coli RNase H areverysimilarstructurally (51). Whereasmodelstructures forboth ofthese enzymes include five1B
strands andfivea helices, oneofthe a helicesismissingfrom HIV RNase H. Intheexperiments presented here,wehavestudieda mutant M-MuLV, whichwecallAC,
withan 11-amino-acid deletion in RT which is predicted to make its RNase H domain resemble structurally that of HIV RT. Theresiduesthatwe removed,593(Ile) through603(Leu)from M-MuLVRT,
are predicted to include the a helix in the M-MuLV enzyme which is not present in HIV RT.We havepreviously
reported
theeffects ofeliminationof RNase Hactivityin the murine systemby
studying
a series of mutants of M-MuLV with linker-insertion mutations in the RTregion
ofthepol
gene(41, 43).
Unliketheseprevious
615
on November 10, 2019 by guest
http://jvi.asm.org/
RTmutants, whose RNase Hactivityis reducedprofoundly
or abolished, we demonstrate here that the AC mutant
retained much of its basic RNase H activitybut was
none-theless unable to sustainviral infectivity. In this report, we
examine thereplicationdefectsof the AC mutant andexpand
ourprevious analyses of the effects of RNase H mutations
on retroviral replication by establishingassays to compare
the extents of minus-strand translocation by wild-type, AC,
and RNase H-null forms of RT. We also report evidence whichsuggeststhat mutations in the RNase Hdomain of RT
can affect elongation properties of the DNA polymerase activityof theenzyme withoutperturbingthebasiccatalytic activity of DNA polymerization, thus strengthening the notion that the DNA polymerase and RNase H activities of
RTacttogether,bothfunctionallyandstructurally, in retro-viral replication.
MATERIALSAND METHODS
Cells and DNA-mediated transformations. All virus-pro-ducingcelllineswere single-cellcloned frompopulationsof
NIH 3T3 cells whichwere subjected to calcium
phosphate-mediatedcotransformation with a mixtureofproviral DNA
and pSV2neo (38, 49). Transient transfection with DEAE-dextran, RTassaysofsupernatant medium, maintenanceof
cells inDulbecco modifiedEaglemediumsupplementedwith
10% newborn calfserum, centrifugal concentrationof viral
particles, XC plaque assays, infection with virus in the
presence of Polybrene, isolation of low-molecular-weight DNA, and Southern blot analysis were all performed as
previously described (12, 15, 26, 32, 37, 42). The cell line
used as a producer of virions containing wild-type RT was
d15401, which harbors a mutation in IN(36).
DNA manipulations. Subcloning into the E. coli RT
expression vector (44) and into pNCA, which contains a
clonedcopyof theM-MuLVgenome(3),andconstructionof
M13 derivatives involved routine techniques (33). The E.
coli expression vector contains the T7 phage Al promoter
and the sequencesencoding mature M-MuLV RTpreceded by a methionine codon (5, 40, 44, 45). Microsequencing of
the purified wild-type RT produced bythis vector revealed
that the N-terminal methionine had been removed (datanot
shown). Thus,thisexpression system producesRTidentical
in sequence to that found in M-MuLVparticles. ARHRT is
a truncated form of M-MuLV RT; its construction will be
described elsewhere (44). It produces a DNA polymerase single-domain form of RT which is missing the
carboxy-terminalRNase Hdomain,and is similartotruncatedforms
of RT which have been described previously (16, 22, 41).
The AC deletion was constructed by
oligonucleotide-medi-ated site-directed mutagenesis (52). Annealingthe oligonu-cleotide, 5'-CCTTCTGATGTGAGATGGGCAGTAGCAA-3', to a single-stranded M13 derivative containing much of
theM-MuLVRTcoding region andthenfollowing standard
site-directed mutagenesis procedures resulted in the lossof
the33nucleotides(nt)whichencodethe 11amino acids from
593 (Ile) through603 (Leu) ofM-MuLV RT.
Preparationandassayofenzymes.RToverexpressedin E.
coliwaspurified essentiallyas wehavedescribedpreviously
(31).AllenzymeswerepurifiedfromE. coliDH5cx, which is
a derivativeof DH1(13). All buffers and methodswerethe
same, with the following modifications. Prior to passage
through the DEAE-cellulose column, 0.6 volume of satu-rated ammonium sulfate (pH 7) was added to the crude
extract supernatant. After 30 min of stirring at 4°C, the precipitated proteins were collected by centrifugation at
16,000 rpm for 30 min in a chilled SS34 rotor,
gently
resuspended in a small volume of buffer M(50mMTris-HCl [pH 7], 1 mM EDTA, 1 mM dithiothreitol, 0.1% Nonidet P-40, 10% glycerol), and desalted by chromatography through a Sephadex G-50 column which had been
equili-brated with buffer Mcontaining 75 mM NaCl. Fractions that contained RTactivitywere pooled, passed through DEAE-cellulose, and then fractionated furtherby
chromatography
on phosphocellulose. The phosphocellulose column frac-tionscontainingthe RTpeakwerepooledand concentrated in Centriprep 10 concentrators (Amicon) according to the manufacturer's instructions anddialyzedinto storage buffer. This materialwasstoredat -80°C forusein the
experiments
in this study. Estimations ofpurity, analyses of RT DNA polymerase activityusingtheoligo(dT)-poly(rA) assay,and unit definitions were as describedpreviously
(31).
RNase H activity was assessed by using an in situ
gel
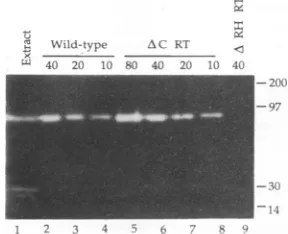
assay as described previously (41). This assay involves electrophoresis of the proteins on sodium dodecyl sulfate-polyacrylamide gels cast with 32P-labeled RNA in RNA: DNA hybrid form, followed by incubation of the gels in severalchanges of a buffer, which allows the renaturation of theprotein and digestion of the radiolabeled hybrid. Thegels are then fixed, dried, and exposed toX-ray film. Levels of RNase Hactivity are estimated by comparing the extent of radioactive clearing, which results from digestion of the labeledRNA.
Endogenous RT reactions. Endogenous reactions using concentrated virus were carried out for 3 h at 40°C in the presence of 100 ,ug ofactinomycinDper ml and 2 mM each dCTP, dATP, and dGTP and 1 mM [a-32P]dTTP at 1 Ci/mmol. These reactions were performed, and their prod-ucts wereprocessed andcollected, as describedpreviously (43).Reactionproducts were incubated in 0.33 N NaOH for 20 min at 55°C before the final ethanol precipitation to removeRNA primers. Reactionproducts were analyzed on 7.5%polyacrylamide gels, and DNAfragments were recov-ered from wet gels as previously described (43). Alkaline agaroseelectrophoresiswasperformed by standardmethods
(33).
Translocationassays. Si assaysfor translocation made use of an unlabeled single-stranded M13 derivative containing the plus strand of a Sacl-Sacl fragment of pNCA, which includes36 ntof U3 upstream ofintactRandU5 regions of M-MuLV (3). This plus-strand DNA was hybridized to radiolabeled endogenous reaction products and digested with S1, and the labeled products were analyzed by using standardprocedures for Si mapping(33).
Therestrictiondigestassay oftranslocationmade use of a similarM13derivativewhichcontained the plus strand of a PstI-EcoRIfragment of pNCA. This fragmentincludes intact U3, R, and U5regionsof the M-MuLVlong terminal repeat (LTR). Portions of radiolabeled endogenous reactions esti-mated to contain less than 10 fmol of replication products were annealed to200fmol of the plus-strand DNA in 10-,ul reaction mixtures containing 75 mM NaCl, 10 mM MgCl2, and 10 mM Tris-HCl (pH 7.5) by incubating the reaction mixtures for 40 min at 66°C and then slowly cooling the mixtures to 25°C over 1 h. A
100-pl
volume of a buffercontaining
50mMNaCl, 10 mM Tris-HCl (pH 8.0), 10 mM MgCl2, and 5 mM 2-mercaptoethanol, and 30 U AvaI was added to each sample, and the reaction mixtures were incubated at 37°C for3 h. The products were then phenolextracted,
ethanol precipitated,andanalyzedby electropho-resis on7.5%polyacrylamide-8 Murea gels. The gels wereon November 10, 2019 by guest
http://jvi.asm.org/
a
FIG. 1. Schematic ribbon drawings ofthe structure of E. coli RNase H (a) and a structural prediction for the HIV RNase H domain (b).
Theposition oftheC helixin the E. colistructure is indicated with abracket. For moredetails, see reference 51.
dried and exposedto X-rayfilm with intensifyingscreens for 2weeksat -80°C.
RESULTS
The C helix is not essential to basic RNase H function. Although the core structures of evolutionarily divergent RNases H are conserved, there are significant differences between the
predicted
structures of various retroviral RNasesH. Thethird ahelix present in the E. coli enzyme, known as the C helix (the first and second helices being designated A and B), is predicted to be present in the M-MuLV enzyme. However, it is missing from the HIV RNaseHstructure andalsopredictedto beabsent from the dimeric RT of Rous sarcoma virus (Fig. 1 [6, 51]). This finding suggested thattheChelixmight be either dispensable for RNase H function orelse responsible for some of the differences between such RNases H as those ofM-MuLV andHIV. In thebacterialenzyme,the regionof this deletion formsanahelixthatispartofaflap oftheenzyme which is not close to the catalytic site but which may contact the RNA:DNAhybridwhenit isengagedat theactive site(51). The absence of this helix in the HIV enzymeresults in theflattening
of this region of the protein relativetothe structure of the E. coli enzyme. It has been suggested that this flattening allows the HIV RNase Hdomain tointeract with someportion
of the two DNApolymerase
domains ofthe intactheterodimcer
and that this interaction both stabilizes thedimerandis essential forproductivesubstrate
binding by theRNase Hdomain (6).The M-MuLV RT mutant in this study is called AC because it has an 11-amino-acid deletion in the enzyme's RNase H domain which removed a
region
predicted to include the C helix.To create the AC mutant,weexamined the published structure ofE. coli RNase H and homologyalignments
made between it andretroviralRNase Hregions
and then chosetodeleteresidues593
(Ile) through
603(Leu)
of M-MuLV RT. The AC deletion was constructed
by
oligonucleotide-mediated
site-directedmutagenesis
andbuilt into an E. coli expressionplasmid encoding
authentic M-MuLV RT (33, 44, 52).Wild-type
and AC M-MuLV RT and atruncated form ofthe enzyme calledARH,
which is missingtheentireRNase Hdomain,
werepurified
to >90%homogeneity from E. coli cells harboring RT expression plasmids byavariation ofthemethod ofRoth et al. (31). The DNA polymerase activities of these enzymes were com-pared by measuring incorporation of TMP, using an
oli-go(dT)-poly(rA)primer-template.
Wild-type, AC, and ARH RT were determined todisplay 44, 99, and 112 U ofDNA polymerase activity per ,ug ofprotein, respectively. Thus, according to this assay, the DNA polymerase activity of theseRTmutantsisnotreduced;rather,itis higher thanthat ofthe wild-type enzyme.RNase Hactivities werecompared in an in situ gelassay (41). The amounts ofdegradation ofRNA:DNA hybrid by 10,20, and 40 UofDNApolymerase activity ofeachenzyme werecompared(Fig. 2). By this comparison,it appears that the
AC
enzyme retains all or nearly all of the RNase H activity of the wild-type enzyme. Thus, whereas the AC mutation results in the removal of the central 1/10 of the RNase H domain, thecatalytic sitehas not beendestroyed, and we have successfully constructed a largely functional RNase H allele ofRT based on the crystal structureofthe related enzyme, E. coliRNase H.Note that we wereunabletoquantifyourpartiallypurified
- (] -)n 1n Qn A fl ') 1f')
-200
-97
-3u .2 3 4 5 b
FIG. 2. In situgel assayforRNase Hactivity.Proteinspurified from E. coliRTexpression strainswere quantifiedasdescribed in Materials and Methods; lane 1 contains wild-type RT in a crude
extractpreparedasdescribedbyHizietal.(17).Numbersatthetop indicatethe numberof units ofRT DNApolymeraseactivityloaded
ineachlane. Sizesinkilodaltons areindicatedattheright.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.154.486.71.254.2] [image:3.612.374.518.548.665.2]enzymes'
RNase Hactivities insolution,
since these enzymepreparations
maycontain someresidual nucleases. Extracts from E. coli cells contain several host-encoded RNase H activities. Whereasnocontaminating
E. coli RNase H activ-ities weredetectable in ourpurified
enzymepreparations
in thein situ assay,solution assayssuggested
thatourARH RT containssignificant
levelsofcontaminating
RNase Hactivity(data
notshown).
Viruses
harboring
the ACmutationarenoninfectious. The AC mutationwasmoved intoaclonedcopyofthe M-MuLV genome, and theinfectivity
oftheresulting
mutantprovirus
was tested in 3T3 cells. No viral
spread
wasdetectable,
either
by
the XCplaque
assay orby assaying
culture media forRT,
when thecellsweretransiently
transfectedwith thisprovirus
via DEAE-dextran.Cell lines that
produced
RT+
virionsharboring
the AC mutation wereestablished. When virions shedby
thesecell lineswereapplied
tofresh 3T3cells, they
werefound tobenoninfectious,
andnolow-molecular-weight (preintegrative)
viralDNAwasdetectedonSouthern blots
(data
notshown). No reversion of the AC mutation was observed during 6 weeks ofpassage ofcells infected with AC virions.There-fore,
we conclude that the AC mutation renders M-MuLV noninfectious.Minus-strand translocation is reduced in AC virions. To
assessthe natureofthe
replication
defectsconferredbythe ACmutation,
wecompared the replication properties of AC virions with those of virions withwild-type
RT and twopreviously
described RNase H-null forms of RT(41, 43).
One of the M-MuLV RT RNase H null alleles
(M4)
is a linker-insertion mutation whichdestroys
RNase Hactivity,
and the other
(H7)
containsaframeshiftearly
in the RNase Hcoding
regionwhich also abolishes RNase Hactivity.
Both the wildtype andall ofthe RNase Hmutantsformed minus-strand strong-stop
DNA
whenreplication products
were examined
by
using detergent-permeabilized
virions in the so-calledendogenous
reaction(Fig. 3). However,
theamounts of
slower-migrating
products
of theendogenous
reaction were
significantly
reduced forall ofthe RNase Hmutants. Since these reactions were
performed
in thepres-ence of
actinomycin
D,
which preventsDNA-dependent
DNA
synthesis,
theseslower-migrating
DNAs should belimnited
to minus-strandproducts.
Since RNase H
activity
is believed to be involved in the translocationof minus-strandstrong-stop DNAtothe 3' end of thegenomic
RNA(necessary
for thecompletion
of minus-strand DNAsynthesis),
wedeveloped
twoassays toassess theextentoftranslocation
(Fig. 4).
Oneofthesewasan S1
protection
assay which allowed determination of theextentofminus-strand translocation
by comparing
the ratio of minus-strandproducts
whosesynthesis
ceased at the stage of minus strand strong stop to those which had been extended after translocation and thus included sequences from the U3region.
The second assay involvedcleavage of the labeledendogenous
reaction products hybridized tosingle-stranded
plus-strand
LTR DNA by a restriction en-zyme which cuts in both the U5 and U3regions.
Thisanalysis yielded
products
a and b for minus-strand strong-stop DNAandproducts
aand b'for translocatedDNA, and theratiob'/(b
+b') gives
thefractionofreplicationproducts that had translocated(Fig.
4).These assays revealed that the null alleles of RNase H translocate toavery low but detectableextent and that the
extentof translocation of minus-strand strong-stop DNA by the AC enzyme is intermediate to that observed with the
wild-type
andmutantenzymes. Translocationby the RNaseI1202
-49 _
1
_1-4-~.zq4imi~ .:
if,i
4-\li un strnLi
[image:4.612.365.500.74.348.2]strnrlg9top
FIG. 3. Endogenous reaction products from wild-type and
RNase H mutant virions analyzedby polyacrylamide gel electro-phoresis. WTindicates the productsfromvirions containing wild-type RT;AC, M4, and H7 are theproducts of virions with RNaseH
mutationswhich are described in the text. Theposition ofmigration of minus-strandstrong-stop DNAisindicated. Marker sizesarein
nucleotides. Note that the discretebandsmigrating between the 181-and 499-nt markers are VL30minus-strandstrong-stop DNAs which
areproduced from copackagedviruslikeRNAs(43).
H-null forms of RT could not be detected whenthe assays were performed on total reactionproducts. Thus, toassess whether any minus-strand translocation had occurred, we fractionated the endogenous reaction products of an RNase H-null allele. DNAs were eluted from gel slicescontaining populations ofhigh-molecular-weight reactionproducts.The isolated DNAs called ii, iii, and iv are populations of molecules approximately 500 to 800, 800 to 1,200, and >1,200 ntlong, respectively (Fig. 4D). When theseisolated DNAs were assayed, very low levelsof translocationwere detectable. We estimate that translocation by the nullalleles is lower than for the wild type by 2 or 3 orders ofmagnitude; in contrast, translocation by AC is approximately 10-fold reduced.
The S1 protection assay of AC endogenous reaction prod-ucts also revealed a prominent set of DNA products less than 40 nt in length which were protected by plus-strand LTR DNA (Fig. 4b). The size and mapping properties of theseproducts suggest that theyareprematurelyterminated tRNA-initiatedminus-strandproducts. As is visible in Fig. 5, there areprominent DNAs smaller than minus-strand strong-stop DNAamong the ACendogenous reaction products that maycontribute to these protected products.
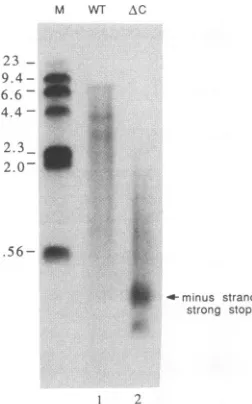
Translocatedminus-strandproducts of AC are shorter than those of the wild type. The amount of longer minus-strand productsin theACreaction appeared less than expected for the measured amount oftranslocation, and the gel system
.1, i .%' 'T .%(' \1_1 11-7
AAW .
on November 10, 2019 by guest
http://jvi.asm.org/
A
provirus
|
;3IR|
U51
U3|RfU-5|
genomic RNA -
-minusstrandstrongstop4 -:
plusstrandfragment f -4 protectedbyfragment 145nt
translocatedDNA
-plusstrandfragment
I--protectedbyfragment 181nt
C Asa I AvaI AvaI AvaI
provirus U3 RU5
IJ_U3
RJU51
genomic RNA
minusstrand strong stop
.4-plus strandfragment
Ava Iproducts b a 31 and114nt
translocated DNA plus strand fragment
Ava Iproducts T2vbv 66and114 nt
D
B
4q.'4
ii-4C4
-.,, _
242
-I1,i
-lransloc)atL --_
minits stratnd
14-
-intts stran'd-strongstop
I1.
i-I....
IV.
AvaIdigil,ts :eolatcd LDNAs
117 T M WTr 117 i ii iii iv
-_
U..
minusstrarl_ N _ _
strongstop
0
'14'lI*l
nr._~ * ~ ~ ~
_ _
..- k
2 I 3 4 v t 7f
FIG. 4. Translocationassays.(A)Schematic explanation oftheS1assayfor translocation.Dottedlines indicate RNA, solid lines indicate DNA, andheavy lines indicate DNAradiolabeledduring itssynthesis in endogenous reactions. (B) Products of theS1 assay. Endogenous reactionproducts hybridizedtoplus-strandDNA areindicated above the lanes.Thepositions ofmigration of digestion products generated fromminus-strandstrong-stopand translocatedminus-strandDNAs areindicated. Marker sizesarein nucleotides.(C) Schematic explanation of the restriction digest assay of translocation. Dotted lines indicate RNA, solid lines indicate DNA, and heavy lines indicate DNA
radiolabeled during its synthesis in endogenous reactions. (D) Products ofthe restriction digestassay. Lanes 1 and2 containundigested endogenous reaction products from virionscontainingtheRNase HmutantH7orwild-type form ofRT. Lanes 3and4contain theproducts of therestrictiondigestassayperformed onunfractionatedendogenous reaction products. Lanes5 to8containtheproductsofrestriction digestassaysperformed onDNAsisolated fromwetgelscontainingRNase H mutantH7endogenous reactionproducts;the isolated DNA
labeled iispurified minus-strandstrong-stop DNA: DNAs ii,iii, and iv arepopulations of molecules whichwere eluted fromgelslices of successively highermolecularweight endogenousreactionproducts. Lanes 5to8containapproximately equivalentportionsofendogenous reactionproducts; theamountsofendogenous reactionproductsassayedinlanes 5to8aremuch greater thanthose in lanes 1to4sothat
minortranslocation productscould be detected. The positions ofmigration oftherestriction digestfragments andof intact minus-strand
strong-stop DNAareindicated;the markersarethesame asthose inpanel B.
used in the assays described above was not
capable
of resolving the sizes of large reaction products.Thus,
it seemedpossible
that AC virions generated translocated reaction products shorter than those produced in virionsharboring wild-type
RT. To compare the sizes of translo-cated minus-strand products,aliquots
containing
similar levels ofradioactivity
from AC andwild-type endogenous
reactions were
analyzed by
denaturing
agarosegel
electro-phoresis(Fig. 5).Whereasasignificantlevel of DNA migrat-ing in the vicinity of the predicted size for fully extended minus-strand productswasobserved in reactionscontaining wild-type RT, translocated AC products
averaged
lessthan one-third this length.Comparisonof theproductsofmutantsACand H7(Fig. 3) showsthat eventhoughtheefficiencyof translocationduring reverse
transcription
is verydifferent,
these viruses make44"P
40on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.91.533.71.492.2]M WT AC 2 3
4
I.4
I-6.6-m
4.42.0 ..
.56- 4.
ALi,
ffi -minus strand
[image:6.612.116.242.71.273.2]strong stop
FIG. 5. Analysisof the size distribution ofendogenousreaction
products ofwild-typeandAC virions.Sampleswerenormalizedso
thatapproximately equalamountsof radioactiveincorporationwere
loaded in each lane of an alkaline agarose gel. The position of
migration of minus-strand strong-stop DNA is indicated. Marker sizesarein kilobasepairs.
similar low levels of high-molecular-weight DNA. In the
caseofAC,the lowlevel oflong productsappears tobe due tolimitedelongationof therelatively efficientlytranslocated products. Perhapsthehigh-molecular-weight material of H7
results from efficientelongationof theveryrareH7 translo-cationproducts. It is alsopossiblethat VL30 DNAs,which
are reverse transcription products of copackaged
non-M-MuLV RNAs, might also contribute to the high-molecular-weight material and thus mask the contribution of the minisculeamount oftranslocatedproducts produced bythe RNase H-null mutants in these assays.
DISCUSSION
On the basis of the crystalstructure of E. coliRNase H,
we have constructed a deletion mutation called AC in the
RNase H domain of M-MuLV RT. The studies presented herewere aimed atcharacterizing thephenotype ofthe AC
mutant. The late stages of the life cycle were apparently
normal for themutant. Cell linesexpressingthe AC mutant
release virions containing genomic RNA and RT activity, andexamination of these virionsbyimmunoprecipitationof metabolicallylabeled proteins showed that the gag
precur-sor was processed correctly (data not shown). However,
viruses containing this mutation were noninfectious when
applied tocells andwere incapable of producing detectable
amounts of preintegrative DNA. The very low titer of AC virus haspreventedadirectanalysis of the processing of AC
polproducts. However, sincecorrectpol processingoccurs
in the RNase H deletions that we have previously studied, we believethat the primary defect of AC is probablynot in the processingof RTfrom itsprecursor(43).
Unlike our previous RNase H linker-insertion mutants,
the AC enzyme, which is missing the central 1/10 of its RNase H domain, retained much of its basic RNase H activity. It seems unlikely that the lethality of AC is due
solely to the relatively modest decrease in basic RNase H
activity
thatthis mutation caused. It has been estimated that there areapproximately
50 times more RT molecules thangenomic
RNAsinvirions(48),
and the DNApolymerase
and RNase Hactivities ofRTcanbe shownto actindependently
in some
settings.
Onemight
thereforeimagine
that theinefficiency
ofthe RNaseHactivity
ofoneAC RT molecule could becompensated
forby
thesubsequent
action of another molecule of RT. Thereis, however,
some contro-versyoverwhetherthepolymerase
andnuclease activities of RTnormally
doactinconcertin in vitroreactions,
andevenlessis known aboutthe
potentially
obligate coupling
of these activitiesduring
normalretroviralreplication (7, 19, 28, 35,
50).
It ispossible
that ACmight
be affected in theputative
coupling
of these twoenzymatic
activities.Alternatively,
even asubtledefectin RNase H
activity
might
be sufficient to account for the observedlethality,
due to cumulative effectsduring replication
of the entire genome. It is alsopossible
that theACmutantis defectiveinspecific
functions ofRT such as thespecific cleavage
of RNA:DNAhybrids
required
forplus-strand
priming
orthe RNA-DNAunwind-ing
activity
that has been described for avianmyeloblastos
virus
RT,
whichmaybe necessaryfor efficient minus-strand translocation(4).
The sensitive assays for translocation
presented
in this report show that a limited amount oftranslocation is ob-servedevenin thecaseofRNase H-null alleles.Though
the action oftrace amounts ofcontaminating
RNase Hactivity
cannot be ruled out, this low level of translocation is
presumably
due to the RNaseH-independent
template
switching
capability
ofRNase H-nullforms ofRTwhichhas been observed inpurified
systems(24).
Examination of thereplication
products
formed inpurified
AC-containing
viri-ons revealedthat theamount of minus-strand translocation waslowerthan thatof wildtypebut much
higher
than thatof the RNase H-nullalleles. The muchhigher
levelof translo-cationobserved withtheAC mutant,which retainsmuch of its RNase Hactivity,
further supports the beliefthat thevirally
encoded RNase Hactivity
isrequired
for efficienttemplate
switching
in M-MuLV.Although
the rate ofDNAsynthesis
on exogenoustem-plates
by
the AC enzyme wasslightly
higher
than that ofwild-type RT,
thetranslocated minus-strandproducts
made indetergent-permeabilized
AC virions were much shorter than those formed in virionsharboring wild-type
RT,
thussuggesting
that minus-strandsynthesis
terminatedprema-turely.
Additionally,
experiments
withpurified
enzymes have shown that the ACandARHenzymes aredefectiveinprocessivity
ofDNAsynthesis
(44).
Intemplate
competition
experiments,
themutantenzymesproduce
shortertemplate-directed
products
than thewild-type
enzymedoes(data
notshown). Thus,
the block toreplication
in the AC mutation may resultfrom subtledeficiencies in the DNApolymerase
activities oftheenzymeasmuchasfrom deficiencies in the RNase H
activity
itself.Weare
currently
constructing
additionalmutations in the RNase Hdomain ofM-MuLV RTtoexamine theimportance
of
regions
which vary among theretroviralRNases Handto testpredictions,
based on thecrystal
structure, of theproperties
associated with variousportions
ofthe enzyme.Studying
otherC-helixmutationsmay furtherdefinethe role ofthisregion. Although
theC helixisnotrequired
for basic RNase Hactivity
in intact M-MuLVRT,itsabsencemay bepermissible
because of structural informationprovided by
the DNA
polymerase
domain.Wild-type
M-MuLV RNaseH,
with the C helixintact,
isvery active whenexpressed
asa
single
domain in E. coli (41).However,
when the ACon November 10, 2019 by guest
http://jvi.asm.org/
mutation was rebuilt into an RNase H single-domain expres-sion vector, no RNase H activity was observed in in situ gel assays, which should detect activity as low as 1% of the wild-type level (2). Since AC was designed to resemble HIV RNase H, this provides an interesting parallel to the diffi-culties that our laboratory and other laboratories have encountered in attempts to producean active single-domain HIV RNase H (6, 16-18, 29).
ACKNOWLEDGMENTS
We thank W. Yang and W. Hendrickson for helpful discussion and advice on the design of the AC mutation and for obtaining the N-terminal sequence ofthe wild-type RT that we expressed inE.
coli, W. Yang and K. Smith for modeling the HIV RNase H
structure, W. Yangfor preparation of Fig. 1, andK. de losSantos for help with construction of the expression plasmids and prepara-tionof the enzymes fromE. coli.
This work was supported by PHS grant CA 30488 to S.P.G. S.W.B. is an NIH predoctoral trainee, and A.T. isafellow of the American CancerSociety.
REFERENCES
1. Baltimore, D. 1970. RNA-dependent DNA polymerase in
viri-onsofRNA tumorviruses. Nature(London) 226:1209-1211.
2. Blain, S. W., A. Telesnitsky, andS.P. Goff. Unpublished data.
3. Colicelli, J.,andS. P. Goff. 1988.Sequenceandspacing
require-mentsofa retrovirusintegration site. J.Mol. Biol. 199:47-59.
4. Collett,M. S., J. P. Leis, M. S. Smith, and A. J. Faras. 1978. Unwinding-like activity associated with retrovirus RNA-di-rected DNApolymerase. J. Virol. 26:498-509.
5. Copeland,T.D.,G. F.Gerard,C. W.Hixson,andS. Oroszlan.
1985. Amino- andcarboxyl-terminal sequence of Moloney
mu-rine leukemia virusreversetranscriptase. Virology143:676-679. 6. Davies, J. F. I., Z. Hostomska, S. R. Jorden, and D. A. Matthews.1991.Crystalstructureof the ribonucleaseHdomain of HIV-1 reversetranscriptase. Science252:88-95.
7. DeStefano, J. J., R. G. Buiser,L. M. Mallaber, T. W. Myers,
R. A.Bambara,and P.J. Fay. 1991.Polymerizationand RNase Hactivities of thereversetranscriptasesfrom avian myeloblas-tosis, humanimmunodeficiency andMoloney murine leukemia viruses are functionally uncoupled. J. Biol. Chem.
266:7423-7431.
8. di Marzo Veronese, F. D., T. D. Copeland, A. L. DeVico, R.
Rahman, S. Oroszlan, R. C. Gallo, and M. G. Sarngadharan. 1986. Characterization ofhighly immunogenic p66/p5l as the
reverse transcriptase of HTLV-IlI/LAV. Science 231:1289-1291.
9. Doolittle,R.F.,D.-F.Feng,M.S.Johnson,and M. A.McClure.
1989.Origin andevolutionaryrelationshipsofretroviruses. Q. Rev. Biol. 64:1-30.
10. Gilboa, E., S. W.Mitra,S. P.Goff, and D.Baltimore. 1980. A
detailed model of reverse transcription and tests of crucial aspects.Cell 18:93-100.
11. Goff, S. P. 1990. Retroviral reverse transcriptase: synthesis,
structureandfunction. J.AIDS 3:817-831.
12. Goff,S. P.,P.Traktman, and D.Baltimore. 1981. Isolation and properties of Moloney murine leukemia virusmutants: useofa
rapid assay forreleaseof virionreversetranscriptase. J. Virol.
38:239-248.
13. Hanahan,D.1983.Studiesontransformation ofEscherichia coli withplasmids. J. Mol. Biol. 166:557-580.
14. Hansen, J., T. Schulze, W. Mellert, and K. Moelling. 1988.
Identification and characterization ofHIV-specificRNAseHby monoclonalantibody. EMBO J. 7:239-243.
15. Hirt, B. 1967. Selective extraction of polyoma DNA from infectedmousecell cultures. J. Mol. Biol. 26:365-369.
16. Hizi, A.,S. H.Hughes,and M.Shararabany. 1990. Mutational analysis oftheribonuclease H activity of human immunodefi-ciencyvirusreverse transcriptase. Virology175:575-580.
17. Hizi, A., C. McGill, and S. H. Hughes. 1988. Expression of
soluble, enzymatically active human immunodeficiency virus
reverse transcriptase in Escherichia coli and analysis of
mu-tants. Proc. Natl. Acad. Sci. USA85:1218-1222.
18. Hostomsky,Z.,Z.Hostomska, G. 0.Hudson, E. W.Woomaw,
and B. R. Nodes. 1991. Reconstitution in vitro of RNase H
activity by using purifiedN-terminalandC-terminal domains of
human immunodeficiency virus type 1 reverse transcriptase.
Proc. Natl. Acad. Sci. USA88:1148-1152.
19. Huber,H.E., J.M.McCoy, J. S.Seehra,andC. C.Richardson. 1989. Humanimmunodeficiencyvirus1reversetranscriptase.J.
Biol. Chem. 264:4669-4678.
20. Johnson,M.S.,M. A.McClure,D.-F.Feng,J.Gray,and R. F. Doolittle. 1986. Computer analysis of retroviral pol genes:
assignment ofenzymatic functions to specific sequences and homologieswithnonviralenzymes. Proc.Natl. Acad. Sci. USA 83:7648-7652.
21. Katayanagi, K.,M.Miyagawa,M.Matsushima,M.Ishikawa,S.
Kanaya, M. Ikehara, T. Matsuzaki, and K. Morikawa. 1990. Three-dimensional structure of ribonuclease H from E. coli.
Nature(London)347:306-309.
22. Kotewicz,M. L., C. M. Sampson, J. M. D'Alessio, and G. F.
Gerard. 1988. Isolation of cloned Moloney murine leukemia virus reverse transcriptase lacking ribonuclease H activity. NucleicAcidsRes. 16:265-277.
23. Lightfoote,M.M., J.E.Coligan,T.M.Folks,A.S.Fauci,M. A.
Martin,andS. Venkatesan.1986.Structuralcharacterization of
reverse transcriptase and endonuclease polypeptides of the
acquired immunodeficiency syndromeretrovirus. J. Virol. 60: 771-775.
24. Luo,G.X.,andJ.Taylor.1990.Templateswitching byreverse
transcriptase duringDNA synthesis.J. Virol. 64:4321-4328.
25. McClure,M.A.,M.S.Johnson,D.-F.Feng,and R. F. Doolittle. 1988. Sequence comparisons of retroviral proteins: relative
ratesofchangeand general phylogeny. Proc. Natl.Acad. Sci. USA85:2469-2473.
26. McCutchan,J.H.,andJ.S.Pagano.1968. Enhancement ofthe
infectivityof simian virus40deoxyribonucleicacid with dieth-ylaminoethyl-dextran. J.Natl. CancerInst. 41:351-357.
27. Moelling, K., D. P. Bolognesi, W. Bauer, W. Busen, H. W.
Passman, and P. Hausen. 1971. Association of viral reverse
transcriptase with an enzyme degrading the RNA moiety of
RNA:DNAhybrids. Nature(London)New Biol. 234:240-243. 28. Oyama, F., R. Kikuchi, R. J. Crouch, and T. Uchida. 1989.
Intrinsicpropertiesofreversetranscriptaseinreverse
transcrip-tion;associated RNase Hisessentiallyregardedas an
endonu-clease. J. Biol. Chem. 264:18808-18817.
29. Prasad,V. R.,andS. P. Goff. 1989. Linker insertion
mutagen-esis of human immunodeficiency virus reverse transcriptase
expressed in bacteria: definition of the minimal polymerase domain. Proc. Natl. Acad. Sci. USA86:3104-3108.
30. Repaske, R.,J. W. Hartley,M. F. Kavlick,R. R. O'Neill,and
J. B. Austin. 1989. Inhibition of RNase H activity and viral replication by single mutations in the 3' region of Moloney murine leukemiavirusreversetranscriptase. J. Virol.
63:1460-1464.
31. Roth, M., N. Tanese, and S. P. Goff. 1985. Purification and
characterization of murine retroviral reverse transcriptase
ex-pressedin E. coli. J. Biol. Chem. 260:9326-9335.
32. Rowe,W.P.,W. E.Pugh,andJ.W.Hartley.1970.Plaqueassay
techniquesfor murine leukemia viruses.Virology42:1136-1139. 33. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold
Spring
Harbor Laboratory, ColdSpringHarbor, N.Y.34. Schatz, O.,F. Cromme, F. Grueninger-Leitch,andS. F.J. Le Grice. 1989. Point mutations in conserved amino acid residues
within the C-terminal domain of HIV-1 reverse
transcriptase
specificallyrepress RNase Hfunction.FEBSLett.257:311-314. 35. Schatz, O., J. Mous, and S. F. Le Grice. 1990. HIV-1
RT-associated ribonucleaseHdisplaysboth endonucleaseand 3'-5'
exonuclease activity. EMBOJ. 9:1171-1176.
36. Schwartzberg,P.,J.Colicelli,and S.P.Goff. 1984.Construction
andanalysis of deletion mutations in the
pol
gene ofMoloney
murine leukemia virus:a new
proviral
functionrequired
fortheestablishmentofintegrated
provirus.
Cell37:1043-1052.on November 10, 2019 by guest
http://jvi.asm.org/
37. Southern, E. M. 1975. Detection of specific sequences among DNAfragments separated by gel electrophoresis. J. Mol. Biol.
98:503-517.
38. Southern, P. J., and P. Berg. 1982. Transformation of
mamma-lian cells to antibiotic resistance with a bacterial gene under controloftheSV40early region promoter.J.Mol. Appl. Genet.
1:327-341.
39. Starnes, M. C., and Y. C. Cheng. 1989. Human
immunodefi-ciency virusreversetranscriptase-associatedRNase Hactivity.
J. Biol. Chem. 264:7073-7077.
40. Studier, F. W., A. H. Rosenberg, M. N. Simon, and J. J. Dunn.
1979. Genetic and physical mapping of the early region of bacteriophage T7 DNA. J. Mol. Biol. 135:917-937.
41. Tanese, N., and S. P. Goff. 1988. Domain structure of the
Moloney MuLVreverse transcriptase: mutational analysisand separateexpression of the polymerase and RNAseHactivities. Proc. Natl. Acad. Sci. USA85:1777-1781.
42. Tanese, N., M. Roth, and S. P. Goff. 1986.Analysis of retroviral
polgene products with antisera raisedagainst fusion proteins expressed in Escherichia coli.J.Virol. 59:328-340.
43. Tanese, N. T., A. Telesnitsky, and S. P. Goff. 1991. Abortive reversetranscription by mutants ofMoloney murine leukemia
virus deficient inthe reverse transcriptase-associated RNase H
function. J. Virol.65:4387-4397.
44. Telesnitsky, A., and S. P.Goff. Unpublished data.
45. Telesnitsky, A. P. W., and M. J. Chamberlin. 1989. Sequences
linked to prokaryotic promoters can affect the efficiency of
downstream termination sites.J. Mol. Biol. 205:315-330.
46. Temin, H. M., and S. Mizutani. 1970. RNA-directed DNA polymerase in virions of Roussarcomavirus. Nature(London) 226:1211-1213.
47. Tisdale, M., T. Schulze, B. A. Larder, and K. Moelling. 1991.
Mutations within the RNase H domain of human
immunodefi-ciency virustype 1 reversetranscriptase abolish virus infectiv-ity. J.Gen. Virol.72:59-66.
48. Varmus,H.,and P. Brown.1989.Retroviruses,p.53-108. In D.
E.Berg and M. M. Howe (ed.), Mobile DNA. American Society forMicrobiology, Washington D.C.
49. Wigler, M., R.Sweet,G. K.Sim,B.Wold,A.Pellicer,E.Lacy, T. Maniatis, S. Silverstein, and R. Axel. 1979. Transformation of
mammaliancells with genes from procaryotes and eucaryotes. Cell 16:777-785.
50. Wohrl, B. M., and K. Moelling. 1990. Interaction of HIV-1 ribonuclease H with polypurine tract containing RNA-DNA
hybrids. Biochemistry29:10141-10147.
51. Yang, W., W. A. Hendrickson, R. J. Crouch, and Y. Satow. 1990. Structure ofribonucleaseH phasedat2 A resolutionby
MAD analysis of the selenomethionyl protein. Science 249:
1398-1405.
52. Zoller, M. J., and M. Smith. 1982. Oligonucleotide-directed mutagenesis using M13-derivedvectors: anefficient and general procedurefor theproductionofpointmutations in anyfragment of DNA. Nucleic Acids Res. 10:6487-6500.