0022-538X/86/090635-11$02.00/0
Copyright 1986, American Society for Microbiology
Pseudorabies Virus Gene Encoding Glycoprotein glll
Is Not
Essential for Growth in Tissue Culture
ALAN K. ROBBINS,' MARY E. WHEALY,' ROGER J. WATSON,2 AND LYNN W. ENQUIST1*
Central Research & DevelopmentDepartment, E. I. duPont deNemours & Co., Wilmington, Delaware 19898,1and
Imperial
Cancer Research FundLaboratories,
St.Bartholomew's
Hospital,
London ECIA7BE,
England2
Received 19March 1986/Accepted29May 1986
We have established that in the Becker strainof pseudorabies virus (PRV), the glycoproteinglllgeneisnot essential for growth in cell culture. This was accomplished by construction and analysis of viral mutants containing twodefineddeletion mutations affectingthe glll gene. These mutations werefirstconstructed in
vitro and introduced intoEscherichia coli expression plasmidstoverifystructureand protein production.Each mutation was then crossed onto PRV by cotransfection of plasmid DNA and parental viral DNA by using glll-specific monoclonal antibodies as selective and screening reagents. One resultant virus strain, PRV-2, contained anin-frame deletion ofa402-base-pair (bp)Sacl fragmentcontained within theglllgene.Another virus strain, PRV-10, contained adeletion ofa 1,480-bpXhoI fragment removing 230 bpof the upstream,
putative transcriptional controlsequences and 87% ofthe gIll coding sequence. The deletion mutants were
compared withparental virusby analysisof virion DNA,glll specific RNA, and proteins reactingwith glll specificantibodies.Uponinfection of PK15cells, the deletionmutantsdidnotproduceanyproteinsthat reacted with two glll specific monoclonal antibodies. However, twospecies of truncated glycosylated proteinswere observed in PRV-2 infected cells that reacted with antiserum raisedagainst bacterially producedgIll protein. PRV-10 producednodetectablegIll-specificRNAorprotein. PRV-10 could be propagated without difficulty in tissue culture. Virus particles lacking gIII were indistinguishable from parental PRV virus particles by analysisof infected-cell thin sections in the electron microscope.We therefore conclude thatexpressionof the gIll genewas notabsolutelyessential for PRVgrowth intissue culture.
Herpesviruses are large-enveloped-DNA viruses that in-fect a wide variety of organisms (28). One general charac-teristic of the members of this diverse virus group is that they allencodea setof several distinct structuralmembrane glycoproteins localized both in the viral envelope and the membranes of infected cells (26, 28). The function ofthe glycoproteins in the virus life cycle, the control of their synthesis, their transit throughthe protein localization ma-chinery of the cell, and their pivotal role in host-virus interactionareactiveareasof research. Oneareaof interest has been the comparison of glycoprotein genes and gene products from a variety of herpesviruses with the idea of findingshared unitsofstructureandperhapsfunction. Even though considerable diversityexists among the herpesvirus glycoproteins in structureandfunction, some similaritiesin DNAand protein sequence have been identified (9, 21, 24, 28, 29, 31, 34). In this report, we extend ouranalysis of a swine herpesvirus (pseudorabies virus [PRV])glycoprotein that shares significant homologytoglycoproteinC of human herpes simplex virus type 1 (HSV-1) and that of HSV-2.
While considerableprogress has been made in the identi-fication and localization of
glycoprotein
genes fora variety ofherpesviruses (3, 6, 9, 11, 13, 15, 17, 20, 22, 25, 31, 33, 35), the functional and genetic analysis of glycoprotein gene expression has advanced more slowly. One reason is that until recently, few good selection or screening techniques existed for the direct analysis ofglycoproteins. However, recombinant DNA technology and monoclonal antibodies have provided new tools for the analysis of the HSV glycoproteins (10, 14, 19, 20, 28). An example of glycopro-tein geneanalysiswith thesenewtechniquesis thatdone by Hollandetal. (14),usingtheglycoprotein C gene of HSV-1.* Correspondingauthor.
Thesestudiesweresuccessfulbecause the HSV-1gCgeneis notessentialforgrowth intissue culture, and mutant isola-tion was relatively straightforward. As demonstrated by Hollandetal.,monoclonal antibodies provedtobeusefulfor selecting infectious virus carrying a variety of mutations in theHSV-1gCgene.Aswewill show in this report,asimilar approach ispossiblein PRV with theglycoprotein glllgene. We are interested in bringing the combined tools of recombinantDNAtechnologyand monoclonalantibodiesto bear onageneticanalysis of herpesvirus glycoprotein struc-ture and function. We have chosen to concentrate on the swine herpesvirus PRV. PRV encodes atleast five distinct families of structural glycoproteins, as defined by studies with glycoprotein-specific monoclonal antibodies (13). The gene for glycoprotein gI is located in the Us region and is apparently not essentialfor growth in tissue culture (2, 17, 18).TheglycoproteingII genesharessignificanthomologyto HSV glycoprotein gB and has been localized in the UL region at mapposition 0.1 (A. K. Robbins, C. Gold, L. W. Enquist,M. E. Whealy,and R. J.Watson, 10th Int. Herpes-virus Workshop, p. 130, 1985;T. Mettenleiter, H. J. Thiel, N. Lukacs, C. Scheurs, and H. J. Rziha, 10th Int. Herpes-virus Workshop, p. 142, 1985). Inaddition to the envelope glycoproteins,anexcretedglycoproteincalled gX is encoded by a gene in the Us region (22). We have identified aPRV glycoprotein gene at map position0.4and suggested that it encoded the gIII glycoprotein (24, 25). The proof for this assertion is the subjectof this report.
Thefunction ofPRVglycoprotein glll is notwell under-stood; however, Hampl etal. (13)have observed that only monoclonalantibodies directed againstthe
glll
protein neu-tralize PRVin vitroby inhibiting adsorption of the virusto cell surfaces. Monoclonalantibodiestoother PRV glycopro-teins were notneutralizing in that study. In this report, we 635on November 10, 2019 by guest
http://jvi.asm.org/
usethe PRVglll-specific monoclonalantibodies ofHamplet al. (13) to isolate infectious virus containing defined dele-tions affecting gIII expression. This analysis enabled us to conclude that (i) the glycoprotein gene we had identified previouslywas indeed the PRV
gIll
gene, and(ii) like HSV gC, the PRV gIll gene was not absolutely essential for growth in tissue culture.MATERIALS AND METHODS
Cell culture andviruses. Swine kidney cells (PK15) were grownin Dulbecco modified Eagle medium containing 10% heat-inactivated fetal bovine serum as described previously
(25).
The Becker strain ofPRVhas also been described previ-ously (25). PRV mutants derived from DNA transfection
experiments
were plaquepurified
two times before stocks weremadefor characterization. Virusstockswerepreparedby
infection of PK15 cells at a multiplicity of infection between 1 x 10-" and 5 x 10-5 PFU per cell.Monoclonal antibodies and monospecific sera directed against PRV gIlI. The
glll-specific
monoclonal antibodies Ml and M16 were obtained from T. Ben-Porat and were made with the Ka strain of PRV (13). These monoclonal antibodies showed essentially the same reactivity with the Becker strain of PRV (unpublished observations; this pa-per). Oneimportant
feature ofthese monoclonal antibodies is that they reacted poorly with denaturedgIll
protein in Westernblotanalysisandnot atallwithglll
fusionproteins produced in Escherichia coli (data not shown). They do neutralize PRV(Becker and Ka strains) in the presenceor absence ofcomplementanddo immunoprecipitateglll
pro-teins from infected cells (13; this paper). ThegIl-specific
monoclonal antibody M2 was also obtained from T. Ben Porat andwas made with the Ka strain ofPRV (13). This monoclonal antibody reacts similarly with the three
gll
protein
species (apparent molecular weights, 110,000,68,000,
and 55,000) of either the Becker or Ka strains of PRV, further establishingthecloserelationship ofthesetwo strains. 490 serum was raised in rabbits against denatured Cro-PRVglll
protein produced in E. coli from expressionplasmid
pALM27 (previously designatedp7/123AM
[24]).490serum reacts with both native and denatured PRV
glll
protein,
as determined by direct staining of plaques (blackplaque
assay [15, 27]),reactivity
in Western blot analysis, andimmunoprecipitation
of proteins from PRV-infected PK15 cells.Construction of recombinant viruses. PK15 cells were cotransfected with 2 ,ug ofPRV DNA and 1 ,ug of
NcoI-digested pALM2 plasmid
DNA or 1jig
ofPstI-digested
pALM10 plasmid
DNAby
theprocedure
of Graham and Van der Eb (12). After a 4-h incubation at 37°C, the transfected cells were washed with growth medium,sub-jected
to a 3-min shock with 15% glycerol, and washedtwice,
and then fresh growth medium was added. After asubsequent
12-h incubation, guinea pig complement(5%)
and the
glll-specific
monoclonal antibodies Ml and M16(final
concentrationof eachantibody,
0.6,ug/ml)
wereadded. Inthe presenceofcomplement,thesemonoclonalantibodies neutralized the infectivity of parental PRV but did not neutralize theinfectivity
of PRV mutants thateither do not express anyglll protein
orexpress certain altered variants ofgIll.
For this report, it isimportant
to understand that neither Ml nor M16 alone ortogether reacts with theglll
variant protein expressed by PRV2 (402-base-pair [bp] in-frame deletionwithin
glll).
The total virusyield from eachtransfection was harvested when complete cytopathic effect was observed, usually about 36 h. Aliquots were plaqued and screened with Ml and M16 monoclonal antibodies by using the black plaque assay (15, 27). Plaques that did not react with glll-specific antibody were picked and purified twice before analysis.
Mutant virus was named by the number of the pALM plasmid that was used to make it; i.e., PRV-2 contains the deletion carried by pALM2 and PRV-10 carries the deletion specified by pALM10.
Preparation of virions and cell extracts for im-munoprecipitations. The procedure used for virion purifica-tion was as described by Ben-Porat et al. (1). The procedure was modified to prepare[3H]glucosamine-labeled virions by growth in the presence of 100 ,uCi of [3H]glucosamine (D-[6-3H(N)]glucosamine hydrochloride; New England Nu-clear Corp., Boston, Mass.) per ml. The techniques for making cell extracts for immunological analysis were de-scribedpreviously (25).
Northern blotanalysis of viral RNA in infected cells. Total cytoplasmic RNA was extracted from uninfected or PRV infected cells at 16 h postinfection as described by Robbins et al. (24) and fractionated by electrophoresis on a 1% agarose gel containing 2 M formaldehyde, 0.02 M MOPS (morpholinopropane sulfonic acid), 5 mM sodium acetate, and 1 mM EDTA(pH 7) (4, 5). The RNA was subsequently transferred toanitrocellulosemembrane (32) and hybridized at42°C for 16 h with DNA probes labeled with32p, by nick translation (23). After hybridization, the membrane was washed four times for 30 min each at 65°C with 0.25x SSC-0.1% sodium dodecyl sulfate (SDS) (lx SSC contains 0.15 Msodium chlorideand 0.015 Msodium citrate[pH 7.0]) and thenanalyzed by autoradiography.
In vitro translation and immunoprecipitation. In vitro translation of total cytoplasmic RNA was carried out by using the rabbit reticulocyte lysate system obtained from New England Nuclear Corp. asrecommended by the man-ufacturer. The 25-,u reaction mixture contained 2.5 ,ug of totalcytoplasmicRNA andL-[35S]methionine.Before immu-noprecipitation, the reaction mixtures were preadsorbed overnight at 4°C with normalrabbit sera to reduce nonspe-cific reactions. An excess of freshly washed Pansorbin Staphylococcus aureus (Calbiochem Behring, La Jolla, Calif.) was added and incubated for 15 min on ice. The reaction mixture was then clarified by centrifugation for 5 min at 12,000 x g in an Eppendorf microfuge, and the supernatant wasdividedand incubated with theappropriate antibodies. After thisincubation,thesampleswereanalyzed asdescribed by Robbinset al. (24).
Purificationandanalysis of viral DNA. PRVviral DNAwas purified from PRV nucleocapsids as previously described (3). DNA so obtained was analyzed by the Southern blot method (30). DNA-DNA hybridizations were doneat 37°C for16 hin60% formamide-10mMPIPES [piperazine-N,N'-bis(2-ethanesulfonic acid) (pH
6.5)]-5
x SSC-sonicated calf thymusDNA(100 ,ug/ml)-5x Denhardtsolution(0.1% each of Ficoll 400 [Pharmacia, Inc., Piscataway, N.J.], bovine serum albumin, and polyvinylpyrollidone [Sigma Chemical Co., St. Louis, Mo.]). DNA probeswereprepared by nick translation with32P-labeled nucleotides(16, 23). The hybrid-ized nitrocellulose membranes were washed four times for 30 min eachat65°Cin 2x SSC(pH 7.0). Thenitrocellulose membranewasthenprepared forautoradiographyonKodak XAR-5 film.Electron microscopy. PK15 cells were infected with PRV at a multiplicity of 10 PFU per cell. Infected cells were
on November 10, 2019 by guest
http://jvi.asm.org/
UL
IRS
US
rRS
0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
MAP
UNITS
11
3
14,
1025
1
~1
2
9
4
3
16
1
8
71
58k13
*Bam HI
*'*@
DamnL .0000 0.~~~~~~~~~~~~~~~~~~~~~~0 0
00.~ ~ ~ 00
0
1.0
2.0
3.0
4D
4.3
KI
LOBASES
PstI
XhoI
I I
SphI
Xhol
Socl
SocI
Xhol
4
4
4
I
--~~
INcol
NcoI
Bom
HI
5'
MRNA
3,
FIG. 1. Map of the PRVgenome,indicatingtheDNA restrictionfragmentsusedtostudytheglll glycoproteingene.(A) Genome of PRV,
depicting the unique long (UL) and uniqueshort(Us) regionsaswellastheinvertedterminal repeatsequencesbracketingUs(IRSandTRs,
respectively). The genomeis divided intomapunits for reference asindicated. (B)BamHI restriction enzymecleavage map of the PRV
genome.Theglllgeneislocatedwithin thetwoadjacentBamHIfragments,BamHI-2 andBamHI-9.(C) A 4.3-kb PstI fragment containing
theglllgeneisexpanded and detailed,with relevant restrictionenzymecleavagesitesnoted. Symbols: , theglllglycoproteingene,the
sequenceof which isgiven byRobbinsetal. (24);-*,themRNAtranscriptforthisgeneand itsorientation. The datainpanel Caretruefor
both the Becker andKaPRVstrains (datanot shown).
incubated for 10 h, collected bycentrifugation, and fixed in buffered 5% glutaldehyde. The infected cells were further treated with 1% osmium tetroxide and dehydrated in
ethanol. Thepreparationwasthen infiltrated and embedded in Spurr(Electron Microscopy Sciences, Fort Washington, Pa.). Thinsections(50 nm) weredouble stained withuranyl
and lead acetate and analyzed by electron microscopy.
Electronmicroscopyandphotography of sectionswasdone
by Richard Hebert, Central Research and Development
Department, E. I. du PontdeNemours &Co., Inc.
Bacterial strains. E. coli MC1000 has the genotype araDl39 A(araABC-leu)7679galUgalKA(lac)X74 rpsL thi. Strain NF1829 is MC1000 carrying an F' plasmid with the lacP mutation which results in overproduction ofthe Lac
repressor. The F' also carries a Lac operon with a TnS
transposon inlacZ.
Construction of plasmids with deletions of the PRV glll
gene.The PRVglycoproteinglllgenewaslocatedat
approx-imately0.4 onthePRVgenome(Fig. 1). Deletions affecting glllwerefirstconstructed inthe E. coliexpressionplasmid
pALM3 (Fig. 2A). In this plasmid, transcription of the Cro-PRV glll hybrid gene was directed from the tac
pro-moterundercontrol of the Lacrepressor. pALM3produced
significant quantities of a Cro-PRV glll fusion protein that accumulated to approximately 3 to 5% of the total cell protein (Fig. 3). As determined by DNA sequence analysis (datanotshown),theamino-terminal 23 amino acids derived from thebacteriophagelambdaCroproteinwerefusedtothe first amino acid (Met) of glll. In pALM3, the coding
se-quence terminated at the natural translation stop signal (TGA) after amino acid 479, followed by approximately 1,000 bpof PRV sequence. By usingthisexpressionplasmid
Pst
I
NcoI
I
on November 10, 2019 by guest
http://jvi.asm.org/
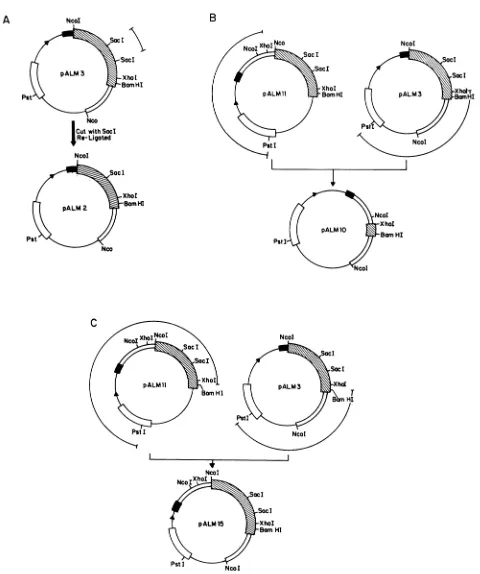
[image:3.612.68.547.72.491.2]A
B
NcoI
Sac!I
XhoI
pALM 2 SamHI
Pst
Nca
NcoI
Xhol
pALM1O Barn HI Pit I
NcI
C
FIG. 2. Construction of plasmids pALM2 (A), pALM10 (B), and pALM15 (C). The plasmid molecules are drawn as circles with the relevantelements and sites indicated. All plasmidsare pBR322 derivatives. Each plasmid has a uniquePstIsite located withintheP-lactamase
gene (L)derivedfrom pBR322. The tacpromoter-operator region 1 is shown, followed by DNA encoding the amino-terminal 23 amino
acidsofthebacteriophagelambdaCro gene (-). The PRVglllglycoprotein gene (g) is shown, with the relevant restriction enzyme sites indicated. The ATG codon in the NcoI site at the junction of Cro andglll defines the firstamino acid of the gIII gene. A double line immediately followingorprecedingthegIllgene indicates contiguous PRV DNA sequences downstream or upstream of theglllgene. DNA fragments removedorligatedtogetherfor plasmidconstruction areindicated as barred lines above the plasmid map. Construction details of eachplasmidareoutlined in Materials andMethods. The figures are not drawn to scale.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.74.554.63.637.2]A
92
--
68-
43-
35-
30-B NcoI SacI Socl
XhoI BomHI
NcoI
ALM 3
NcoI SocI XhoI BBan HI Ncot ALM2
NcoI XhoI BomHI Ncol
ALM 10 H_
FIG. 3. Induction of Cro-PRVgIII fusionproteins from expres-sionplasmids. E. coli NF1829,containing either pALM2, pALM3, orpALM10,wasgrownin L-brothcontaining100 pugofampicillin
per ml.Fusionproteinswereinducedby addition of
isopropyl-P-D-thiogalactopyranoside(final concentration, of1mM).A3-mlsample fromeachinduced culture washarvestedat 4hpostinduction.The Cro-PRVgIII fusion proteins were found primarily in insoluble
aggregates and were prepared as described previously (36). The
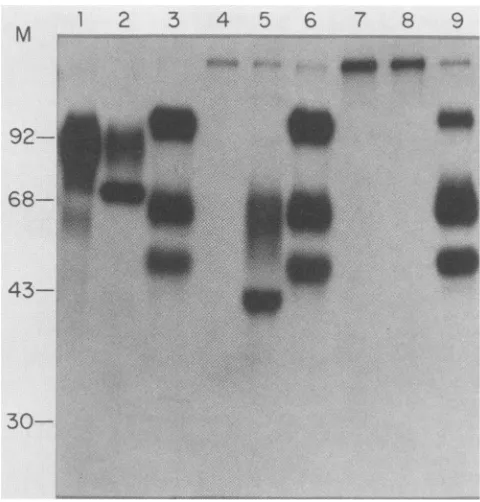
aggregated proteinswerefractionated byelectrophoresison a 10% SDS-polyacrylamide gel and then stained with Coomassie blue. Lane 1, uninduced culture ofpALM3; lanes 2, 3, and 4, induced cultures of pALM3, pALM2, and pALM10, respectively. The positions of the Cro-PRVgIII fusion proteins are indicated by arrows. The positions and molecular masses in kilodaltons of molecularmassstandards runon the samegelareindicated to the leftof the gel. The relevant features ofeachexpressionplasmidare indicated below the gel. Symbolsaredefined inthelegendto Fig. 2.
system, we were able not only to verify the structure and translation products of the mutants before crossing them onto PRV, but also to produce the unglycosylated and unprocessedformsofmutantgIIIfusion proteins in reason-able quantities for use as reagents, e.g., for raising mono-specific, polyvalent antisera.
All plasmids were constructed by standard recombinant DNA techniques (8, 16). The starting plasmid pALM3, described above, was a recombinant of pALM13 and pALM18, both of which were described previously as ptacNB and pK64, respectively (24).
pALM2 (Fig. 2A) contained an in-frame deletion within thegIll gene and produces atruncated fusion protein (Fig. 3). It was constructed by digesting pALM3 with Sacl, religating, and selectinga plasmid that had lost the 402-bp internalSaclfragment. pALM2 containedaunique SacI site defining the 402-bp deletion.
pALM11was needed asan intermediatein the construc-tion of pALM10 and pALM15 (Fig. 2B). Itwasconstructed asfollows:pALM12(formerly p7/123[24])wasdigested with SphI, and the resulting single-stranded ends wereremoved by using the 3'-5' exonuclease activity of the Klenow frag-ment of DNA polymerase I. The DNA was then digested withBamHI, and the 2,500-bp fragmentwasligatedinto the SmaI-BamHI sites ofpHK412 (25).
pALM10 was constructed to contain an XhoI deletion removing mostofthe gIII geneincluding230bpof upstream DNA (Fig. 2B). pALM10 was derived from pALM3 and pALM11 asfollows. DNAfrombothplasmidswasdigested with PstIand XhoI. The 1,840-bpfragment of pALM11 and the 3,670-bp fragment of pALM3 were purified and ligated with T4 DNAligase. Theresultingplasmid,pALM10,lacked all ofthegIllgene exceptfor 186 bp encodingthe carboxy terminus. It also retained the tac promoter-operator and translation elements of thepALM3 expression vector.
pALM15 contained 3,600bp ofPRVDNA, includingthe entire 1,437-bpgIIIcodingsequence (Fig.2C). pALM15isa derivative of pALM3 and pALM11 constructed asfollows. DNA from both plasmids was digested with PstI and BamHI. The 3,100-bp fragment of pALM3 and a 3,400-bp fragment frompALM11 were purified and ligated by using T4 DNA ligase. pALM15 also retained the tac promoter-operatorandtranslationelementsof thepALM3expression vector.
Preparation of Cro-PRV fusion proteins expressed in E. coli. Thetechniques used forproducing andpurifying Cro-PRV fusion proteins expressed in E. coli have been de-scribedpreviously (24, 36).
RESULTS
Analysis of mutant gIll expression plasmids. Two
gIll
deletion mutations wereconstructed in bacterial expression plasmids for ultimateinsertioninto the PRV genome. These mutations included(i)anin-frame deletion ofa402-bp SacI fragment in theglll codingsequence, predicted toresult in production ofatruncated
glll
protein, and(ii) adeletion of a1.48-kilobase(kb)XhoIfragment, removing 87% oftheglll gene, includingtheamino terminus and 230 bp of upstream DNA.This deletionleftbehindthe extreme 186 bpencoding the carboxy terminus ofthe glll gene. The plasmids with these deletions were pALM2 and pALM10, respectively (Fig. 2).A Coomassie-stained SDS-polyacrylamide gel was made of theinsolubleaggregated proteins produced by
isopropyl-,-D-thiogalactopyranoside
induction of cells harboring pALM3(Fig. 3,lane2), pALM2(lane3), andpALM10(lane 4). pALM3 produced a 64-kilodalton (kDa)Cro-PRVgIII
fusionprotein,pALM2producedasmaller fusionprotein (43 kDa), and pALM10 produced no apparent fusion protein uponinduction.Ashas been observedpreviously, there was adiscrepancy between the predicted molecularmass of the fusion
protein
as deduced from the DNA sequence and the observed molecularmasscalculatedfrom migrationin SDS-polyacrylamide gel electrophoresis in that the apparent molecularmassgreater than thepredicted mass (24). This gel artifactwasalso apparentfor proteins extracted fromon November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.55.296.78.450.2]1
2
3
to
23
K
b
-
9.4Kb
-
6.5
K
b
+-4.2Kb
2.3
K
b
1.48
-o1.3
1.1
0.95
-o-2.0Kb
*.
B
Psil XhoI Xhol SacI SocI xhol Pstl
C
phI NcB-r
-I--
t
M
Sphl Ncol NcoI BamHI NcoI
MRNA3-5 MRNA
3.
pALM15
FIG. 4. (A)Southernblotanalysis ofvirion DNAextracted from
parentalPRV (lane 1)and thegIll deletionmutants PRV-2(lane 2)
and PRV-10 (lane 3). VirionDNAwasdigested withPstIandXhoI,
andtheresulting fragmentswereresolved byelectrophoresisina1%
agarosegel andblottedontonitrocelluloseasdescribed in Materials
and Methods. The membrane was hybridized with 32P-labeled
pALM15 DNA and prepared forautoradiography. Thepositions of
molecular size standards areindicated tothe right of the gel, and sizes (in kilobases) of specific PRV fragments hybridizing to the
probeareindicated byarrowstothe leftof thegel. (B)Restriction
map of the 4.3-kbPstI PRVfragment, showing the location of the
gIll gene (SM). The mRNA and direction of transcription are
designated by an arrow below the line. The 3.6-kb SphI-to-NcoI
DNA fragment carried by pALM15 and used as a probe for this
experiment is shown below the map.
infected cells and was further complicated by the 0-and
N-linked glycosylation of the gIll protein.
Constructionandanalysisofinfectious PRVmutants carry-ing deletions of the glll gene. The plasmids pALM2 and
pALM10werecotransfected with PRV DNAasdescribed in
Materials and Methods. Resulting plaques were screened
with the glll-specific monoclonal antibody Ml in the black plaque assay, and white plaques (nonreactive with glll
specific antibody) werepickedandpurified beforeanalysis. Control cotransfections of PRV DNA withnoaddedplasmid or with non-gIll-containing plasmids never yielded white plaques (data not shown). The frequency of spontaneous monoclonal antibody-resistant mutants was less than 10-underourconditions.
Awhiteplaquewasisolated and purified from thepALM2 transfection, and one was likewise obtained from the pALM10 transfection. These mutant viruses were
desig-nated PRV-2 and PRV-10, respectively.
By analyzing the viral DNA, we determined that the deletion constructed in each plasmid had indeed been crossed ontothe virus(Fig. 4). For these experiments, viral DNA wasisolatedfrom nucleocapsids of parental PRV and the gIll-defective mutants PRV-2 and PRV-10. The DNA wasdigested with PstI and XhoI, fractionatedon an agarose gel, transferred to nitrocellulose, and hybridized with a 32P-labeled pALM15 probe. The gIII gene was located approximately in thecenterofa4.3-kb PstI fragment (Fig. 1 and 4). There were three XhoI sites in this PstI fragment, two upstream of the gene and one within the gIll coding sequence near the carboxy terminus. The 1.48-kb XhoI fragment defined by the site 230 bp upstream of the gIll gene and the site within the gIll gene was the critical fragment for this analysis. All DNAsamples weredigested with PstI and XhoI. When pALM15 was used as a probe, the patterns shown in Fig. 4 were observed. The parental pattern is shown in lane 1. Note the twofragments of 1.3 and 0.95 kb and the 1.48-kb internal XhoI fragment. In PRV-2, which should contain a 402-bp deletion within the glIl gene, the 1.3-and 0.95-kb fragments were unchanged, but the 1.48-kb internal XhoI fragment now migrated as a 1.1-kb fragment (Fig. 4, lane 2). Similarly, in PRV-10, which should contain aprecise deletion of the 1.48-kbinternal XhoIfragment, only the 1.3- and 0.95-kb fragments were seen (lane 3). In other analyses with Sacl, we found that PRV-2 contained a precise deletion of the 402-bp internal gIII Sacl fragment, leavinga single SacI site in the truncated gIII gene (data not shown). Thus, the two site specific deletions constructed in vitro were transferred precisely by recombination to PRV.
gIII-specific RNA in cells infected with PRV gIIl deletion mutants. Cytoplasmic RNA transcribed from the gIll gene was analyzed by using the Northern blot technique as described in Materials and Methods. The probe was nick-translated pALM15, which contained the entire gIll geneas well as 1.2 kb of upstream and 1.0 kb of downstream PRV DNA (Fig. 5). Cytoplasmic RNAwas extracted from PK15 cells infected with parental PRV, PRV-2, and PRV-10 and also fromuninfected PK15 cells as acontrol. No hybridiza-tion was observed with control RNAfrom uninfected cells (Fig. 5, lane 1). Asreported byRobbinsetal. (24), a 1.55-kb gIll specific mRNA was observed fromparental PRV infec-tion(Fig. 5, lane 2).Theotherminorbandscorrespondedto PRV-specific RNAs outside the gIll gene and served as internal controls. The proof that the 1.55-kb RNA was indeed gIll specific and that the minor RNAs were not was established by the RNA produced by the gIll deletions PRV-2 and PRV-10, which hybridized to the probe (Fig. 5, lanes 3 and 4). PRV-2 carried a 402-bp deletion within the gIIIgene,and, aspredicted, the 1.55-kb messagewasabsent and replaced by a 1.15-kb transcript (Fig. 5, lane 4). The minor RNA species were unchanged. Similarly, PRV-10 produced no 1.55-kb transcript, as predicted, since the 1.48-kb Xhol deletion removed 87% of the gIll coding sequence as well as 230bp upstream, presumably encoding at least the gIll promoter (Fig. 5). In this case, the minor
A
0
4-10's
IVC
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.62.299.71.497.2]1
2
3
4 5
6
M
-idbi
_I1''A:*
1.75
..
W
1..
,.
4.
B
Psti XhoI XhoI Socl SacI Xhol PstI
Sphl NcoI NcoI BCIM HI NcoI
5 MRNA 3
pALM15
-68
-46
FIG. 6. In vitro translation ofRNA extracted fromPK15 cells infected withparentalPRV ortheglll deletionmutantPRV-2.Total cytoplasmic RNA was isolated from uninfected cells or cells in-fected withparental PRV orthegIll deletion mutant, PRV-2. The
RNA was translated in a rabbit reticulocyte system, and the
products were immunoprecipitated with490 serum asdescribed in MaterialsandMethods.Immunecomplexes collectedbyadsorption toPansorbinS. aureuscellswerefractionatedbyelectrophoresison a10%SDS-polyacrylamide gel, and 35S-labeled polypeptideswere
detectedbyfluorography.Lanes1,2, and 3,gIll-specifictranslation
productsimmunoprecipitatedwith 490 serumfrom uninfectedRNA
andfrom RNAsfrom cellsinfected with PRV andPRV-2,
respec-tively; lanes 4, 5, and 6, total translation products ofRNA from uninfected, PRV-infected and PRV-2-infected cells, respectively.
Arrows indicatetheglll-specific translation products. Positionsof
molecularmass standards (shown in kilodaltons)areindicated.
FIG. 5. (A) Northern blot analysis of glll-specific RNA from
PK15cellsinfected withparentalPRVand thegllldeletionmutants
PRV-2 and PRV-10. Total cytoplasmic RNA was extracted from
uninfected cells (lane 1) orfrom cells infected with parental PRV
(lane 2)orthegllldeletionmutantsPRV-2(lane 4) and PRV-10 (lane
3)at16 hpostinfection. EqualamountsofRNAwerefractionatedon
anagarose-formaldehyde gel and subsequently transferredto
nitro-cellulose. Theblot was thenhybridizedwith 32P-labeled pALM15
DNA. The hybridized membranes were washed and prepared for
autoradiography. The positions of the RNA species exhibiting
hybridizationtotheprobeareindicated. Thetwoglll-specificRNA
speciesseenin lanes2 and 4areindicatedbyarrowstotheleft of the
gel. Other minor, non-glll-specific RNA species areindicated by
arrowsto theright of the gel. RNA sizeswereestimated by using
16S, 18S, 23S, and 28S ribosomal RNA moleculesas standards.(B)
Restriction map of the 4.3-kb PstI PRV fragment, provided for
reference. Symbolsaredescribed inthelegendtoFig. 4.
RNA species (1.75 and 1kb) remainedconstantinquantity and size, but two new species of 2.3 and 1.3 kb were observed thatmust haveresultedfrom the XhoI deletion. It islikely that these minor species originated from sequences
upstreamof thegIIIgene. Thetworemainingminorspecies
thatwere unaltered musthave been encoded by sequences
outside the XhoI deletion and downstream of the glllgene.
Invitrotranslation of late RNA isolated from cells infected withPRVdeletionmutants.PRVgIll-specific primary trans-lation products were identified by immunoprecipitation of
proteins made by in vitro translation of total cytoplasmic RNAasdescribed in Materials andMethods. The RNAwas isolated from cells 16 h after infection with either parental PRVorPRV-2carryingtheSacl gllldeletion(seeMaterials and Methods). The translation products were im-munoprecipitated with 490 serum and analyzed on a 10%
SDS-polyacrylamide gel (Fig. 6). Although the predicted molecularmassof the PRVglllprimarytranslationproduct is approximately 51 kDa(24), the apparent molecular mass oftheinvitro translationproductwas57kDa(Fig. 6,lane2). PRV-2 carriedthe402-bpSacl deletionwhich ispredictedto result in a deletion of 134 amino acids that should yield a truncated protein of 37 kDa. The apparent molecular mass was44 kDa(Fig. 6, lane3). These experiments corroborate the datashown in Fig. 3 withbacterially produced Cro-glIl fusion proteins, in which the apparent molecular mass was greaterthanpredicted. Weconclude that the alteredmobility
A
1
2
3
4
1.55
-1.15 -*
0
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.58.294.74.496.2] [image:7.612.325.549.76.342.2]642 ROBBINS ET AL.
1
2
3
4
5
6
7
8
9
M
...,-:.4.:1w~.
..,-.~..-.~.,.-l,.:.or
....
Oiiit.-a
":.4. -l -.._._
'1r,7...
1-...w!"
u.
... .. :-....*: .:
_
r
_
92-
68-43
.. ..
:.
.:
I
30-_f.:_V:StA___Je:...:<e.v...
FIG. 7. Immunoprecipitation ofPRV-specific proteins from in-fected PK15 cells. PK15 cells were infected with either parental
PRVor oneof thegIll deletionmutants, PRV-2orPRV-10, in the presence of[3H]glucosamine as described in Materials and
Meth-ods. Immunoprecipitated proteins are shown from parental PRV
infections(lanes 1, 2,and 3), PRV-2infections (lanes4, 5,and6)and
PRV-10infections(lanes 7, 8, and 9).Cellextractswerereactedwith
either Ml monoclonal antibody(lanes1, 4,and7), 490serum(lanes
2, 5, and 8),orM2monoclonal antibody(lanes 3, 6,and 9). Immune complexeswere collected byadsorptiontoPansorbin(StaphA)and fractionated by electrophoresis on a10%SDS-polyacrylamide gel.
3H-labeled polypeptides were detected by fluorography. The
posi-tions of molecularmass standards (in kilodaltons) areindicated.
of gIll peptides in SDS-polyacrylamide gels was due to structural features imposed by the primary protein
se-quence.
Immunoprecipitation of proteins produced by glll deletion
mutants. Weanalyzed thegIll-specificproteins produced by parental PRV and the two PRV gIll deletion mutants by immunoprecipitation. Two reagents were critical for this
analysis. One was the monospecific, polyclonal 490 serum made against bacterially produced Cro-PRV gIII fusion protein (see Materials and Methods and reference 24). The other was the PRV gIll-specific monoclonal antibody Ml
(13). Since deletions PRV-2 and PRV-10were both selected
by their inability to reactwith the Ml and M16 monoclonal antibodies,wedidnot expectthemtoimmunoprecipitate the
mutantgIll proteins frominfected cells. On the otherhand,
since the polyvalent 490 serum reacts with denatured and nativegIll protein (24), weexpected it toreactwith altered variants ofgIllthatdidnot reactwith MlandM16.Thedata shown below substantiate thisexpectation.
PK15 cells were infected with parental PRV or with
deletion mutant PRV-2 or PRV-10, labeled with
[3H]glu-cosamine for 16 h, and immunoprecipitated as described previously (24, 25). The gIl-specific monoclonal antibody M2 was included as a control. Ml monoclonal antibody
specifically immunoprecipitatedacharacteristic diffuse band ofglycosylatedgIll proteinatanapparentmolecularmassof
about 92 kDa (Fig. 7, lane 1). The 490 serum
im-munoprecipitated the same material plus a sharper band at an apparent molecular mass of 74 kDa (Fig. 7, lane 2) that presumably represented a gIll protein of a different glycosylation state (perhaps a precursor form). A similar observation was reported for HSV-1 gC (7). Alternatively, the 74-kDa protein may represent a major and specific degradation product of the 92-kDa protein produced during preparation of the cell extracts. Some support for the former hypothesis was obtained from a similar analysis of PRV-2 infected cells (Fig. 7, lanes 4, 5, and 6). PRV-2 is predicted to produce a truncated gIll protein due to the420-bp internal deletion. The Ml monoclonal antibody used in selection showed no specific reaction (Fig. 7, lane 4), but the 490 serum immunoprecipitated two truncated forms of gIll at apparent molecular masses of about 68 (diffuse) and 43 (sharp) kDa (lane 5). The relationship of the two truncated species ofgIll produced by PRV-2 correspondedwell to that ofthe 92- and 74-kDaforms seen for authentic gIll produced after infectionwith parental PRV. These results were more consistent with a precursor-product relationship than with degradation.
Extracts made from PRV-10-infected cells showed no evidence ofimmunoreactivity witheither Ml or 490antisera (Fig. 7, lanes 7 and 8). Similar results were obtained by using the M16 monoclonal antibody (data not shown). The M2 monoclonalantibody controlindicated that the gIl glycopro-tein family was unchanged in eitherdeletion mutant(Fig. 7, lanes 3, 6, and 9). This control indicates that infection and labeling of glycoproteins must have been as efficient as parental virus since equal volumes of infected cell extracts and not equal counts were analyzed.
The experiments shown in Fig. 7 were done with
[3H]glucosamine
labeling. In similar experiments with [35S]methionine and[3H]proline
labels, essentially identical results were obtained (data not shown). Inaddition, results obtained inexperiments with the M16 gIII-specific monoclo-nalantibody wereindistinguishablefrom those obtained with the Ml monoclonal antibody (data not shown).We conclude the following. (i) Late in infection, PRV-2 expressed atleast two glycosylated and truncated forms of gIll consistentwith the 402-bp Sacl deletionintroduced into the gIll gene. Moreover, these two truncated species ap-peared with theconcomitantdisappearance ofthe twoforms of parental gIll (92 and 74kDa). (ii)The deletion carriedby PRV-10 effectively abolished expression of any gIll im-munoreactiveprotein.
Growth ofPRVgIll mutants. The lack of thegIll genein PRV-10 was not entirely without effect since we routinely observed that titers of mutant stocks prepared by low-multiplicity infection were variable and could be 10- to 20-fold lower than comparably prepared parental virus stocks. This result was unexpected since measurements of mRNA (Fig. 5) and the gIl glycoprotein family (Fig. 7) showedessentially nodifferences,other than thoseexpected for the gIll gene, when compared with those of parental PRV. Moreover, in DNAtransfection experiments,parental PRV, PRV-2, and PRV-10 were indistinguishable, giving about 3,000 plaques per
pLg
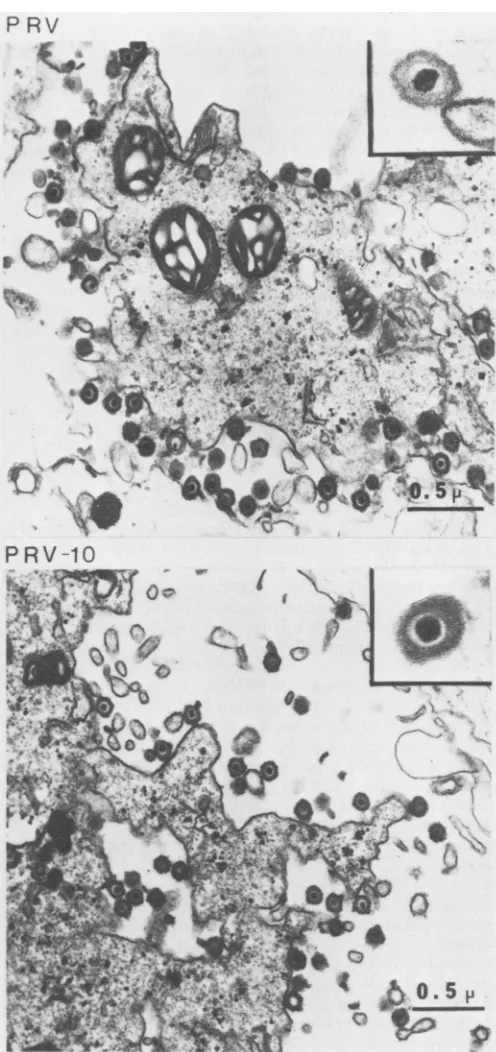
ofinput viral DNA. Finally, in high-multiplicity infections (10 PFU per cell), cytopathic effect was seen at approximately the same time and to the same extent for gIll-defective mutants and parental virus.Virus particles in cells infected with either parental PRV or PRV-10 were visualized in an electron microscope
(Fig.
8). At 10 h postinfection, infected cells were harvested and prepared for electron microscopy as described in Materials and Methods. Typical features of PRV-infected cells were J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.62.302.73.324.2]seen for both virus infections, notably enveloped virions outsidethecellneartheplasma membrane. Asenlargement of a single-enveloped virion of either parental PRV or PRV-10 is also shown in Fig. 8. There was no obvious difference in the morphology of virus particles nor in the features of the infected cells.
DISCUSSION
Inthisreport we describe the construction and character-ization oftwodefineddeletions ofthe PRVgIIIglycoprotein gene. This analysis enabled us to conclude that (i) the glycoprotein gene we had previously identified and which, we suggested, encodedglycoproteingIll (24, 25)wasindeed the PRV gIll gene, and (ii) like its related homolog, glyco-protein C of HSV-1 and HSV-2, the PRVgIIIgene wasnot absolutely essential for growth invitro.
Theidentification oftheglycoproteingene at mapposition 0.4asgIII(Fig. 1) dependedontwospecificreagents: (i)the monoclonal antibodies MI and M16, characterized by
Hampl
etal. (13), which defined the gIIIfamilyof glycopro-teins, and (ii) themonospecific,
polyvalent490serummadeagainst
theputative Cro-glll
fusionprotein expressed
in E. coli. Since theglll-specific
monoclonal antibodies reactedpoorly
with denatured authenticglll,
proteins
madeby
in vitro translation, and bacterially producedproteins
thought to begIII, the directidentification oftheglllgene was notstraightforward.
However, in this report we confirmed its identity by constructing specific deletions that predictably altered the putativegIll
gene and gene product and thendemonstrating
that we could cross these mutations onto PRVby selectingandscreening for recombinants resistantto gIII-specific monoclonal antibodies. With these defined de-letionmutants, itwaspossibletocomparegIIIspecificgene productsinparentaland mutantvirus infectionsaswellas to determine the behavior oftheprimary translation productsby using
in vitro translation.Although the predicted amino acid sequence of
glll
sug-gested
about20%homology
tothe nonessential HSV-1 and HSV-2 gC glycoprotein (24), we had no other reason to believe thatglll
would besimilarily expendable
for PRV. Therefore,ourinitialconcern was thatthesedeletions could be lethalwhenrecombined backontoPRV.Forthatreason,defined, easily
verifiable deletionswereabsolutely essential, because inusing
thegIII-specific
monoclonal antibodies in theselectionandscreening,wehad toavoidsimply selecting spontaneousgIll
mutations that did not react with the monoclonal antibodies. It would be highly unlikely that either thespecific
Sacl orXhoI deletion would arise by a spontaneousevent.The resultswereclear. Both deletionswerecrossedeasily onto PRV, and,
indeed,
the resulting viral mutants were nonreactive withglll-specific
monoclonal antibodies. More-over, thedeletionspermitted unambiguous assignmentof the 92-and 74-kDafamily of glycosylated proteinsasproducts of the same gene because both were altered accordinglyto 68 and 43 kDa by theinternalglll
deletion carriedby PRV-2. Thisentiregroup ofproteinswasnotmadeby the essentially complete gIII deletion carried by PRV-10.Previously,
glll
was reportedas avirionenvelope protein of 98 kDa (13); however, by using our gel system and standards, wecalculated the apparent molecular mass to be 92 kDa. As did Hampl et al. (13), we found the 92-kDa species to be virion associated. However, we found the 74-kDagIII protein only in infected cells (data not shown). We favor the hypothesis that the 74-kDagIII species wasPRV-10
4 ~ . N.---w-41 7.4q a,M a. .f E
FIG. 8. Electron micrographs of infected cells. PK15 cells
in-fected withparentalPRV andPRV-10deleted for theglllgene were
prepared for electron microscopy as outlined in Materials and Methods. Micrographs are shown ofcells infected with parental PRV(top) and PRVcarryingadeletion ofthe glllgene (bottom).
The inset(right-handtopcorner)in each of the micrographs shows
a x80,000magnification ofarepresentative virionfrom the micro-graph. A 0.5-,um size marker is indicatedin the lowerright-hand
cornerofeach micrograph.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.612.311.559.96.622.2]either a precursor form of the 92-kDa protein or a form of gIll destined for a cellular location other than the virus envelope. A similar situation of two distinct glycosylated proteinsis seen for the related gC protein of HSV-1 (7).
Itis interesting that the internal Sacl deletion carried by PRV-2 removed five of the eight potential N-linked glycosylation sites predicted from the DNA sequence (24). However, it appears that both the 68- and 43-kDaforms were labeled with glucosamine to approximately the same extent as were the 92- and 74-kDa forms of glll in a parental infection (Fig. 7). The extent and function of N-linked and 0-linked glycosylation are currently under investigation.
Little is known concerning the role of glll in the infectious cycle of PRV. Hampl et al. (13) have demonstrated that, of themonoclonalantibodies to PRV glycoproteins tested, only those directed against glll prevent infectivity; antibodies against the other glycoproteins are ineffective. Since we wereable to delete thegIII gene and transfer this deletion to infectious virus, it is unlikely that the gIII gene is primarily responsible for adsorption and infection of cells in tissue culture. However, it should be noted that the absence ofglll was notwithout effect. PRV-10 stocks were variable in titer andoften 10- to 20-fold lower than parental virus stocks. At this time it is not clear whether this result is due to the absenceofglllprotein or toeffects of the deletion in PRV-10 on upstream PRV DNA. Complementation experiments must be done to determine whether this effect can be compensated by supplying agood glll gene.
We have observed that the truncated gIII protein ex-pressedby PRV-2 is efficiently inserted into the virus enve-lope (M. E. Whealy, A. K. Robbins, and L. W. Enquist, manuscript in preparation). Therefore, it is apparent that PRV can exist as anominally normal infectious virusin cell culture withnogIII(PRV-10) or withatruncatedform ofglll in the virus envelope (PRV-2).
Ben-Porat et al. (2) have shown that the Bartha and Norden vaccine strains of PRV have deletions in the Us region, removing the coding sequences forglycoprotein gI. These gI-defective viruses grew well in culture but were defectivein the releaseof virions from certaincell types. We have not been exhaustive in oursearch for defects in theglll deletion mutants. Further studies are in progress to test these viral mutants for virus release and host range in cell culture as well as pathogenicity in animals.
As afinalpoint, we have usedE. coliplasmid expression vectorsasintermediates in theconstructionofviralmutants to prepare in quantitythe mutantprotein ofinterest devoid of eucaryotic posttranscriptional modifications. The 490 serumused in this paper isanexampleof suchareagent.We havefound that such proteins aregood immunogens forthe preparationofmonospecific,polyvalentsera(24, 25,36; this report).Since thesebacterially produced fusionproteins are generally denatured wheninjected intoanimals,the antibod-ies produced often react with not only authentic native viral proteins, but also with denatured viral protein precursors andproducts in Western blot analyses (25, 36).
ACKNOWLEDGMENTS
We thank T. Ben-Porat for gifts of monoclonal antibodies and
advice. T. Silhavy provided MC1000 and NF1829. We thank Walt Hagelstein and Dorothy Tinker for excellentgraphicartservice.We areindebtedtoI.P.O'Friel for thesuggestionthat theSacl sites in
gIll were in phase. Electron microscopy was done by Richard
Hebert of theE.I.Dupontde Nemours& Co.Central Research and
Development Department. We also thankGusCambanesfor
excel-lent photography.
LITERATURE CITED
1. Ben-Porat, T., J. M. DeMarchi, and A. S. Kaplan. 1974.
Char-acterization of defectiveinterfering viral particles present ina
populationofpseudorabies virions. Virology 60:29-37.
2. Ben-Porat, T., J.DeMarchi, J.Pendrys, R. A. Veach, and A. S.
Kaplan. 1986. Proteins specified by the short unique region of
the genome of pseudorabiesvirus play a role in the release of
virions from certain cells. J. Virol. 57:191-196.
3. Ben-Porat, T., and A.S. Kaplan. 1970. Synthesisof proteinsin
cellsinfectedwith herpesviruses. V.Viralglycoproteins.
Virol-ogy41:265-273.
4. Berk, A. J., and P. A.Sharp.1977.Sizingand mappingofearly
adenovirus mRNA by gel electrophoresis of S1 endonuclease
digested hybrids. Cell 12:721-732.
5. Berk, A. J., and P. A. Sharp. 1978.Structure of theadenovirus
2early mRNAs. Cell. 14:695-711.
6. Bzik, D. J., B. A. Fox, N. A. DeLuca, and S. Person. 1984.
Nucleotide sequence specifying the glycoproteingene, gB, of
herpessimplex virus type 1. Virology 133:301-314.
7. Cohen, G.H., D. Long, and R. J. Eisenberg. 1980.Synthesisand
processing ofglycoproteins gD and gC of herpes simplex virus
type 1.J. Virol.36:429-439.
8. Denniston, K. S., M. J. Madden, L. W. Enquist, and G.
Vandewoude. 1981. Characterization ofcoliphage lambda
hy-bridscarryingDNAfragments from herpes simplex virus type I
defective interferingparticles. Gene 15:365-318.
9. Dowbenko,D. J., and L. A. Lasky. 1984. Extensive homology
between the herpes simplex virus type 2 glycoprotein F gene
and the herpes simplex virus type 1 glycoprotein C gene. J.
Virol. 52:154-163.
10. Eisenberg,R. J., D. Long, M. Ponce de Leon, J. T. Matthews,
P. G.Spear, M. G. Gibson, L. A. Lasky, P. Berman, E. Golub,
and G. H. Cohen. 1985. Localization of epitopes of herpes
simplexvirus type 1 glycoprotein D. J. Virol. 53:634-644.
11. Frink, R. J., R.Eisenberg, G. Cohen, and E. K. Wagner. 1983.
Detailedanalysis oftheportionof the herpessimplexvirustype
1 genome encodingglycoprotein C. J. Virol.45:634-647.
12. Graham, F. L., and A. S. Van derEb. 1973. A newtechnique for
the assay of infection of human adenovirus 5 DNA. Virology
52:456-467.
13. Hampl, H., T. Ben-Porat, L.Ehrlicher, K.-O. Habermehl, and
A. S. Kaplan. 1984.Characterization of the envelopeproteinsof
pseudorabies virus. J. Virol. 52:583-590.
14. Holland, T. C., F. L. Homa, S. D. Marlin, M. Levine, and J.
Glorioso. 1984. Herpes simplex virus type 1 glycoprotein
C-negative mutants exhibit multiplephenotypes, including
secre-tion oftruncated glycoproteins. J. Virol. 52:566-574.
15. Holland, T. C., R. M. Sandri-Goldin, L. E. Holland, S. D.
Marlin, M. Levine, and J. C. Glorioso. 1983. Physical mapping
of the mutation in an antigenic variant of herpessimplex virus
type 1 by use of an immunoreactive plaque assay. J. Virol.
46:649-652.
16. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular
cloning: alaboratory manual. ColdSpringHarborLaboratory,
Cold SpringHarbor, N.Y.
17. Mettenleiter, T. C., N.Lukacs, and H.-J. Rziha. 1985. Mapping
ofthestructural gene ofpseudorabies virusglycoproteinA and
identification of the two non-glycosylated precursor poly-peptides. J. Virol. 53:52-57.
18. Mettenleiter, T. C., N. Lukacs, and H.-J. Rziha. 1985.
Pseudorabies virus avirulent strains fail to express a major
glycoprotein. J. Virol. 56:307-311.
19. Pancake, B. A., D. P. Aschman, and P. A. Schaffer. 1983.
Genetic and phenotypic analysis of herpes simplex type 1
mutants conditionally resistant to immune cytolysis. J. Virol.
47:568-585.
20. Pellett, P. E., K. G. Kousoulas, L. Pereira, and B. Roizman.
1985. Anatomyoftheherpessimplexvirus 1 strain F
glycopro-tein B gene: primary sequence and predicted protein structure
of the wildtype andofmonoclonal antibody-resistantmutants.
J. Virol. 53:243-253.
21. Pellett, P. E., M. D. Biggin, B.Barrell, and B.Roizman. 1985.
on November 10, 2019 by guest
http://jvi.asm.org/
Epstein-Barr virus genome may encode a protein showing
significant amino acid andpredicted secondary structure
homol-ogy with glycoprotein B of herpes simplex virus 1. J. Virol.
56:807-813.
22. Rea, T. J., J. G. Timmins, G. W. Long, and L. E. Post. 1985.
Mappingand sequence of the gene for the pseudorabies virus
glycoproteinwhich accumulates in the medium of infected cells.
J. Virol. 54:21-29.
23. Rigby, P. W. J., M. Dieckmann, C. Rhodes, and P. Berg. 1977.
Labelingdeoxyribonucleicacid tohighspecific activity invitro
by nick translation with DNA polymerase I. J. Mol. Biol.
113:237-251.
24. Robbins, A. K., R. J. Watson, M. E.Whealy, W. W. Hays, and
L. W. Enquist. 1986. Characterization of a pseudorabiesvirus
glycoprotein gene withhomology to herpessimplex virus type1
and type 2glycoprotein C. J. Virol. 58:339-347.
25. Robbins, A. K., J. H. Weis, L. W.Enquist, and R. J. Watson.
1984. Construction of E. coli expression plasmid libraries:
localization ofapseudorabies virus glycoproteingene. J. Mol.
Appl. Gen. 2:485-496.
26. Roizman, B., L. E. Carmichael, F.Deinhardt, G. de The, A. J.
Nahmias, W. Plowright, F. Rapp, P. Sheldrick, M. Takahashi,
and K. Wolf. 1981. Herpesviridae: definition, provisional
no-menclatureand taxonomy. Intervirology 16:201-217.
27. Smith, K. O., W. L. Kennel, and D. L. Lamm. 1981.
Visualiza-tionof minute centers of viral infection in unfixed cell cultures
by an enzyme linked antibody assay. J. Immunol. Methods
40:297-305.
28. Spear, P. G. 1984. Glycoproteins specified by herpes simplex
viruses, p. 315-356. In B. Roizman (ed.). The herpesviruses.
PlenumPublishingCorp., NewYork.
29. Snowden, B. W., and I. W. Halliburton. 1985. Identification of
cross-reacting glycoproteins of four herpesviruses by western
blotting. J. Gen. Virol. 66:2039-2044.
30. Southern, E. M. 1975. Detection ofspecific sequences among
DNAfragments separated bygel electrophoresis. J.Mol. Biol.
98:503-517.
31. Swain, M. A., R. W. Peet, and D. A. Galloway. 1985.
Charac-terization of the gene encoding herpes simplex virus type 2
glycoprotein C and comparisonwith the type 1 counterpart. J.
Virol. 53:561-569.
32. Thomas, P. S. 1980.Hybridization of denatured RNA and small
fragments transferred to nitrocellulose. Proc. Natl. Acad. Sci. USA 77:5201-5205.
33. Wathan, M. W.,and L. M. K. Wathan. 1984. Isolation,
charac-terization,andphysicalmapping of a pseudorabies virus mutant containing antigenically altered gpSO.J. Virol.51:57-62.
34. Watson, R. J. 1983. DNA sequenceof the herpes simplex virus
type2glycoproteinD gene. Gene 26:307-312.
35. Watson, R. J., J. H. Weis, J. S. Salstrom, and L. W. Enquist.
1982. Herpes simplex virus type 1glycoprotein Dgene:
nucle-otide sequence and expression in Escherichia coli. Science
218:381-384.
36. Watson, R. J., J. H. Weis, J. S. Salstrom, and L. W. Enquist.
1984. Bacterialsynthesis of herpes simplex virustypes 1and 2
glycoproteinDantigens. J. Invest. Derm. 83:102-111.