0022-538X/89/051951-08$02.00/0
Copyright © 1989, American Society for Microbiology
Modified Model for the Switch from
Sendai Virus
Transcription
to
Replication
SILVIA VIDALAND DANIELKOLAKOFSKY*
Department of Microbiology, Geneva MedicalSchool, CMU, CH1211 Geneva, Switzerland
Received 19August1988/Accepted 10 January 1989
RNasemappingwasusedtoestimate the levels of unencapsidatedSendai virusplus-strand RNAswhichcross
theleader-NPjunction relativetoNPmRNA.Significantamountsof leaderreadthroughRNAswerefound in
Zstrain-infected cells,similartothatdescribed forthe poiRmutantofvesicular stomatitisvirus, eventhough
this strain isconsidered wildtype.The levels ofthereadthroughRNAsdetected fellsharply when progressively longer probeswereused, unlike thatofNPmRNA. These studiessuggestthat polymerases which readthrough
the firstjunction terminate shortly afterwards in the absence of concurrent assembly ofthe nascentchain, whereas those which reinitiate at NP continue efficiently to the next junction. Reinitiation appears to be
necessary toconvert the polymerase toa mode inwhichelongation is independent ofconcurrentassembly. Concurrent assembly appears to be required not only for the polymerase to read
thro'ugh
the junctionefficiently, butalso for it tocontinueelongationbetweenjunctions. Sendai virus, amember of the Paramyxoviridae, contains
a nonsegmented minus-strand RNA genome of 15.3 kilo-bases (kb), from which a leader RNAand six mRNAs are
transcribed. The viral polymerase responsible for mRNA synthesis,referredtoasthetranscriptase,isthoughtto enter its nucleocapsid (NC) templateat or. nearthe 3' end andto sequentially synthesize the leader and the NP, P/C, M, F, HN, and L mRNAsby terminating and restartingateach of thegenejunctions. The mRNAsarecappedand
polyadeny-latedduring synthesis, whereas theleaderRNAis unmodi-fied at either end. The minus-strand genome is also a
templatefor thesynthesisofantigenomes,whichare unmod-ifiedfull-length complements ofthegenome whichserve as
intermediates in genome replication. The polymerase
re-sponsibleforantigenomesynthesis,whichwerefertoasthe
replicase, therefore ignores thejunctional stop-start signals that the transcriptase responds to (fora recent review, see
reference 16).
The manner in which this switch from transcription to replicationtakesplace for paramyxoviruses, acentralevent inthe lifecycle ofallminus-strandRNAviruses,isthought tobe similartothatproposed forthe rhabdovirus vesicular stomatitis virus (VSV) (1, 2, 5, 7, 15). Since, like VSV, Sendai virus genomes and antigenomes are found only encapsidated with the nucleocapsid protein (NP) and their synthesis is dependent on on-going protein synthesis
whereas that of mRNA is not, the switch may also be
effected here simply by the intracellular concentration of unassembled NP. In this minimal model, the template and the polymerase for mRNA and antigenome synthesis are
identical; the difference lies in whether the nascent RNA chainisbeing concurrentlyassembledinto NCs.Concurrent assemblywouldsomehow beresponsiblefor thepolymerase
ignoringthejunctional signals, all thatisrequired to switch itfromatranscriptase into areplicase.
The sitefor theinitiation of NC assemblyonVSVleader
RNAhas been mapped tothefirst 14 bases at the 5' end of the chain(7). Polymeraseswhosenascentleaderchainshad notassembled with NPbefore the firstjunctionwasreached
wouldterminate andthen restart atthe beginning ofNPto
* Correspondingauthor.
make all the mRNAs. All junctional signals would be ob-served,as noneof thenascentmRNAs could assembleNP. Polymeraseswhosenascentleader RNAs hadbegun
assem-bly wouldnotobserveanyofthejunctional signals,because
NC assemblywouldfollow thepolymerase, resultingin the synthesis ofa full-length and simultaneously encapsidated
plus-strand RNA. The frequency of genome replication would then bedetermined bythe level of unassembled NP, and the switch would. be part ofa self-regulatory system controllingthe relativeamountsoftranscriptionand replica-tion. We refertothisschemeasthe minimalmodel',since the
switch is effectedentir'elybyunassembledNP,theonlyviral proteinknowntobe requiredfor replicationthat is not also required for transcription.
Theorganizationof the Sendaivirus and VSVgenomesis
also quite similar, including the nature of the junctional signalsfortranscription (11-13). Wewould therefore expect that the minimalmodelwould alsoapplytoSendaivirus,at least in broad outline, even though several of the
experi-mentswhich ledtothemodel havesofaronlybeenreported for VSV. Paramyxoviruses such as Sendaivirus, however, have an overlapping gene (C) not found in rhabdoviruses, contained within thie P gene in an alternate reading frame (12). As the Cproteinhas beenlocalized with NCs intracel-lularly (4, 20),itmayalsoparticipate inRNAsynthesis. We have therefore further examined Sendai virus plus-strand RNAsynthesis to investigatehowclosely theabQve model applies.
MATERIALS ANDMETHODS
Isolation ofcytoplasmicRNAs. Confluent cultures of BHK cells in 10-cm dishes were infected with 20 PFU of Sendai virusHarris (H)orZ strainpercell and maintainedat33°C. Cycloheximidewasadded afteradsorption (1 h)ata
concen-tration of10 p.g/mlwhen indicated.The cellswereharvested at various times by scraping intophosphate-buffered saline andrecoveredby centrifugation. Cytoplasmicextractswere
preparedbyvortexing'107cells in0.5 ml oflysisbuffer(0.15 MNaCl,0.05 M Tris hydrochloride [pH7.4],0.6% Nonidet P-40[NP-40],followedby centrifugationfor 10minat4,000
xg.Theextractwaslayeredonapreformed20to40% CsCl
density gradientandcentrifugedfor 20 hat52,000rpminan
1951
on November 10, 2019 by guest
http://jvi.asm.org/
1952 VIDAL AND KOLAKOFSKY
SW60rotor. TheRNApellet wastakenup in TNE (25mM
Tris chloride [pH 7.4], 50mMNaCl, 1 mM EDTA), precip-itated in ethanol, and dissolved inET (1 mM EDTA, 10 mM Tris chloride [pH 7.4]).
Invitropolymerasereaction.Purified Sendaivirus(50 ,1u,5 mg/ml)grownin LLC-MK2 cellswasused in250-,ul reaction
mixes containing 50 mM HEPES (N-2-hydroxethylpipera-zine-N'-2-ethanesulfonic acid, pH 8.1), 5 mMMgCl2, 5 mM spermidine,0.05% NP-40, 1 mM eachallfourNTPs, 100 U of RNasin. 48 mM Na2HPO4, and 1 mgofpoly-L-glutamic acid perml. The reaction mixwasincubatedat 30°C for 3h and thenbrought to0.5 M NaCl and 1%NP40, heated for2 minat37°C, and centrifugedon a20to40% CsClgradient to obtain the pelleted RNAs asabove.
RNase protection. Approximately 105 cpm ofriboprobe werecoprecipitatedwith 1to9,ug of CsCl pelletRNAand20 ,ug of tRNA, suspended in 9
RI
of ET, heated for 1 min at 90°C, quickchilled, andthenmade 0.3 MNaCl,20 mM Tris hydrochloride (pH 7.4), 2 mM EDTA (2x bufferA)inatotalvolume of10,ul. After annealing for 30 minat70'C, 50 pI of
0.1% sodium dodecyl sulfate (SDS)-40 ,ugof RNase A per
mi-5 U of RNase T1 per ml in 2.5x buffer A was added.
After 45 min atroom temperature, 600 ,ug ofproteinase K
permlandanadditional 0.1% SDSwereadded. After 20min
at30°C, the volumewasraisedto250,ul withTNEand 0.5% SDS, extracted twice with phenol-chloroform, and ethanol precipitated. The remaining RNAsweredissolved in 3 pI of 80% formamide plus dyes, heated for 2 min at 90°C, and separated on a6% polyacrylamide sequencing gel.
Preparation of riboprobes.TheH strain probe(nucleotides [nt] 22to 142)wasderived from pSL3, whichcontains the 3' endof thegenomestartingat nt22in the PstI siteofpBR322 (11). The first site for TaqI is at nt 142. This PstI-TaqI fragmentwasinserted intopSP65atthe PstI andAcclsites. After linearization with Hindlil, SP6 polymerase reactions
were carried outwith 0.5 mMATP, CTP, and GTP and50 ,uCi of[32P]UTP(10). Equalvolumesofformamide anddyes
were then added, and the radioactive transcript was
sepa-rateddirectly on a6% polyacrylamide sequencing gel. The
transcriptwas localized by autoradiography,andafter exci-sion of theband itwaselutedbyagitating overnightat4°Cih TNE.
The Z strain probes were prepared from clone B36 (21)
which contains the 3' end of thegenome startingatnt 1 plus 22 extra bases inserted into the BamHI site of pBR322. Three of theriboprobe plasmids containingtheentire leader sequence plus variable portions of the NP gene were ob-tainedby BamHI-NcoI (nt 358), RsaI (nt 234),and TaqI (nt 142) digestion, followed by insertion into the BamHI and AccI/HincII sitesofSP65 (nt1to142)orpGEM3 (nt 1to358 and 1 to 234). When the second enzyme was NcoI, it was
filled in with theKlenow enzyme before BamHI digestion.
The Sall site at nt58 was created by site-directed muta-genesis in clone 1-142 with EcoRI and HindIIIto create the gap and the synthetic oligodeoxynucleotide 3'-AATCCCA GCTGCATAGGTG-5' (the two base changes are
under-lined). The procedure used was essentially as described
before (10). The 81-bp BamHI-SalI fragment containingthe first 58ntofthe Sendai virusgenome wastheninserted into
the same sites of pGEM3. All riboprobes were prepared
simultaneouslywiththesamelabeled andunlabeled
nucleo-tide triphosphates by using DNAs linearized immediately after theinserts toensure thesame specific activity.
The plasmid containing the NP-PC junction (nt 1638 to
1812)wasderived from B36by digestionfirst with AccI and NcoI. The 459-bp fragment was isolated and digested with
TaqI, and the 174-bp NcoI-TaqI fragmentwas isolated,and the endswereblunted with the Klenow enzymeand inserted into the SmaIsite ofpGEM4.
RESULTS
Sendai virus genomesand
antigenomes
arefoundonly inNCstructures, with abuoyant density of 1.31
g/ml
in CsCldensity gradients
(9). Whencytoplasmic
extractsof infected cells arecentrifuged
on suchgradients,
thepellet
fractioncontains
only
theunencapsidated
viralRNAs,
i.e.,
the mRNAs and some of the leader RNAs (6), without theircomplements.ThesepelletedRNAsaresuitable for
measur-ingthe amountsof thevarious viral
transcripts
whichcross a genejunction by RNase mapping, sinceself-annealing
ofthe viral RNAs or annealingof the minus-strand
riboprobe
withantigenome RNA should not occur.Wefirstexamined the
transcripts
which crossthe leader-NPjunctionwithaminus-strandriboprobe
containing
nt1to 142 (Fig. 1). When similarpreparations
of CsClpellet
RNA from H strain infectionswereexamined with 3'-end-labeledgenomeRNAas a
probe
(17),leader RNAs of31,55,
and 65 ntwerefound, thepredominant
55-ntspecies
having
termi-nated at the junction. Protected
fragments
consistent withthese species were also weakly seen with the
internally
labeled riboprobe(Fig.
2, marked1),
which detects NP mRNA (87-nt band) and leader-NP readthroughtranscripts
(142-ntband)aswell. (Inthisexperiment,
asmallamountof theintact 240-ntriboprobe,
ofwhichonlythe central 142ntarevirus
specific,
surviveddigestion
in allcases).Theorigin
ofthese bands isdefinedby
theirexpected lengths,
by
theirability
toannealtoriboprobes specific
toeither the leaderor NP mRNAregionswhen thegel
is converted intoaNorthern blot (notshown),
and whether theirmobility
isunchanged
whenanestedsetofriboprobesis used(Fig.
3A).The leader RNAs are minorspecies
compared with the NP mRNA in thesesamples,possibly
because theyareless stableorhave beenencapsidated
(6)atnd
are removedduring
preparation.
Themoststriking
result, however, is therelativeamountofleader-NP
readthroughtranscripts.The
142-nt bandpresumably
results from atranscript
which initiatedatnt1and extendedtoatleastnt142andwas not encapsidated, asjuldged
by
CsCldensity
gradient
cen-trifugation.
Thesetranscripts
are notantigenomes
whichhad somehowescaped
theseparation protocol,
asriboprobes
from theopposite
end of the genome could not detect anyplus-strand
RNA
other than 3' end of the LmRNA,
eventhough the L mRNA was 20-fold less abundant than NP mRNA(notshown). Wealso examined therelativeamounts
ofthese
transcripts
during
the timecourse of the infection. RNAwasprepared
from cells harvestedat4h aswellas 18 hpostinfection
and at 4 h from cultures incubated in the presenceofcycloheximide.
Theinhibition ofprotein
synthe-sis prevented genome replication but not
transcription,
so that under these conditions the viral RNAs werepredomi-nantly the result ofprimary
transcription.
When all thesesamples were
compared
(Fig. 2)
and theresults were quan-titated bydensitometry
and normalized for the number of uridinesin eachfragment,thereadthroughRNArepresented
39% ofthe NP mRNA at 4 h when new genomesynthesis
waspreventedwith
cycloheximide,
43%at4h withoutdrug,
and 19% at18 h.Unencapsidated
leaderreadthrough
RNAs are thus present atrelatively
high
levels relative to NP mRNA throughout the infection.The
readthrough
transcripts appeared
tobetooabundant and stable to be truereplication
intermediates,
which areJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
1 358
1 234
1 142
1 58
/
1638 1812
NP
PC.
I
FIG. 1. Schematic diagramofriboprobes used and themodifiedmodel. The middle line represents the 3' orleft end of theminus-strand genome withtheleader. NP,and PCregionsseparatedbyverticalbarsrepresenting thejunctions.Mappositionsarenumbered from this end
orthe 5' endof theantigenome. Above areshown the Zstrain riboprobes used. Theextentsof the viral sequences areindicatedby heavy lines and mappositions;thethin lines at the endsindicate transcribed vector sequences. Below are shown thetwosituations ofplus-strand RNAsynthesisfoundin Z strain-infected cells in the absence of concurrentassembly. In each case thepolymeraseis shownjustafterhaving crossed the firstjunction, butwithorwithouthavingreinitiated, and the consequences for continued elongation.
expectedtobeverytransientspecies.Their levels relativeto NPmRNA were reduced by onlytwofold between 4 and 18
h, whereas NP mRNAs increased by more than 30-fold
between these times. J3ona fide replication intermediates describedforVSV(14)werefourd torepresent onlyasmall fractionof the RNAmade ina5-minpulse of[3H]uridine and were difficult to see at all after longer labeling. They were
also considerably larger (2,000 to 11,000 nt) and, more
importantly, wereassembled with N protein, astheirRNAs were resistant to RNase. The pelleted Sendai virus readthrough transcripts, onthe otherhand,wereonlyafew
hundred nucleotides in length, sensitive to RNase (not shown), and madeatthesamerate relative toNPmRNA in the
absencp
of viralproteinsynthesis. They therefore donot require assembly with NP for theirsynthesis either during primary transcription in vivo or in vitro (see below). True Sendaivirusreplication intermediates mustofcoursealso bepresentintheseinfections, but they would be expectedtobe retained in the CsCI gradient, along with mature genomes
and antigenomes.
Polymerases which read through the leader-NPjunction. Thislevel ofleaderreadthrough RNAs was unexpected, as
nascent chains which had not begun assembling with NP
shouldhaveterminatedefficientlyatthe firstjunction. Itwas
thereforeof interesttocharacterize further the polymerases which made readthrough RNAs; e.g., did they continue
alongthetemplate until the nextjunction, which mightnow
beobserved, ordid they somehow sensethe absenceof NC assembly on the nascent chain and abort prematurely? For this experiment, a nested set of minus-strand riboprobes
containing nt 1 to58, 142, 234, and 358were used (Fig. 1). Themappingswereagain carriedout atmorethanoneRNA concentration to ensure that the results were proportional,
asforFig. 2. Thegel from onesuchexperiment is shown in Fig. 3A. Riboprobe 1-58 only detected the leaderregion plus
3 nt, but the other three detected both NP mRNA and readthrough RNAs. Besides riboprobe 1-142, described be-fore, riboprobes 1-234 and 1-358 detected protected
frag-ments of 180 and 302 nt (NP mRNA) and 234 and 358 nt (leader-NP readthrough), respectively. In this experiment,
the bands were cut out and counted, and the results were plottedascountsperuridineperminuteineachfragment,so that therelativeamountsof eachspecies could becompared directly(Fig. 3B).Except forriboprobe 1-358, astraightline could be drawnthrough the origin with at least two of the threepoints,andtriplingtheprotectingRNAateach step led to three times as many counts protected. As the relative amountsofthevarious RNAsaregiven bytheslopesofthe lines, it is clear that there is a decreasing abundance of readthrough RNAsasafunction of how far their3' ends had extended. This can also be seen in Fig. 3C, in which the various readthrough RNAs from the lowest concentration point used for all four probes (from three separate
experi-ments) are plotted as the percentage of the NP mRNA
moll.--L
-- - - - -I
I I
4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.51.557.56.385.2]1954 VIDAL AND KOLAKOFSKY
4h
INFECTION M 4h CHX 18h
pg RNA 8 4 8 4 8 48
642
240 PROBE
172
*. ..l
.0
_-.s.142 /NPI
-87
leader(I
T
_4
FIG. 2. Detection of Sendai virus plus-strand RNAs in CsCl pellet RNA by riboprobe 1-142. CsCl pellet RNA from
mock-infected(M)orvirus-infected cells harvestedat4 h withorwithout cycloheximide (CHX) added after virus adsorption or 18 h were
assayedforplus-strand RNAs witha minus-strandriboprobe
con-taining positionsnt1to142. TheamountoftheprotectingRNA used
in each lane is indicated above. The four right-hand lanes show riboprobesrun as lengthmarkers(small arrows). The large arrows
show the viral RNAs detected.Three leader RNAsareindicatedby the letter1.
detected by the same probe. There was no tendency for RNAs which had initiated at nt 56(NP mRNA)todecrease inabundance,asmeasuredby probeswhich extend
progres-sively downstream, whereas the readthrough RNAs de-creased from 19% of NP mRNA at nt 58 to 3% at nt 358. Furthermore, less than 10% of the polymerases which started the NP mRNA failed tofinish it (Fig. 4A), whereas
we have been unable to detect any complete leader-NP readthroughRNAsbyNorthernanalysiswithleader-specific riboprobes (not shown).
In theseexperiments, themappingswerecarried outwith
more than one concentration ofprotecting RNA to ensure
that the riboprobe was not limiting fortwo reasons. First,
under conditions of limiting riboprobe, formation of the longer hybrids mightoccurpreferentially.Moreimportantly, under these conditionsariboprobe mightbesimultaneously
hybridized to botha leader and an NP mRNA without any gapinbetween,asleader RNAmayextendto nt55 and NP begins at nt 56. Such a composite hybrid might not be sensitive to RNase attack and appear as a readthrough
transcriptin thisassay.Themoretheriboprobeis inexcess,
however,the less likely it is thatacomposite hybrid would
form. As shown in Fig. 3, the ratio ofreadthrough RNAto
NP mRNA did not
change
even when nine times lessprotecting
RNA from agiven sample
wasused,
and wemeasuredmorerather than less
readthrough
RNArelativeto NP mRNA when therewas 30times less viral RNA in thesample
at 4 h relative to 18 hpostinfection (Fig. 2).
These results are inconsistentwith themeasurementofcomposite
hybrids,
as is thefinding
thatreadthrough
RNAs become less abundant relative to NP mRNA whenprogressively
longer
riboprobes
areused.Finally,
asthe sequence ontheriboprobe
preceding
thejunction
isAAAA,
in someexperi-ments we used RNase
T2
instead ofRNases A andT1,
butthis did not
change
the results(not shown).
As thereadthrough
RNAs do not appear to be artifacts of themethod,
these results indicatethatpolymerases
which start at nt 1andreadthrough
thefirstjunction
terminatehetero-geneously
shortly
thereafter,
whereasthose whichreinitiateat NPcontinue at
high frequency
tothe nextjunction.
Zand Hstrains ofSendai virus. Several strains of Sendaivirusare
available,
and the onewe have described sofar is the Zstrain,
theonly
onewhichhas beenclonedcompletely
(21, 22).
The sequence ofthe
H strain with whichwe have worked in the past has been cloned uptothe L gene,butourclone closesttotheleft endstartsat nt22
(11). However,
asthese strainsare99% conserved atthenucleotide
level,
the Z strainriboprobes
should also be able to measure the Hstrain
transcripts.
Whenequal
amountsofCsCl
pellet
RNAfromZand Hstrain-infectedcellswereusedtoprotectthe Z
strain
riboprobe 1-142,
thesesamples
werefoundtocontain similaramountsofNPmRNAbut verydifferentamountsofleader
readthrough
RNAs(Fig. 4A). By
directcounting
asfor
Fig.
3,
theZstrainsample
wasfoundtocontain 12.2%asmuch
readthrough
RNAsasNPmRNAs,
ingood
agreementwith the
14%
averageinFig. 3C,
but the H strainsample
wasfound to contain 20 times less
(0.6%).
To ensure that thisdifferencewas not due tothe
slight
differences in sequence between the two strains in thisregion,
the amount ofreadthrough
RNA in H strain CsCIpellet
RNA wasmea-sured withanHstrain
riboprobe
containing
nt22to142. Asshown in
Fig. 4B,
thishomologous
probe again
detectedless than 1%readthrough
RNAinthe in vivosample
relativeto NP mRNA under conditionsinwhich theproducts
ofan Hvirion
polymerase
reaction werefoundtocontain consider-able amounts ofreadthrough
RNA. Theability
of the Zstrain
polymerase
toreadthrough
the firstjunction
at anunexpectedly high
frequency
in vivo is thus not sharedby
the H strain.SincetheZand Hstrain
polymerases
readthrough
thefirstjunction
at such differentfrequencies,
we examined thereadthrough
frequency
at the nextjunction (NP-PC),
by
using
a Z strainriboprobe
containing
positions
nt 1638 to 1812(the
NP mRNA ends at nt1736,
and PCbegins
at nt1740).
Asshown
inFig.
4A, thisprobe
detectedroughly
equal
amountsof NP(100 nt)
and PC(71
nt) mRNAs,
aswell asNP-PCreadthrough
RNAs(174
nt)
inequal
amountsofZ and HCsClpellet
RNA.However,
the level ofreadthrough
atthis
junction by
theZand Hpolymerases
wasidentical
to thatofthe Hpolymerase
at the firstjunction (0.6
and0.5%,
respectively).
The Z strainpolymerase
therefore readsthrough
the first and secondjunctions at differentfrequen-cies,
whereas the Hstrainpolymerase
readsthrough
bothat the samelowfrequency.
Invitro studies. The level of
transcripts
from the leader-NPregion
thatwe measured in vivoaresteady-state levels,
determined
by
turnoveraswellassynthesis.
Moreover,
fortranscripts
whichcontain theleader sequences, turnoverin thiscontext includesencapsidation
as wellasdegradation.
J. VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.86.283.71.382.2]A PROBE 1-358 1-234 1-142 1-58 pgRNAM.7 2 6 M .7 2 6 M.726 M 1 3 1
INP 358_ NP 302 1,NP
234-NP 180
'NP 142 ~d- 9
NP 87
leaderi)
_
56-142 X
NP mRNA
FIG. 3. Quantitation of leaderreadthrough and NP mRNAswith 9PROBES a nested set of riboprobes. (A) Three different amounts of CsCl 431 pellet RNA (18 h) eachathreefold increase,were used toprotect riboprobescontaining positions 1to58, 142, 234, and358(Fig. 1),
307 and theRNAsremainingafterdigestion wereseparated by electro-phoresis. Theautoradiogram fromone suchexperiment is shown; * - 240 the lengths (innucleotides) ofthe various probes used andRNAs detected are indicated on the sides. The probes used in each experimentareindicatedabove,asisthe amountofprotectingRNA for each lane. M refersto 6 or9 ,ugofRNA from mock-infected cells. The firsteight lanes containing the larger probeswereexposed for shorter times than those containing the smaller probes. (B) * - 104 Bandsrepresenting the leader readthroughRNAs
(1/NP)
andthe NP mRNAs (NP) were excised from a parallel gel which contained blank lanes in between, and their Cerenkov radiation was deter-mined in a beta counter. After background subtraction (30 cpm total), thecountspresent ineachband weredividedbythenumber of uridines and plotted as afunction of the amountofprotecting RNAforthe threesmallestriboprobes. Onlythe NP mRNAbands from riboprobe 1-142 are shown. (C) Results obtained with the lowestamountofprotectingRNAfromthree separateexperiments, P - 58 NP one of which is shown in panels A and B, are plotted according to the 3' end of the plus-strand fragment protected in the various hybrids.Tonormalizethevaluesforthe NPmRNA,the averageof the valuesfrom thethree riboprobes in each experiment (115, 34, and 189cpm/Uprotected)wasset to100, and the results from each riboprobeareplotted relativetothis value. Thereadthrough RNAs, ontheotherhand,areplotted relativetothe NP mRNAdetectedby the sameprobein the sameexperiment,exceptforthat detectedby riboprobe 1-58, whichisplotted relativetotheaverage value for the NP mRNA. Large and small crossesand circles indicatethemean and individual values forNP mRNA and the leader readthrough RNAs,respectively.1-58
z
a
cc
z cc
FE
0.
0
z
1-142 5
cc
a-0u
C
120-100
80
60-40
20
READ -THR U
20F
n
u0
:i o
E-v
CL
uJ 10
z
a
0.
u)
1-234
.71 2 3
pg CsCI PELLET RNA
Wetherefore alsoexaminedreadthrough in vitro with puri-fied Sendai virions, as degradation of the uncapped RNAs containing the leader region might be less important here than in vivo, and encapsidation of these transcripts should not take place. RNA levels may therefore more closely
approximate their rates of synthesis in vitro. Polymerase reactionmixes containing equal amounts of Z and H strain virionsweremade,and theproductswereexamined with the
Z strain riboprobe 1-142 asbefore. To distinguish between
MAPPOSITION (NUCLEOTIDE) 3 END OF FRAGMENT
RNAmadedenovoand thatpresentasvirioncontaminants,
mock reaction mixes containing excess EDTA were
proc-essedinparallel. As showninFig. 5, the results in vitrowere
basically similar to those in vivo, but there were some
notable differences. The leader RNAs, which were very
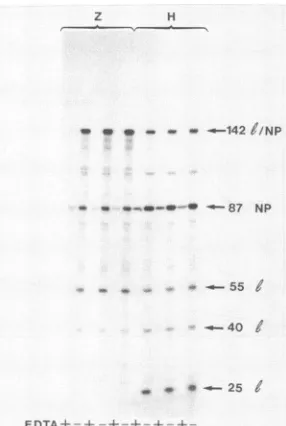
minorspeciesinvivo,weremoreprominentinvitro.Z strain
virions made two leader RNAs which migrated at
approxi-mately 55 and 40 nt in length, and H virions made an
additional leader band at 25 nt (which was also present in
vivo [not shown]). When the results were quantitated by
densitometry and normalized for uridine content and the
signaldue tocontaminatingNP mRNA(EDTA reaction)was
subtracted,therewas62 + 4%asmuch leaderasNP mRNA
in the Z reaction whentheleader bands were summed, and
B
0
LC
U
:L
z
10
C:
CL
0
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.285.554.381.561.2]1956 VIDAL AND KOLAKOFSKY
B:
TI Ti TI
RNase
A A A A A A I/iNP NP/PC JNP
3 1 3 1 3 1
_4I_P 120 /NP
NP 100-_-o
71.
PC 71 _-4
40
STRAIN H
_." _l .~
AR
4,..T..
*
0
--- 87 NPP
;;-z
qbf Im
45,I o ---87
[image:6.612.99.530.72.355.2]MOCK INF POL
FIG. 4. Estimation of readthrough frequencyat the leader-NP and NP-PCjunctions in strain Z- and H-infected cells. (A) Equal and increasingamountsof CsCl pellet RNA from strain Z and H infectionswereusedtoprotectriboprobes whichspanthe leader-NP and NP-PC junctions(Fig. 1). The length (in nucleotides) of the protected fragments and their originareshownonthe sides. (B) CsCl pellet RNA from
mock- andstrainH-infected cells (20 ,ug)aswellastheproducts from 10,ulofavirionpolymerase reactionwereusedtoprotectanHstrain riboprobe containingnt22to142. Thehybridsweredigested with RNase A aloneorplus RNaseTl.Thelengths and origins of theprotected
bandsare indicatedonthe side.
120± 13%intheHreaction. LeaderRNAswerethus made
inroughly equimolaramountsrelativetoNP mRNAinvitro,
asexpected, althoughmore leaderRNAwas made intheH
reaction. The equimolar levels of leaders andNPmRNAs in vitro suggest that the scarcity of leader RNA in vivo is predominantly due to turnover. Another difference is that the Z and H polymerases read through more frequently in vitro than in vivo. In this experiment, leader readthrough RNAswhichextendedto nt 142 orbeyondwere 106 ± 12%
asabundant asNPmRNA in the Z reactions andonly38 ±
4%asabundantintheHreaction.Thus,similartothe results in vivo, the Z polymerase reads through more frequently
than H, but the difference is only 3-fold, as opposed to 20-fold invivo. Inaddition, similar results wereobtained in
vitro when the reaction products were radioactive and the
riboprobe was not, and here the readthroughRNAs cannot be the result ofcomposite hybrids.
The apparentdifference in readthrough frequencyinvivo and invitro of both strains isunlikely to be dueentirely to more turnover in vivo, as we measured only 39%
read-through with the Z strain in vivo at 4 h even when protein
synthesis was inhibited to prevent encapsidation.
Further-more it is difficult to accept that the H strain difference of 63-fold (0.6 versus 38%) is only a question ofturnover. It appearsmorelikelythatthis differencereflects thepeculiar conditions of the in vitro reaction. Forexample, the virion reactions take place within envelopes containing the M protein, which may be a transcriptional inhibitor. Further-more, the optimum conditions for the in vitro reaction are
Z H
_ -w__142I/NP
-_-a -W ---87 NP
.._,,,,, .. - 5 e
-40e
2
^,,_ 5 e"
E DTA+ -+-+
+±+-FIG. 5. Polymerase readthrough in Z and H virion reactions.
Triplicate nonradioactiveinvitropolymerase reactionswerecarried
outwith bothstrainsasdescribed in Materials and Methods. Mock
reactionswerealsodone inparallel,whichcontainedanadditional
20 mM EDTA as indicated, to prevent de novo synthesis. The
products from 10 of each reaction were analyzed by RNase
mapping with riboprobe 1-142. The lengths of the leader RNAs
(script l) wereestimated relativetothose ofriboprobes of known
length (not shown) andaretherefore approximate. A:
PROBE NP/PC jgRNA 3 1
NP/PC174-_
NP
J. VIROL.
dO--- 142 I/N P
*
o
9
:sl;:.0 .0.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.370.513.425.638.2]quite different from those of VSV. For Sendai virus, the replacement of Tris with HEPES buffer, chloride or acetate with phosphate as counter ions, and the addition of poly-L-glutamic acid, led to a 500-fold stimulation of activity. All or any of the above could account for the differences between readthrough in vivo and in vitro. We also note that whereas parallel reactions were remarkably reproducible (Fig. 5), readthrough levels varied significantly from one set of reactions to the next, even with the same preparation of virus, for reasons that are unclear (not shown).
DISCUSSION
RNase mapping is well suited to examine transcripts which cross gene junctions, as all three possibletranscripts can be measured simultaneously with a single probe. How-ever, as it is unreliable when thecomplementary RNA is also present and leader readthrough RNAs cannot be distin-guished from antigenomes by this method, we have re-stricted ourselves to characterizing the CsCl pellet RNAs. We therefore do not know what fraction of the leader readthrough RNAs are represented by this material. How-ever, by comparing their levels to those of the NP mRNA, whichquantitatively pellets in CsCl gradients and is thought to be the most stable of these RNAs, we can get a minimum estimate of their rates of synthesis. The time course of their accumulation during Z strain infections suggests that their levels at 18 h, when most of ourmeasurements were made, do in fact underestimate their rate ofsynthesis relative to NP mRNA, but only by about twofold.
We were surprised to find such large amounts of leader readthrough RNAs in Z-infected cells which had not been encapsidated but not in H straininfections. The presence of easily measurable amounts of the readthrough RNAs al-lowed us to measure how far their 3' ends had extended. According to the minimal model, polymerases which read through the firstjunction would be expected to continue on to the next. Concurrent assembly is supposedly only re-quired for the polymerase to disregard the junctional signals andshould have no effectbetween junctions. This test of the model has clearly not held up for the Z strain, as these replicases aborted synthesis shortly after having crossed the first junction, even though they are traversing the same template sequences as the transcriptases. Z strain tran-scriptases and replicases can thus be distinguished even in the absence of concurrent assembly.
Very few, if any, of the polymerases which read through thefirstjunction continue on to the next. Readthrough of the secondjunction can then only take place with polymerases which had reinitiated at the first, i.e., transcriptases. As the polymerases of the Z and H strains are indistinguishable by their frequency ofreadthrough at the second junction (0.6 and 0.5%, respectively) but vary 20-fold in the frequency with which they read through the firstjunction, this again suggests that thesepolymerases must somehow be different, independent of assembly.
The reason for the difference in these readthrough fre-quencies at the first junction remains unknown. However, a remarkably similar situation has previously been described forVSV. Perrault and co-workers, using a selection protocol forheat resistance, isolated mutants (polR) with enhanced ability to read through the leader-N junction but not subse-quent junctions (18, 19). The enhanced ability to read through the junction is reproducible in vitro, and here it maps to the N protein of the template rather than the exchangeable polymerase components (L and NS). The
VSVpolRmutants stressthepoint that the assembled Nor NPprotein, which isanintegralpartofthetemplate, is thus an integral part of the polymerase complex as well. The differences between the Z and H polymerases could then also lie in the template component ofthecomplex, but any component of this complex could of course be equally
responsible for the readthrough phenotype. This might now also include theSendai virus equivalent of the simian virus 5 and measles virus V proteins (23; R. Cattaneo, K. Kaelin,
and K. Baczko, Cell, in press), which does in fact existas
predicted (unpublished).
Moreimportantly, whatdifferencesbetween thereplicase
andtranscriptase can accountforthefindingthat the former requires concurrent assembly tocontinuedown thetemplate when it has crossed the first junction, whereas the latterdoes not? And to what extent does the minimal model still apply to Sendai virus? A key element of this model is that the switch from transcription to replication is controlled not at the level of chain initiation, but during elongation. This would also appear to be true for Sendai virus, as leader readthrough RNA synthesis continues similarly to NP mRNA synthesis in the presence ofcycloheximide (Fig. 2).
In that case, transcriptases and replicases cannot be distin-guished until the polymerase has crossed the firstjunction. It would appear that the act of crossing thejunction is respon-sible for this difference. Before the first junction, only one form of the polymerase can be distinguished, that which has initiated at nt 1. After this junction there are two forms; transcriptases which have reinitiated at NP and whose nascentchains no longer carry the leader sequences and are capped, and replicaseswhich have not reinitiated and whose nascent chains still contain the leader, with a 5' triphos-phate. Any or all of these may account for the difference in how they continue elongation without assembly.
Exchange or modification of polymerase subunits during reinitiation at the junction may also be involved. In the case of VSV polymerase reactions in vitro (3, 19), wild-type (wt) N mRNA synthesis requires high ATP concentrations for half-maximal synthesis, whereas leader synthesis takes place efficiently at lower ATP concentrations. In the polR mutants, however, N mRNA synthesis behaves similarly to leader synthesis with respect to ATP. These results are consistent with different phosphorylation states of the poly-merases which initiate leader and N mRNA synthesis, and these differences could take place during reinitiation at the first junction. In the case of paramyxoviruses, it has been found that in a persistent measles virus infection of human brain, a single gene (M) could be highly and specifically mutated whereas these mutations stopped precisely at the flanking PC junction (R. Cattaneo, A. Schmid, K. Baczko, V. ter Meulen, and M. A. Billeter, Cell, in press). One explanation they advance is that during replication a highly mutagenic polymerase copied the M but not the flanking genes. This suggests that polymerases could also be ex-changed at genejunctions during replication.
The minimal model clearly requires some modification to account for the Z straininfections. Amodified model which more adequatelydescribes both Sendai virus Z and H as well as VSV polR and wt is one in which the polymerase, in the absence of concurrent assembly, cannot continue for very long on its template unless it has reinitiated. Howfrequently it would stop at or read through the junction without assembly could of course becontrolled genetically. Concur-rent assembly would be required here not only for the polymerase to read through junctions, but also for itto read throughcontinuously. The leader-NP junctionwouldonly be
on November 10, 2019 by guest
http://jvi.asm.org/
1958 VIDAL AND KOLAKOFSKY
the first ofmany places where the polymerase would termi-natewithout assembly if it had not reinitiated. The intracel-lular level of unassembled NP would still be the key element in the switch and would remain the rate-determining one. It would still act by initiating NC assembly on the nascent leader sequences, thereby controlling the rate with which polymerases continue without interruption to the end of the template. Initiation of nascent-chain assembly before the
polymerasehad reached the junction would result in efficient readthrough regardless of the basal rate of readthrough in the absence of assembly. It should be noted that our data do not rule out the possibility that the polymerases (and/or tem-plates) are committed to either the transcription or replica-tion mode before initiareplica-tion, but these differences only be-come apparent atthe first junction. This possibility appears less likely, however, as virions would be expected to contain predominantly those in the transcription mode, whereas in vitro they read through the first junction more frequently than is found in vivo (Fig. 5).
Finally, in Z versus H strain infections, termination at the first junction is relatively inefficient, yet this appears to have only amodest effect, as the Z strain grows only a little less
quickly than H (again similar to polR and wt VSV). As a
majority ofthe polymerases which read through terminate within 100 nt, some of them may move back to the junction and reinitiate, as suggested for the respiratory syncytial virus polymerase at the 22K-L junction (8). However, the modest effect seen in one-step growth curves in culture couldof course have been important in the natural evolution of thisstrain, which is considered to beas wt asthe H strain.
ACKNOWLEDGMENTS
Wethank HiroshiShibuta (Tokyo) forthe Zstrain of Sendai virus and cloneB36andJosephCurran (Geneva) for experimentaladvice. This work was supported by a grant from the Swiss National Science Fund.
LITERATURE CITED
1. Arnheiter, H., N. L. Davis, G. Wertz, M. Schubert, and R. A. Lazzarini. 1985. Role of nucleocapsid protein in regulating vesicularstomatitis virus RNAsynthesis. Cell41:259-267. 2. Banerjee, A. K. 1987. Transcription andreplication of
rhabdo-viruses. Microbiol. Rev.51:66-87.
3. Beckes, J. D., A. A. Haller, and J. Perrault. 1987. Differential effect ofATPconcentrationonsynthesis ofvesicularstomatitis virus leaderRNAs andmRNAs. J. Virol. 61:3470-3478. 4. Bellini, W. J., G. Englund, S. Rozenblatt, H. Arnheiter, and
C. D. Richardson. 1985. Measles virus P gene codes for two proteins. J.Virol. 53:908-919.
5. Blumberg, B. M., M. Leppert, andD. Kolakofsky. 1981. Inter-action of VSV leader RNA and nucleocapsid protein may control VSV genome replication. Cell23:837-845.
6. Blumberg, B. M., and D. Kolakofsky. 1981. Intracellular vesic-ular stomatitis virus leader RNAs are found in nucleocapsid structures. J. Virol. 40:568-576.
7. Blumberg, B. M., C. Giorgi, and D. Kolakofsky. 1983. Nprotein of vesicular stomatitis virus selectively encapsidates leader
RNA in vitro. Cell 32:559-567.
8. Collins, P. L., R. A. Olmsted, M. K. Spriggs, P. R. Johnson, and A.J.Buckler-White. 1987.Geneoverlap and site-specific atten-uationoftranscriptionof the viralpolymerase L gene of human respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 84: 5134-5138.
9. Compans, R. W., and P. W. Choppin. 1967. Isolation and properties of the helical nucleocapsid of theparainfluenza virus SV5. Proc. Natl. Acad. Sci. USA57:949-956.
10. Curran, J. A., and D. Kolakofsky. 1988. Ribosomal initiation fromanACG codon intheSendai virus P/CmRNA. EMBO J. 7:245-251.
11. Dowling, P. C., C. Giorgi, L. Roux, L. A. Dethlefsen, M. E. Galantowicz, B.M.Blumberg, and D. Kolakofsky. 1983. Molec-ular cloning of the 3'-proximal third of Sendai virus genome. Proc. Natl. Acad. Sci. USA 80:5213-5216.
12. Giorgi, C., B. M. Blumberg, and D. Kolakofsky. 1983. Sendai virus contains overlapping genes expressed from a single mRNA.Cell35:829-836.
13. Gupta, K. C., and D. W. Kingsbury. 1984.Completesequences ofthe intergenic and mRNA start signals in the Sendai virus genome: homologies with the genome of vesicular stomatitis virus.Nucleic AcidsRes. 12:3829-3841.
14. Hill, V. M., C. C.Simonsen, and D. F. Summers. 1979. Charac-terization of vesicular stomatitis virus replicating complexes isolated inrenografin gradients. Virology99:75-83.
15. Kolakofsky, D., and B. M. Blumberg. 1982. A modelfor the control ofnon-segmented negative strandvirusesgenome rep-lication,p. 203-213. In B. W. J.Mahy,A.C.Minson,and G. K. Darby (ed.), Virus Persistence Symposium 33. Society for GeneralMicrobiology Ltd., Cambridge UniversityPress, Cam-bridge.
16. Kolakofsky, D., and L. Roux. 1987. The molecularbiology of paramyxoviruses,p. 277-279. In P.Bercoff(ed.),The molecular basis ofviralreplication. Plenum Publishing Corp.,NewYork. 17. Leppert, M., L.Rittenhouse,J. Perrault, D. F.Summers,and D. Kolakofsky. 1979. Plus and minus strand leaderRNAsin nega-tive strandvirus-infected cells. Cell 18:735-747.
18. Perrault, J., G. M. Clinton, and M. A. McClure. 1983. RNP template of vesicularstomatitisvirusregulatestranscription and replication functions. Cell35:175-185.
19. Perrault, J., and P. W. McClear. 1984. ATP dependence of vesicularstomatitisvirustranscription initiation andmodulation by mutationin the nucleocapsid protein.J. Virol. 51:635-642. 20. Portner, A., K. C. Gupta, J. M.Seyer, E. H. Beachey, and D. W.
Kingsbury. 1986. Localization and characterization of Sendai virus nonstructural C and C' proteins by antibodies against syntheticpeptides.Virus Res. 6:109-121.
21. Shioda, T., Y. Hidaka, T. Kanda, H. Shibuta, A. Nomoto, andK.
Iwasaki. 1983.Sequence of3,687 nucleotides fromthe 3'endof Sendai virus genome RNA and the predicted amino acid
se-quence of viral NP, P and C proteins. Nucleic Acids Res. 11:7317-7338.
22. Shioda, T., K. Iwasaki, and H. Shibuta. 1986. Determination of the complete nucleotidesequenceoftheSendai virus genome RNAand thepredicted amino acidsequencesoftheF, HN,and Lproteins. Nucleic Acids Res. 14:1545-1563.
23. Thomas, S. M., R. A. Lamb, and R. G. Paterson. 1988. Two mRNAs thatdifferbytwonontemplatednucleotides encode the aminocoterminalproteinsPand V of theparamyxovirus SV5. Cell 54:891-902.
J. VIROL.