0022-538X/09/$08.00⫹0 doi:10.1128/JVI.00685-09
Copyright © 2009, American Society for Microbiology. All Rights Reserved.
A Conformational Switch in Human Immunodeficiency Virus gp41
Revealed by the Structures of Overlapping Epitopes
Recognized by Neutralizing Antibodies
䌤
Robert Pejchal,
1Johannes S. Gach,
2Florence M. Brunel,
3Rosa M. Cardoso,
1Robyn L. Stanfield,
1Philip E. Dawson,
3,4Dennis R. Burton,
1,2Michael B. Zwick,
2* and Ian A. Wilson
1,4*
Department of Molecular Biology,1Department of Immunology and Microbial Science,2Department of Chemistry and Cell Biology,3
and Skaggs Institute for Chemical Biology,4The Scripps Research Institute, La Jolla, California 92037
Received 2 April 2009/Accepted 4 June 2009
The membrane-proximal external region (MPER) of the human immunodeficiency virus (HIV) envelope glyco-protein (gp41) is critical for viral fusion and infectivity and is the target of three of the five known broadly neutralizing HIV type 1 (HIV-1) antibodies, 2F5, Z13, and 4E10. Here, we report the crystal structure of the Fab fragment of Z13e1, an affinity-enhanced variant of monoclonal antibody Z13, in complex with a 12-residue peptide corresponding to the core epitope (W670NWFDITN677) at 1.8-Å resolution. The bound peptide adopts an S-shaped conformation composed of two tandem, perpendicular helical turns. This conformation differs strikingly from the
␣-helical structure adopted by an overlapping MPER peptide bound to 4E10. Z13e1 binds to an elbow in the MPER at the membrane interface, making relatively few interactions with conserved aromatics (Trp672 and Phe673) that are critical for 4E10 recognition. The comparison of the Z13e1 and 4E10 epitope structures reveals a conformational switch such that neutralization can occur by the recognition of the different conformations and faces of the largely amphipathic MPER. The Z13e1 structure provides significant new insights into the dynamic nature of the MPER, which likely is critical for membrane fusion, and it has significant implications for mechanisms of HIV-1 neutral-ization by MPER antibodies and for the design of HIV-1 immunogens.
The continued spread of human immunodeficiency virus (HIV) worldwide and, in particular, in sub-Saharan Africa, where an estimated 22 million people currently are living with HIV/AIDS, underscores the urgent need for a preventative vaccine. However, despite nearly 25 years of intense interna-tional research, a vaccine is not yet available. Passive immuni-zation with broadly neutralizing antibodies can confer steriliz-ing protection against infection in animal models (4, 12, 39–41, 51, 64), providing encouragement for the development of an antibody-inducing component of an HIV type 1 (HIV-1) vac-cine. Such a vaccine should elicit neutralizing antibodies with activity against the broadest range of primary circulating iso-lates. However, a lack of understanding of how to raise potent, cross-reactive antibodies by immunization, the so-called neu-tralizing antibody problem, is a major hurdle in this effort (6, 24, 72). Thus, an understanding of the structure and presen-tation of neutralizing epitopes on the virus and the antibodies that recognize them is vital for vaccine development.
The targets of antibody neutralization are the surface enve-lope (Env) glycoprotein trimers (gp120/gp41) that mediate the fusion of the viral membrane with that of the host. The ma-jority of antibodies elicited during natural infection or immu-nization show limited or no cross-reactivity against diverse
isolates. However, a few rare, broadly neutralizing, monoclonal antibodies have been isolated from HIV-1-infected individuals and exhibit activity against a wide range of isolates by binding to functionally conserved epitopes exposed on native gp120/ gp41 trimers. These epitopes include the CD4 binding site, recognized by antibody b12, and a relatively well-conserved cluster of N-linked glycans, located on the outer domain of gp120, that is recognized by antibody 2G12 (12, 13, 71, 76). V3-directed antibodies, which are common in natural infec-tion, also are able to sporadically neutralize across clades, as exemplified by 447-52D and F425-B4e8 (7, 16, 49, 66). The identification of three broadly neutralizing antibodies, 2F5, Z13, and 4E10, that target the conserved tryptophan-rich membrane-proximal external region (MPER) of gp41 has im-plicated this region as a highly promising vaccine target and has, therefore, spurred interest in its structural characteriza-tion (15, 35, 45, 47, 48, 50, 80).
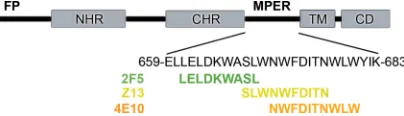
The MPER plays a critical, but not fully understood, role in membrane fusion and is situated between the C-terminal hep-tad repeat (CHR) and the transmembrane domain (TM) of gp41 (Fig. 1). Following the binding of gp120 to the cell surface receptors CD4 and CXCR4/CCR5, the gp41 glycoprotein un-dergoes a series of conformational changes that trigger the membrane fusion activity. Notably, a relatively long-lived pre-hairpin intermediate of gp41 is formed, in which the coiled-coil of the N-terminal heptad repeats (NHR) extends so as to enable the fusion peptides to embed into the target membrane. In the postfusion or fusogenic state, the CHR and NHR reas-semble into an antiparallel 6-helix bundle in a process that drives membrane fusion (18). The MPER contains several functionally conserved tryptophan residues that are critical for membrane fusion and viral entry, although the structural basis
* Corresponding author. Mailing address for I. A. Wilson: Depart-ment of Molecular Biology, BCC206, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla, CA 92037. Phone: (858) 784-9706. Fax: (858) 794-2980. E-mail: [email protected]. Mailing address for M. B. Zwick: Department of Immunology and Microbial Science, IMM2, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla, CA 92037. Phone: (858) 784-2833. Fax: (858) 784-8360. E-mail: [email protected].
䌤Published ahead of print on 10 June 2009.
8451
on November 8, 2019 by guest
http://jvi.asm.org/
for their specific role has not been firmly established (22, 44, 58). Their mutation to alanine leads to the attenuation of viral infectivity, which is most pronounced for Trp666 and Trp672 (numbered according to the HXB2 isolate) (46, 58, 78). In addition, peptides based on the MPER can induce membrane leakage (68). Such membrane-disrupting properties of the MPER have been suggested to be functionally important in the expansion of the fusion pore created after receptor engage-ment (42, 44, 58, 68, 77).
From initial explorations using solution nuclear magnetic resonance, the structure of a 19-residue MPER peptide (resi-dues 665 to 683) was found to be helical in dodecylphospho-choline micelles, with the hydrophobic and hydrophilic resi-dues distributed evenly around the helix axis (62). Another study found that an MPER peptide comprising residues 659 to 671 adopts a 310-helix in water (10). More recently, the struc-ture of an MPER peptide (residues 662 to 683) in liposomes was elucidated by a combination of nuclear magnetic reso-nance and spin-label electron paramagnetic resoreso-nance (69), and it was found to adopt a kinked, amphipathic structure composed of two helices connected by a short hinge (Phe673 and Asn674). Crystal structures of Fab 2F5 in complex with a 7-mer (E662LDKWAS668) and 17-mer encompassing residues
654 to 670 previously had revealed a mostly extended confor-mation characterized by a central -turn involving Asp664, Lys665, and Trp666 (47, 48). This motif is the key recognition determinant for 2F5 and becomes deeply buried in the anti-body combining site, suggesting that it is exposed at some stage in viral entry (45, 47, 78). The crystal structure of Fab 4E10 in complex with peptide-spanning residues W670NWFDITNW678
revealed an amphipathic␣-helical structure with a narrow hy-drophilic face (15). The N terminus of the 4E10 epitope forms a 310-helix that transitions into a regular ␣-helix at residue Asp674 and continues to Lys683, which constitutes the end of the gp41 ectodomain (14). Thus, while the structure of the MPER within functional, membrane-embedded Env trimers is not known, the observation that unconstrained peptides are able to adopt more than one defined structure suggests an inherent degree of flexibility.
Like 4E10, Z13 was identified from an HIV-1-infected indi-vidual, the former being isolated from an immortalized B-cell line and the latter from a bone marrow RNA phage display library (80). The epitope of MAb Z13 spans residues S668LW
NWFDITN677, as determined by peptide mapping, scanning
mutagenesis, and antibody competition studies (46, 80). This region lies between the 2F5 and 4E10 epitopes but overlaps more closely with 4E10 (Fig. 1). 4E10 and Z13 are both able to
neutralize primary as well as laboratory-adapted isolates; nev-ertheless, Z13 is not as broadly neutralizing as 4E10, which has the greatest breadth of any HIV-1 antibody described to date (9). Z13e1 is an affinity-enhanced variant of Z13 and was evolved by randomizing the complementarity determining re-gion (CDR) L3 loop sequence to identify tighter-binding mu-tants using phage display (46). Z13e1 displays higher affinity for both peptide and recombinant gp41 substrates, as well as increased neutralization potency, suggesting that the L3 mu-tations optimize binding to the linear MPER epitope. The neutralization breadth of Z13e1 is limited by the requirement for Asn671 and Asp674 in the MPER, which are approximately 71 and 58% conserved, respectively, among sequences in the Los Alamos HIV sequence database (80). Based on the clear relationship between Env trimer binding and neutralization, the neutralizing activity of Z13e1 derives from binding to a functional trimer (8, 20, 25, 43, 52, 55, 60, 73, 74). While Z13e1 and 4E10 have identical affinities for optimized linear peptides, Z13e1 is still about an order of magnitude less potent than 4E10 against a variety of primary isolates. Although the occlu-sion of the Z13e1 epitope on virion-associated trimers is thought to be the major limitation (46), the structural basis for the lower potency of Z13e1 relative to those of 2F5 and 4E10 is unclear.
Whereas neutralization by 4E10 depends critically on Trp672 and Phe673, Z13e1 instead requires the flanking Asn671 and Asp674 residues (46). Based on a helical model of the MPER, it was predicted that Z13e1 binds the narrow hydrophilic face that displays Asn671, Asp674, and Asn677 that is opposite that recognized by 4E10. As Z13e1 and 4E10 bind to functional trimers, both epitopes must be exposed at some stage before membrane fusion (20). To examine how Z13e1 recognizes its MPER epitope, we determined the crystal structure of Fab Z13e1 in complex with a 12-residue peptide corresponding to the core epitope with C-terminal flanking lysines to aid peptide solubility (W670NWFDITN677KKKK).
The crystal structure at 1.8-Å resolution uncovers a conforma-tion of the MPER that is distinct from that visualized in com-plex with 4E10. Our findings show that Z13e1 and 4E10 rec-ognize different conformers of the MPER and reveal a novel conformational switch that is relevant for HIV-1 neutralization and membrane fusion.
MATERIALS AND METHODS
Preparation and crystallization of Fab Z13e1.Recombinant immunoglobulin
G1() [IgG1()] for Z13e1 was overexpressed in CHO-K1 cells and purified by
protein A affinity chromatography (GE Healthcare) as previously described (46). Fab Z13e1 was generated by the digestion of IgG with endoproteinase Lys-C by following a modification of the protocol for IgG 2F5 (47). In brief, IgG Z13e1 (15 mg/ml) was dialyzed against 50 mM Tris-Cl, pH 8.0, 350 mM NaCl, and treated with 100 mM dithiothreitol (1 h at 37°C). Following reduction, Z13e1 was dialyzed against 25 mM Tris-Cl, pH 8.0, followed by dialysis against 25 mM Tris-Cl, pH 8.0, 2 mM iodoacetamide (48 h at 4°C). Alkylated Z13e1 was dialyzed against 25 mM Tris-Cl, 1 mM EDTA, pH 8.5, and digested with 0.005
g/l sequencing-grade Lys-C (Roche Applied Sciences) for 4 h at 37°C.
Diges-tion was stopped by the addiDiges-tion of 1 mM TLCK (N␣-p-tosyl-L-lysine
[image:2.585.62.264.69.127.2]chloro-methyl ketone) and 0.4 mM leupeptin (Roche Applied Sciences). The mixture was loaded on HiTrap Protein A Fast Flow (GE Healthcare). Flowthrough fractions containing Fab were pooled and loaded on MonoS 10/30 GL (GE Healthcare) and eluted with a 0 to 0.5 M NaCl gradient in 50 mM sodium acetate, pH 5.0. Fab fractions were concentrated and further purified by size exclusion on a Superdex 200 10/300 GL (GE Healthcare) in 20 mM Tris-Cl, 200
FIG. 1. Major features of gp41 include the fusion peptide (FP), NHR, CHR, TM, and cytoplasmic domain (CD). The MPER is lo-cated between the CHR and TM regions of gp41. The core epitopes of 2F5 (green), Z13e1 (yellow), and 4E10 (orange) are indicated. The epitope of Z13e1 is located between those of 2F5 and 4E10, but it overlaps more closely with 4E10.
on November 8, 2019 by guest
http://jvi.asm.org/
mM NaCl, pH 8.0. The Fab peak was pooled and concentrated in size-exclusion buffer to 30 mg/ml, as determined with the Pierce bicinchoninic acid assay.
Peptide 178-1 was synthesized as previously described (11) and dissolved in 10 mM Tris-Cl, pH 8.0, to a concentration of 50 mg/ml. Crystals of Fab Z13e1 in complex with peptide were obtained by cocrystallization. Z13e1 complex with peptide 178-1 was formed by the addition of peptide at a final concentration of 5 mg/ml to 27 mg/ml Fab prior to crystallization. Initial and optimized crystalli-zation conditions were identified by a nanoliter screening format using the IAVI/JCSG/TSRI CrystalMation robotic system. Diffraction-quality crystals were obtained by equilibrating drops of protein mixed 1:1 with 20 to 22% polyethylene glycol 3350 (PEG 3350) and 200 mM potassium fluoride (KF) in sitting-drop vapor diffusion wells at 22°C for 3 days, followed by streak seeding and 2 days of growth. Crystals were harvested from mother liquor and trans-ferred to a holding solution consisting of 20% PEG 3350, 150 mM KF, 100 mM NaCl, and 12.5 mM Tris-Cl, pH 8.0, and then to a cryoprotective solution consisting of 27.5% PEG 3350, 150 mM KF, 100 mM NaCl, 12.5 mM Tris-Cl, pH 8.0, and 12% glycerol and plunged into liquid nitrogen. Data were collected at beamline 11-1 at the Stanford Synchrotron Radiation Lightsource (SSRL) with crystals maintained at 100 K using a liquid nitrogen vapor stream. Diffraction data were processed with XDS (32) and CCP4 (17).
Structure determination and refinement.The Fab Z13e1 peptide 178-1 com-plex was determined by molecular replacement using the program EPMR (34), with Fab 17b (Protein Data Bank code 2NY1) as the search model (76). The constant regions of Fab 17b were placed first, followed by the variable domains with truncated CDR loops. Rigid body refinement was carried out by treating
each domain (VL, CL, VH, and CH1) as a rigid body, and the resulting model
subsequently was refined by one round of torsion angle simulated annealing by slow cooling from 5,000 K in phenix.refine (1), which was used in all subsequent refinement steps. The model then was developed by alternating rounds of man-ual rebuilding with the program COOT (23) and cycles of positional and tem-perature factor adjustment. Bulk solvent and anisotropic temtem-perature factor correction were carried out, as was TLS (T, translation; L, libration; S, screw-motion) refinement in later rounds. Following simulated annealing refinement, clearly interpretable density was observed for the CDR loops and bound peptide, which were built manually in COOT. Water molecules were added manually by inspection of difference Fourier maps and refined. The model quality was as-sessed using the JCSG quality control server, which includes MolProbity for
assessing model geometry (21). The analysis of the ionization state of HisH50
and
the hydrogen bonding network involving TrpH47, TyrH100G, HisH50, and AspP674
was undertaken with Protonate3D (36) implemented in the Molecular Operating Environment suite (Chemical Computing Group). Final refinement statistics are
summarized in Table 1. Crystals of Fab⬘derived from pepsin digestion could be
grown by cross-seeding and diffracted to approximately the same resolution. As this structure shows no apparent differences from the Fab obtained by Lys-C digestion, only the latter structure is reported here.
Mutagenesis and crude Fab preparation. Z13e1 Fab mutants were con-structed in phagemid cloning vector pComb3X, which encodes the wild-type Z13e1 light and heavy chains, by use of the QuikChange XL mutagenesis kit (Stratagene). All mutants were verified by sequencing. The preparation of crude Fab supernatants was carried out as previously described (79), with the following modifications. The mutant clones and wild-type Z13e1 Fab were transformed
separately intoEscherichia coliXL1-Blue cells (Stratagene), and single colonies
were used to inoculate 10 ml super broth medium containing 50 g/ml of
carbenicillin and 10g/ml of tetracycline. Cultures were incubated overnight at
25°C and harvested by centrifugation the following day. Cell pellets were lysed by being resuspended in 1 ml CelLytic B buffer (Sigma) and incubated for 20 min at room temperature. Lysates were clarified by centrifugation at 14,000 rpm and
stored at⫺80°C prior to use. Triplicate crude Fab supernatants were prepared
to lessen the effect of batch variation and used directly for enzyme-linked im-munosorbent assay (ELISA), as described below.
Crude Fab ELISA.Ninety-six-well plates (Costar) were coated with 2g/ml
goat anti-human F(ab⬘)2(Pierce), 2g/ml of gp41-MBP, and 2g/ml of peptide
178-1 in phosphate-buffered saline (PBS), and the plates were incubated over-night at 4°C. Wells then were washed with TPBS (PBS containing 0.05% Tween 20) and blocked with 4% nonfat dry milk at room temperature for 1 h.
Subse-quently, the wells were washed and 50l of crude Fab supernatant was serially
diluted in TPBS containing 1% nonfat dry milk. After 1 h, the plates were washed again and further incubated with peroxidase-conjugated goat anti-human Fab (Pierce) diluted 1:1,000 in TPBS containing 1% nonfat dry milk. Finally, the wells were developed with tetramethylbenzidine substrate (Pierce). Optical densities (at 450 nm) were determined with a microplate reader (Molecular Devices). The concentration of Fab was determined by anti-Fab ELISA (full curve, threefold dilution series) using a standard curve generated with parental Fab Z13e1.
Apparent affinities were determined from the antibody concentration at half-maximal binding. All samples were tested at least twice, and the mean was taken as the final reported value.
Envelope mutation and neutralization.The envelope mutations were intro-duced into the pSVIIIexE7pA-JR2 template (78) by a QuikChange XL
mutagen-esis kit. After sequence verification, the pseudotyped HIV-1JR2mutants,
com-petent for a single round of infection, were generated in HEK 293T cells with the
luciferase reporter plasmid pSG3⌬env andenvcomplementation, as described
previously (78). The pseudotyped virus then was assayed for neutralization using TZM-bl cells as target cells (46). Serially diluted antibody was mixed 1:1 with the pseudoviruses and incubated for 1 h at 37°C prior to addition to target cells. After 48 h of incubation at 37°C, the luminescence in relative light units was measured using an Orion microplate luminometer (Berthold Detection Sys-tems). The extent of virus neutralization was determined as the percent reduc-tion of viral infectivity compared to that of an antibody-free control. All exper-iments were performed in triplicate and repeated at least twice.
Protein structure accession number.Coordinates and structure factors for Fab Z13e1 in complex with peptide 178-1 have been deposited in the Research Collaboratory for Structural Bioinformatics’ Protein Data Bank (http://www.pdb .org) under accession code 3FN0.
RESULTS
[image:3.585.300.541.87.398.2]Structure determination of Fab Z13e1. To examine the recognition of the MPER by Z13e1 and gain insight into the
TABLE 1. X-ray diffraction and model refinement statistics for the Z13e1-peptide complexa
Statistic Data
Crystal
Space group ...P41212
Unit cell constants (A˚ ,°)...a⫽b⫽106.9;c⫽88.8; ␣,,␥ ⫽90
Data set statistics
Resolutionb(A˚ )...30.0–1.80 (1.83–1.80) No. of observations...690,654 (51,453) No. of unique reflections ...48,156 (3,528) Mosaicity (°) ...0.22
Completeness (%) ...99.8 (100.0) Multiplicitiy...14.3 (14.6) I/I...32.2 (5.3) Rsym(%) ...4.7 (55.3)
Refinement statistics
Rcryst(%) ...21.0 Rfree(%)...25.2 No. of protein atoms ...3,210 No. of peptide atoms...83 No. of water molecules ...230 Wilson B value ...29.2 Avg B value (A˚2) ...40.4 Light chain ...38.0 Heavy chain ...43.0 Peptide...41.7 Water ...38.8 RMS deviation for bond
lengths (A˚ ) ...0.016 RMS deviation for bond
angles (°) ...1.55
Ramachandran plotc(%)...97.0 (0.0)
aR
sym⫽ ⌺PIi⫺ ⬍Ii⬎P/⌺PIiP, whereIi is the scaled intensity of theith
measurement, and ⬍Ii⬎ is the mean intensity for that reflection. Rcryst ⫽
⌺储FobsP⫺PFcalc储/⌺PFobsP, whereFcalcandFobsare the calculated and observed
structure factor amplitudes, respectively.Rfree⫽as forRcryst, but for 5% of the
total reflections chosen at random and omitted from refinement. RMS, root mean square.
bStatistics for the highest resolution shell are in parentheses.
cPercentage of residues in the favored region of the Ramachandran plot
generated by MolProbity (21) (outliers are in parentheses).
on November 8, 2019 by guest
http://jvi.asm.org/
differences between Z13e1 and 4E10, the crystal structure of Fab Z13e1 was determined in complex with an MPER pep-tide to 1.8-Å resolution. The 12-residue peppep-tide includes 8 native gp41 residues corresponding to the core epitope of Z13 (W670NWFDITN677) followed by a polylysine solubility
tag. In a previous study, this peptide was named 178-1 and was a member of a series designed to assess the boundaries of Z13e1 and 4E10 recognition (11, 46). The peptide is numbered according to the HXB2 isolate (TrpP670, AsnP671, TrpP672,
PheP673, AspP674, IleP675, ThrP676, and AsnP677) and contains a
P chain identifier.
IgG1() was secreted from a stable CHO-K1 cell line trans-fected with the vector pIgG-Z13e1 (46). Fab Z13e1 was ppared by endoproteinase Lys-C digestion by following the re-ductive alkylation protocol previously described for 2F5 (47). Crystals of the complex were grown by the incubation of Fab (27 mg/ml) with peptide 178-1 (5 mg/ml), followed by sitting-drop vapor diffusion. Crystals normally grew after several days at 22°C, but only in a subset of equivalent drops. Diffraction-quality crystals were obtained by streak seeding and belong to tetragonal space group P41212. The structure was solved by molecular replacement and refined to a finalRcrystof 21.0%
(Rfree⫽25.2%) at 1.8-Å resolution. The final model contains Fab residues L3 to L211 (light chain) and H1 to H227 (heavy chain), numbered according to the nomenclature of Kabat et al. (31), and peptide residues P670 to 678. Only backbone atoms are visible for LysP678, which corresponds to a Trp in the
native sequence, and the rest of the polylysine tag is disor-dered. All CDR loops are ordered except for the apex of H3, which largely is disordered. The final refined model has excel-lent geometry for the Fab and the peptide with all residues, except for AlaL51(65), in the most favored regions (Table 1).
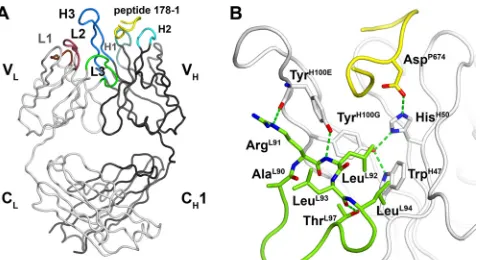
Overall structure of Fab Z13e1.The crystal structure of Fab Z13e1 reveals the canonical immunoglobulin ß-sandwich fold with an elbow angle of 168°. CDR L1 adopts canonical struc-ture 2, L2 canonical strucstruc-ture 1, and H2 canonical strucstruc-ture 2. H1 adopts canonical structure 1, but the peptide bond between IleH30and AsnH31is flipped to allow interaction of the
back-bone amide of AsnH31 with the MPER peptide. Thus, the
hydrogen bond normally present between the carbonyl of res-idue 29 and amide of resres-idue 31 is absent. The conformation and location of the CDR loops and bound peptide are depicted in Fig. 2A.
Conformation and role of L3. In Z13e1, L3 contains the affinity-enhancing mutations that were selected by phage dis-play and adopts a novel conformation. The L3 sequence AL90
RLLLPQL96(underlined residues are mutated from the
wild-type Z13 light chain sequence QL90RSDWPRL96) is more
hydrophobic, particularly in the stretch of tandem Leu residues that replace SerL92and AspL93. As the canonical structures for
L3 are dependent on the hydrogen bonding capability of the GlnL90 side chain (80% conserved in sequences) (2, 70),
mutation to Ala allows the loop to adopt a noncanonical con-formation. The most striking feature is that residues L95 to L97 now loop out from the combining site and are stabilized by a hydrogen bond between the main chain carbonyl of LeuL93
and the side chain hydroxyl of ThrL97(Fig. 2B).
Z13 can dimerize promiscuously with three lambda chain variants in addition to thelight chain, which gives the highest activity. However, the lambda chains have different canonical
L3 structures and do not contain GlnL90, suggesting that the
wild-typeL3 sequence does not contribute directly to binding or is not optimally packed against the heavy chain (80). The best L3 sequence match among antibodies of known structure is Fab 80R (Protein Data Bank entry 2GHW), which differs by only two residues from Z13wt L3: AspL933Asn and
ArgL963Pro. The L3 of Fab80R adopts a distorted canonical
1 conformation, which suggests that Z13wt also is likely to adopt a canonical 1 conformation, consistently with expecta-tions based on the observed frequency (2). However, modeling of the Z13wt L3 loop based on the 80R conformation results in numerous close contacts, particularly with ArgL91, TrpL94, and
ArgL96, implying that some local adjustment must take place in
the ligand-bound structure.
L3 is not involved in any direct contacts with the gp41 pep-tide, for which all Fab interactions are with the heavy chain. The noncanonical conformation allows LeuL92to drop down in
the bottom of the combining site and pack closely between the aromatic side chains of TyrH100Gand TrpH47, optimizing the
interface between VLand VH(Fig. 2). LeuL92does provide a
somewhat long van der Waals contact to ThrP676, but this
interaction is unlikely to contribute significantly to binding affinity (Tables 2 and 3). To assess the importance of LeuL92,
this residue was mutated to Ala, and the resulting affinity for peptide was determined (Table 4). In addition, a double re-version to the wild-type sequence was constructed by the mutations LeuL923Ser and LeuL933Asp. The LeuL923Ala
single mutant and LeuL923Ser, LeuL933Asp double mutant
[image:4.585.303.544.67.197.2]showed 2.3 and 1.9% binding relative to that of Z13e1 in peptide ELISA, respectively. Although the reversions may not be compatible with the Z13e1 L3 loop structure, these results illustrate the importance of LeuL92for optimal binding.
FIG. 2. Crystal structure of Z13e1 with bound peptide. (A) Over-view of peptide-Fab Z13e1 interactions showing the location of light and heavy chain CDR loops. Heavy chains and light chains are shown as tube representations. The locations of CDR loops L1 (brown), L2 (purple), L3 (green), H1 (marine), H2 (cyan), and H3 (blue) are shown with respect to bound peptide 178-1 (yellow). (B) Z13e1 mutations (from wild-type Z13) in L3 result in a novel L3 conformation and optimize the packing of the light chain H3 and TrpH47against heavy chain H3 and TrpH47. Residues 95 to 97 loop out to accommodate a lowering of residues 91 to 94. LeuL94 packs against TrpH47, and a hydrogen bond from TyrH100Eis made to be the backbone amide of LeuL92, further stabilizing the interface between V
Land VH. Leu L93is oriented toward L2, but the resulting hydrophobic pocket is not well packed. The light chain is shown in green, the heavy chain in whitish gray, and gp41 peptide in yellow.
on November 8, 2019 by guest
http://jvi.asm.org/
Conformation and role of H3.Z13e1 has a relatively long 17-residue H3 that encodes mainly hydrophobic, aromatic, and flexible residues (VH95AIGVSGFLNYYYYMDVH102) (80).
H3 displays the common ß-bulge torso, stabilized by a salt bridge between ArgH94 and AspH101 and a hydrogen bond
between the side chain of TrpH103 and carbonyl oxygen of
MetH100I (2). The apex of H3 is composed of GlyH100A,
PheH100B, LeuH100C, and AsnH100Dand has the highest B
val-ues of the model. This degree of disorder is consistent with the expected flexibility and observed lack of interactions with pep-tide 178-1, although some of the long CDR H3s in other human antibodies are remarkably well ordered (15, 47, 59, 66). As in 4E10, the tip of Z13e1 H3 bends away from the binding site and does not interact with bound peptide. Only residues at the H3 base contact peptide, including IleH97, GlyH98, ValH99,
TyrH100E, TyrH100F, and TyrH100G(Table 2). Small and flexible
residues (GlyH98, ValH99, SerH100, and GlyH100A) precede the
two hydrophobic residues, PheH100Band LeuH100C, at the tip
of the loop and may provide sufficient flexibility to adapt to the membrane-bound epitope.
Conformation of gp41 MPER peptide bound to Z13e1.Fab Z13e1 was cocrystallized with peptide 178-1 (W670NWFDI
TN677KKKK), which includes most of the residues in the
Z13e1 epitope but lacks Ser668 and Leu669 (11). Peptide 178-1 (Kd [dissociation constant], ⬃40 M) was critical for obtaining crystals, as the free N-terminal amine on TrpP670is
required for crystal packing. Peptides extended by the addition of N-terminal Ser668 and Leu669 or C-terminal Trp678 and Leu679 did not yield crystals with the Fab.
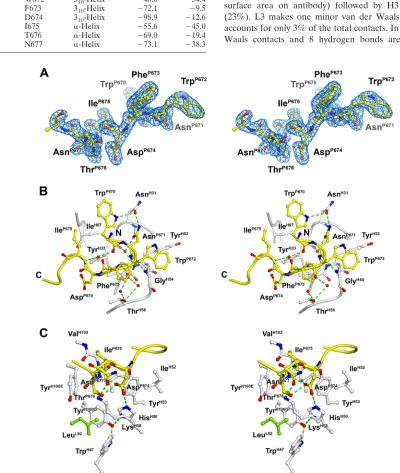
The peptide is bound in a meandering S-shape conforma-tion, with residues 670 to 671 displaying approximately helical backbone angles and residues 674 to 677 adopting a standard type I -turn or ␣-helical conformation (Table 5). The N-terminal turn orients the side chains of residues TrpP670,
AsnP671, TrpP672, and PheP673, along with their corresponding
backbone amides, in the same direction, suggesting a helical turn conformation. The C-terminal portion of the peptide adopts a type I-turn that also resembles a single helical turn, and it is stabilized by a hydrogen bond between the carbonyl of AspP674and the amide of AsnP677and via a side
chain-medi-ated hydrogen bond between the AspP674carboxyl and ThrP676
hydroxyl. The polar AspP674, ThrP676, and AsnP677residues are
clustered together (Fig. 3), forming a prominent hydrophilic surface. The S-shaped conformation allows the close approach of the aliphatic side chain of IleP675to the indole of TrpP670,
with their respective C␣atoms separated by only 5.3 Å, defin-ing a sharp bend in the peptide.
Structural basis for Z13e1 specificity.The combining site of Z13e1 buries 429 Å2 of surface area on the peptide and a
[image:5.585.303.543.88.233.2]corresponding 473 Å2on the antibody, which are within the
TABLE 2. Direct contacts between Fab Z13e1 and gp41 MPER peptide
gp41 MPER peptide
residue and contact typea Fab Z13e1 residue or atom Distance
(A˚ )
van der Waals contacts
TrpP670 AsnH31, TyrH33, IleH97, IleH52 AsnP671 IleH30, AsnH31, IleH52,
TyrH53, GlyH54 TrpP672 IleH52, TyrH53, GlyH54 PheP673 TyrH33, IleH52 AspP674 TyrH33, HisH50, IleH52,
ThrH56, LysH58 IleP675 TyrH33, IleH97, GlyH98,
ValH99, TyrH100E ThrP676 LeuL92, HisH50, LysH58,
TyrH100E, TyrH100G
AsnP677 LysH58
Hydrogen bond contacts
TrpP670-O TyrH33-OH 2.63
TrpP670-Nε1 AsnH31-O␦1 3.27
AsnP671-O␦1 AsnH31-N 2.81
AsnP671-N␦2 AsnH31-O␦1 2.67
AsnP671-O GlyH54-N 2.78
AspP674-O␦2 HisH50-Nε2 2.82
IleP675-N TyrH33-OH 2.94
AsnP677-O␦1 LysH58-N 2.74
aHydrogen bonds and van der Waals contacts were identified with CONTACSYM
(63).
TABLE 3. Buried surface areas in the Z13e1 combining sitea
gp41 residue
Buried
SA(A˚2
) Z13e1 residue
Buried
SA(A˚2
)
TrpP670 79.3 LeuL92 15.1
AsnP671 68.1 IleH30 12.8
TrpP672 54.4 AsnH31 37.0
PheP673 9.0 TyrH32 6.7
AspP674 51.8 TyrH33 50.2
IleP675 69.4 HisH50 16.3
ThrP676 67.5 IleH52 41.3
AsnP677 26.4 TyrH53 41.6
LysP678 3.2 GlyH54 31.2
ThrH56 27.1
ThrH57 2.9
LysH58 38.2
ArgH71 2.5
IleH97 37.8
GlyH98 21.0
ValH99 17.2
AsnH100D 7.3 TyrH100E 55.4 TyrH100F 1.1 TyrH100G 10.3
Total 429.1 473.0
a
[image:5.585.43.281.88.327.2]Buried surface areas (SA) were calculated with MS (19).
TABLE 4. Mutagenesis of key residues in Fab Z13e1 that contact MPER peptide
Mutation Region
% Apparent affinity relative to Z13e1
Peptide gp41
Wild type 100.0 100.0
LeuL923Ala CDR L3 2.3 1.1
LeuL923Ser/LeuL933Asp CDR L3 1.9 0.4
AsnH313Ala CDR H1 8.9 15.6
TyrH333Ala CDR H1 ⬍0.1 ⬍0.1
TyrH333Phe CDR H1 0.4 0.2
HisH503Gln FR H2 3.5 3.6
HisH503Ala FR H2 ⬍0.1 ⬍0.1
IleH523Ala CDR H2 0.9 0.5
PheH100B3Asp CDR H3 2.2 0.5 TyrH100G3Ala CDR H3 2.1 0.7
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.42.283.495.715.2]range expected for Fab-peptide complexes (400 to 700 Å2)
(67), and this compares favorably with the 451-Å2buried
sur-face area for the equivalent peptide epitope of 4E10 (15). Since peptide 178-1 lacks Ser668 and Leu669, which contribute significantly to binding affinity and may have specific interac-tions with the antibody, the buried surface area for the full epitope is expected to be slightly greater. As in 4E10, some residual available surface area in the combining site, particu-larly for H3, L2, and L3, may indicate that residues outside the core epitope are involved in binding.
Z13e1 uses all three heavy-chain CDR loops to bind peptide, with the greatest contribution from H2 (42% of total buried surface area on antibody) followed by H3 (32%) and H1 (23%). L3 makes one minor van der Waals interaction that accounts for only 3% of the total contacts. In total, 80 van der Waals contacts and 8 hydrogen bonds are made between
TABLE 5. Backbone torsion angles of MPER residues 670 to 677 bound to Z13e1 and 4E10
Antibody Residue Structure Phi (°) Psi (°)
Z13e1 W670 Turn ⫺18.7
Z13e1 N671 Turn ⫺76.3 ⫺21.4
Z13e1 W672 Turn ⫺119.6 ⫺23.8
Z13e1 F673 Turn ⫺130.1 130.6
Z13e1 D674 Turn ⫺64.8 109.0
Z13e1 I675 Turn ⫺60.0 ⫺12.9
Z13e1 T676 Turn ⫺98.6 5.6
Z13e1 N677 Turn ⫺80.6 168.4
4E10 W670 47.8
4E10 N671 Coil ⫺87.8 116.0
4E10 W672 310-Helix ⫺48.6 ⫺34.4
4E10 F673 310-Helix ⫺72.1 ⫺9.5
4E10 D674 310-Helix ⫺98.9 ⫺12.6
4E10 I675 ␣-Helix ⫺55.6 ⫺45.0
4E10 T676 ␣-Helix ⫺69.0 ⫺19.4
[image:6.585.77.477.203.676.2]4E10 N677 ␣-Helix ⫺73.1 ⫺38.3
FIG. 3. Combining site of Fab Z13e1. (A) Final 2Fo-Fc electron density for bound peptide 178-1 contoured at 1. Clear density is seen for all of the peptide epitope residues 670 to 677, whereas the C-terminal polylysine solubility tag is not ordered. (B) Interactions between the conserved aromatic residues TrpP670, TrpP672, and PheP673 and the Z13e1 heavy chain. The bound gp41 peptide 178-1 (yellow) is shown as a tube representation, with select side chain and backbone atoms as stick representations. (C) Interactions between MPER polar residues AspP674, ThrP676, and AsnP677(hydrophilic face) and the Z13e1 heavy chain.
on November 8, 2019 by guest
http://jvi.asm.org/
Z13e1 and peptide (Table 2). IleH97 (H3), GlyH98 (H3),
AsnH31 (H1), and IleP675 (peptide) contact one face of the
TrpP670indole. The indole nitrogen of TrpP670donates a
hy-drogen bond to the side chain carbonyl of AsnH31. AsnP671is a
highly contacted residue, forming hydrogen bonds with the side chain and backbone amide of AsnH31, a water-mediated
hydrogen bond with the backbone amide of TyrH53, and a
hydrogen bond between its backbone carbonyl and the amide of GlyH54. In contrast, TrpP672and PheP673make relatively few
side chain contacts with Z13e1 (Fig. 3B). The TrpP672indole
makes van der Waals contacts with TyrH53and GlyH54in H2,
whereas PheP673 is completely solvent exposed, although its
backbone carbonyl makes two water-mediated hydrogen bonds to ThrH56(Fig. 3B).
The N- and C-terminal halves of the peptide are bridged by the hydroxyl of TyrH33, which hydrogen bonds to the carbonyl
of TrpP670 and amide of IleP675. The hydrophilic cluster of
AspP674, ThrP676, and AsnP677inserts into the combining site,
resulting in the burial of the AspP674and ThrP676side chains
(Fig. 3C). AspP674 accepts hydrogen bonds from HisH50 Nε2
and a water molecule coordinated by the backbone carbonyl of ThrH57. IleP675is packed tightly against IleH97, GlyH98, TyrH33,
and TyrH100E. TrpP670and ValH99, whose side chain is largely
disordered, also contact IleP675. The ThrP676side chain, which
packs against TyrH100E, involves LysH58in hydrogen bonding
interactions. AsnP677is the last ordered residue in the peptide
and interacts only with theε-amine of LysH58.
Selected positions in Z13e1 and the MPER were mutated to assess the effect on binding and neutralization. Z13e1 mutants were tested for binding to peptide 178-1 and recombinant gp41 (a maltose binding protein fusion) in an ELISA format (Table 4), whereas mutations on the HIV-1 envelope protein itself were incorporated into a pseudotyped HIV-1JR2virus (Table
6) (46). TyrH33hydrogen bonds with the backbone atoms of
TrpP670and IleP675, both of which are invariant residues. The
mutation of TyrH333Ala or TyrH333Phe, which preserves
drophobic packing in the combining site but removes the hy-drogen bonding interactions to the side chain, knock out bind-ing (⬍0.1%). IleH52 packs against TyrH33 and provides
numerous van der Waals contacts to backbone atoms of the peptide. The mutation of IleH52to Ala results in⬍1% binding.
We tested additional mutations in the extended H3 tip. Al-though PheH100Bdoes not contact peptide, the mutation of this
residue or the deletion of the entire apex of H3 (data not shown) resulted in a 99% reduction in binding, a phenotype
reminiscent of an analogous mutation in 2F5 H3 (79). Overall, the mutagenesis studies strongly support the key roles of the antibody-antigen contacts established by the crystal structure. Neutralization by Z13e1 is critically dependent on Asp674 (46, 80). HIV-1 isolates with Asp (58% conserved) or Glu (1.1%) at 674 are neutralized by Z13e1, whereas those with Asn (12.6%), Ser (22.7%), or Thr (1.6%) are not (46). These results imply that a carboxylate at 674 is necessary for recog-nition. The structure reveals a complex hydrogen bonding net-work between the AspP674carboxylate and the antibody (Fig.
2B and 3C). In the unbound state, HisH50N␦1 would be
ex-pected to accept a hydrogen bond from the TyrH100Ghydroxyl,
as TyrH100Gmust accept a hydrogen bond from the indole NH
of TrpH47. However, upon binding to the AspP674carboxylate,
HisH50N␦1 could deprotonate TyrH100Gto form a
three-cen-tered, delocalized salt bridge. The equilibrium between the neutral and charged states of HisH50could serve to delocalize
the charge of AspP674by a proton relay mechanism. However,
in the major protonation state, AspP674and HisH50likely
in-teract through a hydrogen bond. The negative charge on AspP674 is presumed to be delocalized between O␦1, which
accepts two hydrogen bonds from ThrP676, and O␦2, which in
turn hydrogen bonds with HisH50. An asparagine at position
674 would require a hydrogen bond between its side chain NH2 and the protonated Nε2 of HisH50, which would destabilize the
complex. In isolates bearing a Ser at 674, no interaction with HisH50is possible. Thus, the structure shows that only a
car-boxylate can favorably interact with HisH50.
The neutralization of HIV-1JR2bearing the most commonly observed substitutions of Asn671 also was examined; Ser671 and Asp671 variants were neutralized as effectively as wild-type JR2, whereas the Thr671 variant was resistant (Table 6). To-gether, Asn671, Ser671, and Asp671 are observed in approxi-mately 94% of isolates, with Thr671 accounting for fewer than 6% of sequenced envelopes. Thr671 may not adopt the appro-priate rotamer to interact with AsnH31and, thus, would confer
neutralization resistance. The Ala substitution for 671 elimi-nates all three hydrogen bonds observed in the crystal struc-ture. The mutation of AsnH31to Ala resulted in 8.9% binding
relative to that of Z13e1 (Table 4). Thus, the structure clearly shows why Asn6713Ala and Asp6743Ala substitutions result in neutralization resistance to Z13e1 and helps explain the neutralization profiles of common variants (46, 80). These re-sults extend the previous Ala-scanning studies and indicate that the requirement for a carboxylate at 674 is the limiting factor in Z13e1 neutralization breadth.
DISCUSSION
The crystal structure of Z13e1 unexpectedly has revealed a strikingly different conformation of the gp41 MPER than that observed previously and gives insights into why this antibody differs substantially from the other MPER broadly neutralizing antibodies, 2F5 and 4E10, despite the recognition of an overlapping epitope. Peptide Ala scanning previously had as-signed the Z13e1 core epitope as S668LWNWFDITN677, which
overlaps with those of 2F5 (L661ELDKWASL669) and 4E10
(N671WFDITNWLW680). The Z13e1 epitope consists of two
[image:7.585.44.284.81.170.2]helical turns that are perpendicular to one another, and it
TABLE 6. Neutralization of HIV-1JR2mutantsa
Mutation gp41 sequence
IC50(g/ml)
Z13e1 4E10
Wild type L W N W F D I S N W L W Y I K 71.2 8.6
Ser6763Thr L W N W F D I T N W L W Y I K 69.5 7.5
Asn6713Ser L W S W F D I S N W L W Y I K 61.5 5.6
Asn6713Asp L W D W F D I S N W L W Y I K 55.4 5.4
Asn6713Thr L W T W F D I S N W L W Y I K ⬎200b
10.0
Phe6733Leu L W N W L D I S N W L W Y I K 38.6 ⬎100b
Asp6743Glu L W N W F E I S N W L W Y I K 91.9 15.0
a
Mutated residues are shown in boldface type. IC50, 50% inhibitory
concen-tration.
b
Neutralization was not observed at the highest concentration tested (200
g/ml for Z13e1 and 100g/ml for 4E10).
on November 8, 2019 by guest
http://jvi.asm.org/
differs from the more extended␣-helical conformation of the slightly downstream 4E10 epitope.
Our studies help explain and also extend the previous map-ping and mutagenesis aimed at defining the neutralization breadth of Z13e1 (46, 80). The mutagenesis of pseudotyped HIV-1JR2 in single-round infectivity assays revealed that
Asn671 and Asp674 are critical for Z13e1 neutralization. Clearly, Z13e1 is limited by its dependence on Asp674, which is deeply buried and interacts specifically with a histidine on the framework region of the antibody. Accordingly, the muta-tion of HisH50 is one of the most destructive, in terms of
binding affinity, of those analyzed in this study (Table 4). Z13e1 also makes numerous interactions with the peptide backbone, which are insensitive to sequence variation, and recognizes the side chains of invariant residues Trp670, Trp672, and Ile675, although these can be individually mutated to Ala without disrupting neutralization (46). Surprisingly, Z13e1 is tolerant of native sequence variation at Asn671 and insensitive to the common Ser substitution of Thr676 (Table 6). Thus, the structure reveals both the basis of specificity in this broadly neutralizing antibody and the limitations of its breadth of recognition for other clades and strains.
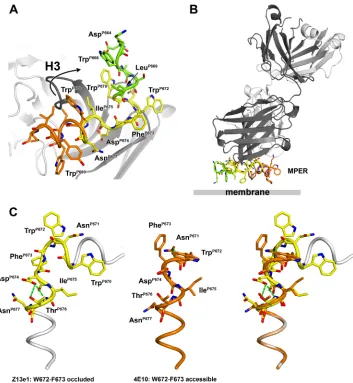
To gain insight into how Z13e1 interacts with the MPER as a whole, we modeled the natural N- and C-terminal extensions of the core epitope on the Z13e1 core epitope structure (Fig. 4A). We drew upon the reported observation (30) that crystal packing influences the conformation of Trp670 in the 2F5 structure (30, 47). New crystal forms of Fab 2F5 with different peptides show that the C terminus (W666ASLW670) of the 2F5
epitope can adopt a helical turn (30). As W670NWF673in the
Z13e1 structure is compatible with a helical turn, the C termi-nus of the 2F5 peptide epitope structure can be connected with the N terminus of the Z13e1 peptide epitope structure to create a short helix composed of residues 666 to 672 (Fig. 4A). Water molecules are found near the modeled main-chain car-bonyl groups of Ser668 and Leu669, indicating that the hydro-gen bonding requirements of peptide 178-1 are not fully met and, hence, are consistent with a truncated helix. The resulting model suggests that N-terminally extended peptides that in-clude residues 666 to 669 have increased affinity for Z13e1 due to the propagation of this helix and perhaps also mediate interactions with the apex of H3 (Fig. 4A). Similarly, the C terminus of the epitope may be modeled by appending resi-dues 678 to 683, as defined in the crystal structure of 4E10 (14). The resulting composite MPER structure consists of two helices bent by⬃90° at Phe673 and reveals that Z13e1 binds in the elbow region of the MPER. The composite model is largely, although not completely, amphipathic and suggests an orientation for the MPER with respect to the membrane, as depicted in Fig. 4B. The majority of hydrophobic and mainly aromatic residues would insert into, or interact closely with, the membrane, including Trp666, Leu669, Trp672, Phe673, Trp678, Tyr681, and Ile682. The hydrophilic residues are lo-calized on the opposite face; importantly, Asp664 and Asp674 both point away from the membrane. Trp670, Ile675, and Leu679 constitute hydrophobic residues that are not predicted to insert deeply into the membrane but form a hydrophobic cluster on the interior of the elbow. Although Trp670 and Ile675 are accessible to Z13e1, they could interact with phos-pholipid or other residues in gp41 in the native trimer, in which
case Z13e1 may displace these endogenous contacts. Such fea-tures are reminiscent of membrane-interacting strucfea-tures (26), particularly the membrane binding domain of PGHS-1 cyclo-oxygenase (53, 54).
A comparison of the 4E10 and Z13e1 structures yields sig-nificant insights into structural flexibility of the MPER. Trp672 and Phe673 are the most highly contacted residues in the 4E10 epitope, with buried surface areas of 123 and 108 Å2,
respec-tively. 4E10 completely buries these side chains, which also contact each other through a T-stacking (edge-to-face) ar-rangement of the aromatic rings. In contrast, Phe673 is the least contacted residue in the Z13e1 complex, with only water-mediated interactions with the peptide backbone and a buried surface area of just 9 Å2. Trp672 has more extensive
interac-tions with Z13e1 than Phe673, primarily via backbone hydro-gen bonding and van der Waals contacts with the accessible face of the indole ring, burying 54 Å2of total surface area in
the Z13e1 combining site. In support of the different confor-mations of the MPER in both crystal structures, Z13e1 effec-tively neutralizes a Phe6733Leu variant, a known 4E10 escape mutation (27, 78), whereas neutralization of this mutant by 4E10 is compromised considerably (Table 6). Thus, a large discrepancy is found in the inferred degree of exposure of these key residues on native trimers.
The composite model suggests an explanation and a mech-anism for how the Z13e1 and 4E10 epitope conformations can be readily interconverted to one another. In the Z13e1 con-formation, the C-terminal helical turn is capped by Asp674 (Fig. 4C and Table 5), which is 58% conserved and can be replaced by serine (23%) or asparagine (13%), both of which are common helix-capping residues (56, 57). In contrast, when 4E10 binds to the MPER, the C-terminal helix is extended and, instead, is capped by Asn671 (Table 5), which is 71% con-served and also can be replaced only by the helix-capping residues serine (22%) and threonine (6%). Thus, these resi-dues appear to act as a switch that enables the extension and stabilization of a longer C-helix in which Trp672 and Phe673 rotate out of the membrane. Therefore, Z13e1 binds a Trp672/ Phe673-occluded structure, while 4E10 ultimately recognizes a Trp672/Phe673-accessible conformation, although it is not clear whether these residues would be accessible in the initial encounter complex (69).
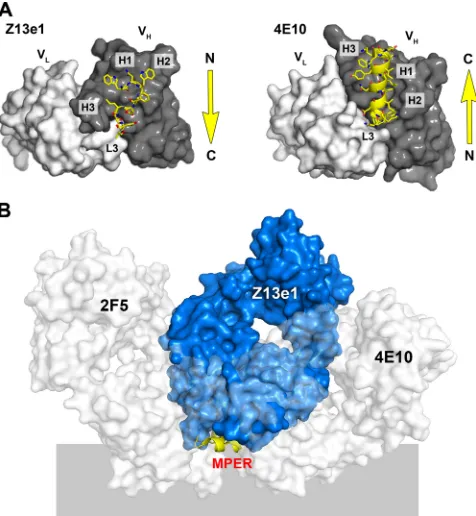
In addition to their distinct structures, 4E10 and Z13e1 also bind to opposite orientations of the MPER (Fig. 5A). With Z13e1, the N terminus of the peptide contacts H3 and H1 and extends toward L3. In contrast, the peptide interaction with 4E10 begins at L3 and ends at H3. As a consequence, the light chains are oriented on opposite sides of the MPER when the peptide structures are superposed (Fig. 5B). Both antibodies are oriented on one side of the putative membrane plane; however, the angle of the approach of the two antibodies is different. Since the MPER is thought to be difficult to access, it is surprising that residues 670 to 677 would be so well ex-posed as to allow two such widely different angles of approach. The epitope structure superposition may, therefore, indicate that these two MPER conformations are presented differently on the trimer and are associated with unique temporal and/or steric constraints. Alternatively, rearrangement of the MPER after initial antibody recognition could account for the appar-ent divergappar-ent approach and oriappar-entation of the two antibodies.
on November 8, 2019 by guest
http://jvi.asm.org/
Such a mechanism seems less likely for Z13e1, which binds less effectively to the hydrophobic residues presumed to be oc-cluded. If 4E10 does extract Trp672 and Phe673 from the membrane, as recently suggested (69), it likely takes advantage of a readily accessible conformation of the MPER.
The strict conservation of helix-capping residues at 671 and 674 suggests that the two alternate conformations are impor-tant for viral fitness and likely represent a conformational switch that plays a role in membrane fusion. In the 4E10 helical conformation of the MPER epitope, the backbone is
[image:9.585.116.469.66.449.2]seques-tered and the peptide presents an extensive hydrophobic face, with the conserved tryptophan residues distributed in a collar around the helical axis. As such, the helix may be able to interact with more than one hydrophobic surface, which could occur, for example, during the close apposition of viral and target membranes, as has been suggested (5). The elbow struc-ture of the MPER bound by Z13e1 is more clearly amphipathic and would be expected to interact with only a single hydro-phobic surface, presumably the viral membrane in the prefu-sion conformation. Interconverprefu-sion between the two forms,
FIG. 4. gp41 MPER and recognition by antibodies Z13e1 and 4E10. (A) A composite model of the MPER was constructed by the fusion of the 2F5 (green), Z13e1 (yellow), and 4E10 (orange) epitope structures. The fusion of the 2F5 and Z13e1 epitopes results in an N-terminal helix in the MPER consisting of residues 666 to 672. The C terminus of the MPER is modeled using residues 678 to 683 from the 4E10 crystal structure, resulting in a C-terminal helix. The composite model for the MPER consists of two consecutive helices from residues 666 to 683 that are kinked by approximately 90° around Phe673. The apex of H3 (GlyH100A, PheH100B, LeuH100C, and AsnH100D) may adopt a conformation in the presence of the complete epitope different from that observed in complex with peptide 178-1. (B) Proposed recognition of the MPER by Z13e1 with respect to the viral membrane. The composite MPER structure is largely amphipathic; the proposed membrane-facing side of the MPER contains only hydrophobic and mainly aromatic residues, which are expected to insert into the membrane. The MPER surface facing the solvent is largely, though not completely, hydrophilic and includes residues that become deeply inserted into the Z13e1 combining site. No specific immersion depth is implied. (C) On the left is a composite model of the MPER in tube representation with the Z13e1 epitope structure shown in yellow sticks. The view is from the perspective of the antibody looking down toward the membrane surface (perpendicular to the view seen in panel B). Z13e1 binds a Trp672/Phe673-occluded conformation and contacts the hydrophilic face of the MPER and hydrophobic residues in the interior of the elbow region. The Z13e1 conformation is characterized by the capping of the C-terminal helix by Asp674. The structure in the middle shows that 4E10 binds a Trp672/Phe673-accessible conformation that is characterized by the capping of the extended C-terminal helix by Asn671. The extension of the helix then would result in the extraction of Trp672 and Phe673 out of the membrane toward the accessible face of the MPER. On the right is the superposition of the Z13e1 composite MPER model with 4E10 MPER conformation.
on November 8, 2019 by guest
http://jvi.asm.org/
perhaps upon activation by receptor binding, may modulate tertiary contacts with other regions in gp41, or even gp120, or interactions of the MPER with lipids. Moreover, the two con-formations must differ in the elbow angle between the N- and C-helices of the MPER, which could affect the curvature of the viral membrane. Thus, the two MPER conformations may have distinct roles in membrane fusion, at different stages, due to differential interactions with the viral membrane or via the modulation of contacts made elsewhere in the trimer.
In summary, the crystal structure of Z13e1 with bound gp41 MPER peptide reveals a novel conformation of the MPER that is relevant for neutralization. Analysis of the complex also sheds light on the mechanism of neutralization and the struc-tural basis of specificity by this broadly neutralizing human
HIV-1 antibody and further suggests substantial structural plasticity in this conserved region of gp41. Neutralizing anti-bodies to the MPER are rare, as only three broadly neutraliz-ing monoclonal antibodies have been described, and serum-mapping studies have suggested that the hydrophobic MPER is not very immunogenic (8, 27, 33, 37, 38, 75). It also has been suggested that 4E10 is polyspecific and reacts with cardiolipin, whereby B-cell regulation (tolerance control) may normally delete MPER-targeting antibodies. Although this view remains controversial (3, 28, 29, 37, 61), the recognition of hydrophobic sequences that would result in equally hydrophobic and sticky binding sites, such as those in 4E10, may be problematic for the immune system and for autoimmunity. On the other hand, it is encouraging that human antibodies can be elicited that are as disparate in their fine specificity for the MPER as 4E10 and Z13e1, which neutralize by binding to very different conforma-tions of the MPER and to different subsets of residues. It also is possible that even more conformations relevant for neutral-ization exist but have not yet been structurally characterized. The structural characterization of novel MPER-targeting an-tibodies, particularly those elicited by non-B clades (8, 27), will be important for understanding the structural repertoire of the MPER. Immunogens based on known conformations and con-siderations of accessibility, including the masking of the hydro-phobic surfaces, may be superior to grafts of unrestrained peptides. Definitive evidence about the full extent of the con-formational transitions that occur during membrane fusion and how the MPER is presented in its native context on the membrane surface will require the structure of the prefusion gp120/gp41 trimer, as well as the complete postfusion structure of gp41, including the MPER, perhaps embedded in a lipid environment. However, as such structures already have proven difficult to obtain, the determination of MPER structures bound to neutralizing antibodies, such as described here, is extremely valuable and critical for the structure-assisted design of HIV vaccines and immunogens.
ACKNOWLEDGMENTS
We thank the staff of the Stanford Synchrotron Radiation Light-source (Beamline 11-1) for beamline support and assistance with data collection, A. Hessell for valuable technical assistance, D. Marciano and M. Elsliger of the JCSG for assistance with the IAVI/TSRI/JCSG CrystalMation robot, and R. Pantophlet and D. Leaman for useful discussions.
This work was supported by NIH grants GM46192 (I.A.W. and R.L.S.), AI69993 (M.B.Z.), and AI33292 (D.R.B.), NIH/NIAID NRSA fellowship AI74372 (R.P.), Austrian Science Fund J2845-B13 (J.S.G.), and American Foundation for AIDS Research fellowship 106427-34-RFHF (R.M.C.). The JCSG is supported by NIH NIGMS (U54 GM074898). The Scripps Research Institute thanks the Interna-tional AIDS Vaccine Initiative for its scientific and development sup-port and financial assistance.
This is manuscript no. 19745-MB from The Scripps Research Insti-tute.
The authors have no conflicting financial interests.
REFERENCES
1.Adams, P. D., R. W. Grosse-Kunstleve, L. W. Hung, T. R. Ioerger, A. J. McCoy, N. W. Moriarty, R. J. Read, J. C. Sacchettini, N. K. Sauter, and T. C. Terwilliger.2002. PHENIX: building new software for automated
crystallo-graphic structure determination. Acta Crystallogr. D Biol. Crystallogr.58:
1948–1954.
2.Al-Lazikani, B., A. M. Lesk, and C. Chothia.1997. Standard conformations
for the canonical structures of immunoglobulins. J. Mol. Biol.273:927–948.
[image:10.585.44.282.69.327.2]3.Alam, S. M., M. McAdams, D. Boren, M. Rak, R. M. Scearce, F. Gao, Z. T.
FIG. 5. Angle of approach of 2F5, Z13e1, and 4E10 to the MPER. (A) Orientation of the MPER bound by Z13e1 and 4E10. (Left) Z13e1 heavy chain (dark gray) and light chain (whitish gray) are shown in molecular surface representation. Bound peptide (yellow) is shown in stick representation and extends along the heavy chain from the N terminus, located between H1 and H3, toward the C terminus, which terminates at L3. (Right) Depiction of 4E10 variable domains, as described for Z13e1. In 4E10, the bound peptide extends from L3 along a groove in VHthat terminates between H3 and H1. Z13e1 and 4E10 bind their peptide epitopes in reverse, resulting in the splaying of their respective light chains to opposite sides of the MPER (see panel B). (B) Superposition of Fab 2F5 and Fab 4E10 onto the Z13e1 MPER composite model. The three Fabs and the composite MPER model are shown as molecular surfaces and a cartoon representation, respec-tively. 2F5 and 4E10 are colored whitish gray, Z13e1 is colored blue, and the MPER is colored yellow. The superposition indicates that the predicted angles of the approach of 2F5 and 4E10 are too high and would lead to severe clashes with the membrane. The epitopes of 2F5 and 4E10 are, therefore, likely to be presented differently on the trimer than that of Z13e1, mediated by flexibility of the elbow region of the MPER. Alternatively, 2F5 and 4E10 may have binding modes, perhaps extracting their complete epitopes from the membrane (69), that are different from that of Z13e1, for which the postbinding manipulation of the MPER is less likely.