0022-538X/08/$08.00⫹0 doi:10.1128/JVI.01537-08
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Equine Infectious Anemia Virus Resists the Antiretroviral
Activity of Equine APOBEC3 Proteins through a
Packaging-Independent Mechanism
䌤
Hal P. Bogerd,
1Rebecca L. Tallmadge,
2J. Lindsay Oaks,
2Susan Carpenter,
2and Bryan R. Cullen
1*
Department of Molecular Genetics and Microbiology and Center for Virology, Duke University Medical Center, Durham,
North Carolina 27710,1and Department of Veterinary Microbiology and Pathology, Washington State University,
Pullman, Washington 991642
Received 21 July 2008/Accepted 19 September 2008
Equine infectious anemia virus (EIAV), uniquely among lentiviruses, does not encode avifgene product. Other lentiviruses, including human immunodeficiency virus type 1 (HIV-1), use Vif to neutralize members of the APOBEC3 (A3) family of intrinsic immunity factors that would otherwise inhibit viral infectivity. This suggests either that equine cells infected by EIAV in vivo do not express active A3 proteins or that EIAV has developed a novel mechanism to avoid inhibition by equine A3 (eA3). Here, we demonstrate that horses encode six distinct A3 proteins, four of which contain a single copy of the cytidine deaminase (CDA) consensus active site and two of which contain two CDA motifs. This represents a level of complexity previously seen only in primates. Phylogenetic analysis of equine single-CDA A3 proteins revealed two proteins related to human A3A (hA3A), one related to hA3C, and one related to hA3H. Both equine double-CDA proteins are similar to hA3F and were named eA3F1 and eA3F2. Analysis of eA3F1 and eA3F2 expression in vivo shows that the mRNAs encoding these proteins are widely expressed, including in cells that are natural EIAV targets. Both eA3F1 and eA3F2 inhibit retrotransposon mobility, while eA3F1 is a potent inhibitor of a Vif-deficient HIV-1 mutant and induces extensive editing of HIV-1 reverse transcripts. However, both eA3F1 and eA3F2 are weak inhibitors of EIAV. Surprisingly, eA3F1 and eA3F2 were packaged into EIAV and HIV-1 virions as effectively as hA3G, although only the latter inhibited EIAV infectivity. Moreover, all three proteins bound both the HIV-1 and EIAV nucleocapsid protein specifically in vitro. It therefore appears that EIAV has evolved a novel mechanism to specifically neutralize the biological activities of the cognate eA3F1 and eA3F2 proteins at a step subsequent to virion incorporation.
Equine infectious anemia virus (EIAV) is a macrophage tropic lentivirus that causes a lifelong persistent infection in horses and other equids (62, 63). Experimental infection of horses induces a reproducible clinical disease course and pro-vides a useful model to examine mechanisms of immune con-trol and virus persistence during long-term lentivirus infection (37, 45). Upon infection, most horses suffer an early episode of acute disease, including fever, viremia, and thrombocytopenia associated with high levels of virus replication in tissue mac-rophages (31, 51, 63). Resolution of acute disease is concurrent with the appearance of virus-specific cytotoxic T lymphocyte and neutralizing antibody and a decrease in plasma viremia (29, 46, 66). Recurrent episodes of high-titer plasma viremia and associated disease often occur within the first year after initial infection but generally abate in frequency and severity with time. Thereafter, most horses enter a lifelong clinically unapparent stage with no evident compromise to their long-term health. Notably, these horses do not eliminate the virus but become lifelong carriers. EIAV is similar to other lentivi-ruses, such as human immunodeficiency virus type 1 (HIV-1), in its overall genome organization; however, EIAV is
geneti-cally the simplest lentivirus and contains only three regulatory/ accessory genes:tat,rev, andS2. The Tat and Rev proteins of EIAV are functionally homologous to HIV-1 Tat and Rev but differ somewhat in the organization of functional domains and the RNA structures that regulate viral gene expression (3, 10, 11, 24, 35, 36). Of particular interest, EIAV is unique among lentiviruses in that it lacks avifgene.
The APOBEC3 (A3) family of cytidine deaminases (CDAs) was first identified in humans, where seven distinctA3genes are found in close proximity on human chromosome 22 (15, 32). The ability of A3 proteins to function as potent inhibitors of retroviral infectivity was first defined for human A3G (hA3G), which inhibits the infectivity of HIV-1 variants lacking an intactvifgene (HIV-1⌬Vif) (16, 64). Conversely, wild-type HIV-1 is largely unaffected by hA3G expression. Subsequent research has demonstrated that the HIV-1 Vif protein directly binds to hA3G and induces its degradation via the proteasome (14, 42, 65, 72). In the absence of Vif, hA3G interacts with the nucleocapsid (NC) domain of the HIV-1 Gag polyprotein and is specifically packaged into progeny virion particles (2, 12, 60, 73). Upon subsequent infection of a susceptible cell, hA3G interferes with retroviral reverse transcription, at least in part by inducing the deamination of dC residues on the proviral DNA minus strand, resulting in dU residues that then template the introduction of A residues, instead of G residues, on the proviral plus strand (30, 40, 71, 74). While massive mutagenesis of reverse transcripts by hA3G is clearly an important part of
* Corresponding author. Mailing address: Duke University Medical Center, Room 426, CARL Building, P.O. Box 3025, Durham, NC 27710. Phone: (919) 684-3369. Fax: (919) 681-8979. E-mail: bryan .cullen@duke.edu.
䌤Published ahead of print on 25 September 2008.
11889
on November 8, 2019 by guest
http://jvi.asm.org/
this protein’s inhibitory activity (47, 61), evidence also exists that hA3G can inhibit retroviral infectivity in the absence of detectable editing (50).
In addition to hA3G, humans also encode six other A3 family members, named hA3A, hA3B, hA3C, hA3D/E, hA3F, and hA3H (15, 32, 52). These proteins can be subdivided into three proteins that contain a single-CDA active site consensus sequence (hA3A, hA3C, and hA3H) and four that contain two tandem CDA domains (hA3B, hA3D/E, hA3F, and hA3G). At least three hA3 proteins can function as potent inhibitors of a range of retroviruses, i.e., hA3B, hA3F, and hA3G (4, 20, 38, 70), and these are all double-CDA-domain proteins. On the other hand, single-CDA-domain proteins, such as hA3A, can function as efficient inhibitors of retrotransposon mobility (7, 13). In the case of hA3G, mutational analysis has revealed that the amino-terminal CDA is enzymatically inactive and func-tions to recruit hA3G into retroviral virion particles, while the enzymatically active carboxy-terminal CDA is required for in-hibition of infectivity (28, 49). Recent data suggest that the segregation of A3 proteins into a virion-packaging domain and an inhibitory domain may greatly facilitate their antiretroviral activities (27).
There is considerable interest in understanding the selective forces that shape the evolution and activity ofA3 genes. A number ofA3genes have been identified in nonprimate spe-cies, and several of these have been characterized for antiviral activity against a range of viruses and/or retroelements. At least one double-CDA-domain A3 protein with antiviral activ-ity has been identified in mice, cows, sheep, and pigs (33, 34, 41, 70). The mouse genome contains one A3 gene (15, 69); however, genomic characterization of theA3 genes in cows, sheep, and pigs is incomplete. In cats, where theA3locus has been examined in more detail, there are four distinctA3genes encoding five A3 proteins. Three of the A3 proteins were shown to be active against feline foamy virus, whereas the other two proteins were active against feline immunodeficiency virus and feline leukemia virus (39, 48). Sequences of equine and canineA3genes have been included in phylogenetic stud-ies (15, 52), but no functional data are available.
While HIV-1 can block the inhibitory activities of hA3G and hA3F by using Vif to induce their degradation (16), other retroviruses have evolved other mechanisms to permit their replication to occur unimpeded by host cell A3 proteins. In the case of foamy viruses, the viral Bet protein has been found to directly bind to hA3G and to sequester this protein away from progeny virions without inducing its degradation (39, 58). Hu-man T-cell leukemia virus type I and Mason-Pfizer monkey virus have both been reported to selectively exclude A3 pro-teins encoded by their healthy host species from virion parti-cles (17, 19), although Mason-Pfizer monkey virus packages, and is inhibited by, murine A3 (mA3). Finally, murine leuke-mia virus (MLV) has also been reported to discriminate against its cognate A3 protein, mA3, but not against the het-erologous hA3G protein in virion incorporation (1, 20, 34). MLV has also been reported to be inhibited less effectively by virion-incorporated mA3 than by hA3G, even when virions containing equivalent levels of each protein were analyzed (9, 57).
In this paper, we characterize the equineA3gene locus and ask how the lentivirus EIAV, which uniquely lacks avifgene,
is able to grow in the presence of these equineA3gene prod-ucts. We report that horses express six distinctA3genes, four of which encode single-CDA domains and two of which, equine A3F1 (eA3F1) and eA3F2, contain double-CDA do-main proteins. While eA3F1 is a potent inhibitor of several heterologous retroviruses, including HIV-1⌬Vif, both eA3F1 and eA3F2 are weak inhibitors of EIAV. Inhibition of HIV-1 infectivity by eA3F1 is associated with high levels of editing of HIV-1 reverse transcripts. Both eA3F1 and eA3F2 are enzy-matically active CDAs, and both can function as effective in-hibitors of retrotransposon mobility. Moreover, mRNAs en-coding both eA3F1 and eA3F2 are expressed at readily detectable levels in vivo in tissues that support EIAV replica-tion, including macrophages. Surprisingly, we demonstrate that eA3F1 and eA3F2 are incorporated into EIAV virions as ef-fectively as hA3G, although only the heterologous hA3G pro-tein can inhibit EIAV infectivity.
MATERIALS AND METHODS
Characterization of the equineA3gene locus.The human double-CDA
do-mainA3genes with known antiviral activities (hA3B, hA3F, and hA3G) were
used to perform BLASTN and/or TBLASTN searches of the NCBI equine sequence databases. A total of 59 expressed sequence tag (EST) clones were identified from peripheral blood lymphocyte libraries (Table 1). Representative clones were obtained from the respective libraries, sequenced in full, and used to BLAST the horse genome sequence through NCBI (EquCab1; accession number NW_001799702). This identified four distinct genes on chromosome 28, each
containing a single-CDA domain. Two additional equineA3genes containing
double-CDA domains,EcA3F1andEcA3F2, were identified following reverse
transcription-PCR (RT-PCR) amplification of total RNA isolated from both fetal equine kidney (FEK) cells and equine peripheral blood mononuclear cells
(PBMCs). Subsequently, partial cDNAs ofEcA3F1andEcA3F2were also
ob-tained by RT-PCR amplification using RNA samples derived from additional unrelated horses.
To determine phylogenetic relationships among the A3 family members, the equine deaminase domains were aligned with CDA domains of several species, including hA3A (NM_145699), hA3B (NM_004900), hA3C (NM_014508), hA3D/E (NM_152426), hA3F (NM_145298), hA3G (NM_021822), hA3H (NM_181773), cow A3F (DQ974646), sheep A3F (DQ974645), pig A3F (DQ97 4647), mA3 (NM_030255), rat A3 (NM_001033703), cat A3Ca (EU109281), cat A3Cb (EU109281), cat A3Cc (EU109281), cat A3H (EU109281), hAID (NM_ 020661), hAPOBEC1 (NM_001644), and hAPOBEC2 (NM_006789). Phyloge-netic trees were constructed by the neighbor-joining method using a p-distance model, and the reliability of branching orders was assessed by bootstrap analysis using 1,000 replicates with MEGA4 software (68). Phylogenetic analyses were also performed with equine APOBEC family members AID, APOBEC1, and APOBEC2 to ensure the sequences identified were indeed A3 family members.
Analysis of A3 mRNA expression patterns.Tissues (brain, kidney, liver, bone marrow, lung, lymph node, and spleen) were collected postmortem from a
clinically healthy pony, snap-frozen in liquid nitrogen, and stored at⫺80°C until
use. PBMCs were obtained from a clinically healthy Arabian horse and isolated by density gradient centrifugation (Histopaque; Sigma). Monocytes and mono-cyte-derived macrophages (MDMs) were derived from the whole blood of a clinically healthy Arabian horse using previously described differential adherence methods (56). Equine dermal (ED) cells were obtained from the ATCC (CCL57), and FEK cells were isolated and cultured from primary cells as pre-viously described (55). Total RNA was extracted using TRIzol reagent
(Invitro-gen). RNA pellets were resuspended in 80l of water and 2l of RNase
inhibitor (RNaseOUT; Invitrogen) and treated with DNase (Turbo DNase-free
kit; Applied Biosystems). cDNA was produced from 1g of total RNA by
random hexamer priming and 200 U of MLV reverse transcriptase (Invitrogen) for 1 h at 37°C. Reactions were terminated by being boiled for 5 min, and the
cDNA was stored at⫺20°C until used. Reverse transcription efficiency was
verified in the cDNA preparations by quantitative real-time PCR for 18S rRNA (23).
For analyses of mRNA expression, 1l of cDNA (approximately 40 ng) was
amplified with eA3F1- or eA3F2-specific primers designed to exclusively amplify double-CDA-containing transcripts. The sequences of the eA3F1 primers were
on November 8, 2019 by guest
http://jvi.asm.org/
5⬘-CTGGCCGTGATGTTGCG-3⬘ and 5⬘-GCAGTCTCTGAAATCCCA-3⬘, amplifying a 579-bp product. The sequences of the eA3F2 gene primers were
5⬘-CATGGTCTTCAGGGATTTCAG-3⬘ and 5⬘-GAAGCGCTCACTTGAGA
ATC-3⬘, resulting in a 683-bp product. Expression plasmids encoding eA3F1 or
eA3F2 were included as controls for specificity. The-actin primers were 5⬘-G
CTCGTCGTCGACAACGGCT-3⬘ and 5⬘-CAAACATGATCTGGGTCATCT
TCTC-3⬘. Amplification reactions consisted of 20 mM Tris-HCl (pH 8.4), 50 mM
KCl, 1.5 mM MgCl2, 0.2 mM deoxynucleoside triphosphates, 0.2M of each
primer, and 2.5 U ofTaqDNA polymerase (Invitrogen, Carlsbad, CA). Cycling
conditions were as follows: initial denaturation at 95°C for 2 min and 40 cycles of 95°C for 30 s, 60°C for 10 s, and 72°C for 1 min, followed by a final extension of 72°C for 10 min. One-fifth of the reaction mixture volume was run on 1% agarose gel and stained with ethidium bromide for visualization.
Construction of molecular clones.Total RNA was isolated from FEK cells, and equine PBMCs were stimulated with phorbol 12-myristate 13-acetate using TRI reagent (Sigma). First-strand cDNAs were synthesized using total RNA from both FEK cells and equine PBMCs, using an oligo(dT) primer and Super-Script II reverse transcriptase (Invitrogen). Primer pairs were designed to am-plify the predicted A3 cDNAs identified in the NCBI’s Horse Genome Resource. The primer pairs for the predicted equine gene XM_001499905.1 (sense primer,
5⬘-gcgcGGTACCaccatggagaagttggatcct-3⬘; antisense primer, 5⬘-gcgcCAATTGct
tgagaatctcctcaagg-3⬘) and the XM_001499905.1 sense primer when paired with
an antisense primer based on equine gene sequence for XM_001499895.1 (5⬘-g
cgcCAATTGcttgagaaggtcctcaagctttctggccaggagat-3⬘) produced PCR products of
⬃1.1 kb. Capital letters indicate introduced Asp718 and MfeI restriction enzyme
sites. The PCR products were digested with Asp718 and MfeI and cloned in frame into pcDNA3-HA (digested with Asp718/EcoRI) to generate the
expres-sion plasmids peA3F1-HA and peA3F2-HA. TheeA3F1andeA3F2 cDNA
sequences were then determined. The eA3 expression cassettes, including the influenza hemagglutinin (HA) tag, were also transferred as HindIII/XhoI
frag-ments into the pK vector, which does not contain aneoselection cassette, to
generate pK/eA3F1-HA and pK/eA3F2-HA.
The following mammalian expression plasmids have been previously de-scribed: pEV53B (53); pUNC-SIN6.1CLW-1 (54); pHIT/G (60);
pNL4-3⌬Vif⌬Env, phA3G-HA, and pNL-Luc-HXB⌬Vif (6); pK/hA3G-HA and pK/
hA3A-HA (7); phA3A-HA (70); pSIV-AGM-Luc⌬Vif (41); pNCS (25); pDJ33/
440N1neoTNF
(18); and pCMVMus-6DneoTNF
(22). pFB-Luc was obtained from Stratagene.
A bacterial expression plasmid, pGEX4T-EIAV NC, expressing glutathione
S-transferase (GST) fused to 62 amino acids of EIAV Gag, spanning the two zinc
fingers of the NC domain (GGPLKAAQTCYNCGKPHLSSQCRAPKVCFKC KQPGHFSKQCRSVPKNGKQGAQGRPQKQTF; critical zinc binding resi-dues are underlined), was constructed by amplification of the relevant region of
the EIAVgag gene. This EIAV sequence was cloned in frame into the
BamHI/XhoI sites of pGEX4T. The pGEX4T-HIV NC plasmid, which en-codes HIV-1 NC fused to GST, has been previously described (5).
Cell culture.HeLa and 293T cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum and gentamicin (Gibco).
Viral production and luciferase assay.A total of 3⫻105293T cells were
transfected with 62 ng, 125 ng, or 250 ng of an A3 expression plasmid, with
pcDNA3 filler plasmid added to a total of 1g. In addition, cells were also
cotransfected with pNL-Luc-HXB⌬Vif (2 g), pSIV-AGM-Luc⌬Vif (2 g),
pNCS(1g), and pFB-LUC (1g) or pEV53B (1g) and pUNC-SIN6.1CLW-1
(1g). All viruses were pseudotyped by the addition of 200 ng of the vesicular
stomatitis virus G (VSV-G) expression plasmid pHIT/G to the transfection cocktail. Forty-four hours posttransfection, virus-containing supernatants were
collected, passed through 0.45-m-pore-sized filters and used to infect naïve
293T cells. At this time, viral producer cells were lysed in sodium dodecyl sulfate
(SDS)-acrylamide gel loading buffer containing-mercaptoethanol, and this
lysate was then analyzed by Western blot analysis for A3 and HIV-1 or EIAV Gag protein levels. Twenty-four hours postinfection, the infected 293T cells were lysed in passive lysis buffer (Promega) and luciferase activity was determined as previously described, using Promega’s luciferase assay system.
Protein purification and binding assays. Escherichia colistrain BL21 was transformed with pGEX4T, pGEX4T-HIV NC, or pGEX4T-EIAV NC. Protein
expression was induced by the addition of 1 mM isopropyl--D
-thiogalactopyr-anoside (IPTG) (Invitrogen). Protein purification and A3:Gag binding assays were performed as previously described (5). All A3 proteins were produced by
transfecting 10g of expression plasmid into 1.5⫻106293T cells using the
calcium phosphate method. At 44 h posttransfection, cells were lysed in 4 ml of 150 mM NaCl, 50 mM Tris (pH 7.5), and 0.5% NP-40 (binding buffer) and centrifuged to remove insoluble matter. One milliliter of the A3-containing
clarified supernatant was incubated with⬃250 ng of partially purified
recombi-nant GST, GST-HIV NC, or GST-EIAV NC, and 50l of washed
[image:3.585.44.539.80.345.2]glutathione-agarose beads at 4°C for 60 min. The glutathione-agarose beads/protein complexes were then
TABLE 1. GenBank accession numbers of equine APOBEC sequences
Gene Genome position
NW_001799702
Genome prediction
accession no. EST accession no.
EcA3A1 35,786,967–35,790,916 None CD464249, CD464288, CD464383, CD464405,
CD464506, CD464621, CD464667, CD464792, CD469998, CD470856, CD470941, CD471095, CD471110, CD471542, CD471720, CD471898, CD472043, CD472049, CD472086,a
CD472090, CD472206, CD472250, CD535895, CD536010, CD536673, CD469635, CD469656, CD469716, CD528537
EcA3A2 35,812,886–35,816574 XM_001499871 CD464369, CD465467, CD468714, CD469225,
CD469582, CD469636, CD469721,a
CD469966, CD470274, CD470596, CD470661, CD470697, CD470713, CD470714, CD470721, CD470816, CD470940, CD471162, CD471262, CD471678, CD471717, CD471750, CD471911, CD472071, CD472096
EcA3F1 35,834,317–35,839,741 XM_001499884 None
EcA3F2 35,846,468–35,851,888 XM_001499895 and
XM_001499905
None
EcA3C 35,858,593–35,860,628 XM_001501833 DN505376,aDN506153, DN506543a
EcA3H 35,865,424–35,869,931 XM_001501833 DN504650,aDN505866
EcAID XM_001493186
EcAPOBEC1 XM_001493159
EcAPOBEC2 XM_001500838
aObtained and fully sequenced EST clone.
on November 8, 2019 by guest
http://jvi.asm.org/
washed four times with binding buffer, and bound proteins were eluted with 100
l of SDS-acrylamide gel loading buffer containing-mercaptoethanol. Input
and bound fractions were then analyzed by Western blot analysis, as previously described (5, 6).
Packaging of hA3G, eA3F2, and eA3F1 into HIV-1 and EIAV virions.A total
of 1.5⫻106293T cells in a 10-cm dish were transfected by the calcium phosphate
method with 1.5g of an A3 expression plasmid, 4.5g pcDNA3 filler, and
either 12g of the pNL4-3⌬Vif⌬Env HIV-1 proviral clone or 6g of pEV53B
and 6g of pUNC-SIN6.1CLW-1. At 48 h posttransfection, virus-containing
supernatants were collected, filtered, and layered over binding buffer supple-mented with 20% sucrose. Virus-producing cells were lysed in SDS-acrylamide
gel loading buffer containing-mercaptoethanol and used for Western blot
analysis of the levels of A3 and viral Gag expression. Sucrose gradients were spun at 40,000 rpm for 90 min. Supernatants were discarded, and the pelleted virus
was resuspended in SDS-acrylamide gel loading buffer containing
-mercapto-ethanol. The lysate and purified virions were then analyzed by Western blot analysis.
Editing of HIV-1 proviral DNA.HIV-1 virions were produced in transfected 293T cells as described above and used to infect naïve 293T cells. Twenty-four hours postinfection, duplicate samples were either analyzed for luciferase activity to confirm inhibition of infectivity (data not shown) or lysed and total DNA was isolated using a DNeasy kit (Qiagen). Purified DNA was then digested with DpnI
to cleave any contaminating plasmid DNA. The 3⬘end of the virally encoded
luciferase reporter gene was amplified by PCR, cloned, and sequenced.
Western blot analyses.All Western blots were developed using the Lumi-Light Western blotting substrate (Roche), as previously described. Proteins were de-tected using the following reagents: HA-tagged proteins dede-tected with a mouse monoclonal HA antibody (Covance), followed by a secondary goat anti-mouse horseradish peroxidase (HRP)-conjugated antibody (Amersham). HIV-1 Gag was detected using a rabbit polyclonal anti-p24 antiserum (Division of AIDS, NIAID, NIH; produced by BioMolecular Technologies), followed by a secondary goat anti-rabbit HRP-conjugated antibody (Amersham). EIAV Gag was detected using serum from an EIAV-infected horse, followed by a secondary goat anti-horse HRP-conjugated antiserum (GeneTex, Inc.)
Bacterial mutator assay.A3 cDNAs, including the C-terminal HA tag, were excised from the relevant pK-based plasmid by cleavage with Asp718 and XhoI and subcloned into Asp718 and SalI sites present in the bacterial expression
plasmid pTrc99A (AP Biosciences). The uracil DNA glycosylase-deficientE. coli
strain BW310 (28) was transformed with the pTrc99A parental plasmid and vectors encoding the various A3 cDNAs. Transformed bacteria were then se-lected overnight on LB plates containing ampicillin. Twenty colonies were pooled into 2 ml of LB medium plus ampicillin plus 1 mM IPTG and cultures grown overnight at 37°C. One hundred microliters of the saturated culture was
then plated on LB plates containing 100g/ml of rifampin, and the total number
of rifampin-resistant (Rifr
) colonies per plate was counted 24 h later. To verify
protein expression, 100l of the saturated IPTG-induced culture was lysed and
analyzed by Western blot analysis as described above.
Retrotransposition assays.The retrotransposition assays used have been
pre-viously described (7, 22). Briefly, 3⫻105
HeLa cells were seeded into 35-mm
culture dishes and then transfected with 2g of reporter plasmid (the
intracis-ternal A particle [IAP] retrotransposition indicator plasmid pDJ33/
440N1neoTNF, the MusD retrotransposition indicator plasmid
pCMVMus-6DneoTNF
, or the controlneoexpression plasmid pcDNA3) and 2g of the
control pK parental plasmid, pK/hA3A-HA, pK/hA3G-HA, pK/eA3F2-HA, or pK/eA3F1-HA. At 72 h posttransfection, the cells were transferred to a 10-cm
dish and subjected to selection with 700g/ml G418 (Geneticin) for an
addi-tional 12 days. Neomycin-resistant (Neor
) colonies were then stained with crystal violet and counted.
Subcellular localization of the eA3F2 and eA3F1 proteins.HeLa cells were
transfected with 2g of either peA3F1-HA or peA3F2-HA. At 44 h
posttrans-fection, the cells were permeabilized (8), and eA3F1 and eA3F2 were visualized by staining with an anti-HA mouse monoclonal antibody, followed by goat anti-mouse antiserum conjugated to fluorescein isothiocyanate. Nuclei were identified by staining with Hoechst stain, as previously described (8).
RESULTS
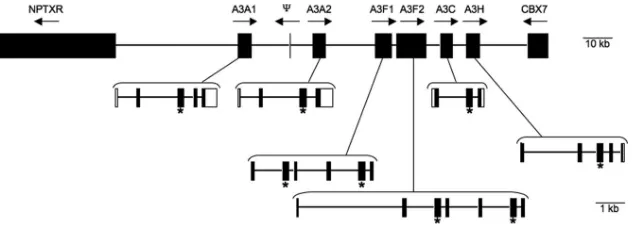
Identifying the equineA3 genome locus and expressed se-quences.The number of A3 genes in mammalian species is quite variable, ranging from a single gene in mice to seven genes in humans (15, 32, 52). Up to fourA3genes have been reported in other nonprimate species, including dogs, cats, cows, pigs, and sheep (15, 33, 48, 52). The recent availability of the horse genome sequence allowed a detailed physical char-acterization of the equineA3gene locus. Searches of equine EST and genome databases identified a number of genes ho-mologous to the hA3G gene. RT-PCR and sequencing were used to delineate gene boundaries and verify gene identity and expression in equine lymphoid cells. Collectively, these analy-ses identified six equine A3 genes that map in a cluster on equine chromosome 28. The six genes are arranged in a head-to-tail orientation and span 83,126 bp (Fig. 1). With the ex-ception of a single exon in the opposite orientation (Fig. 1), no otherA3-like genes were found in this locus, and no A3 se-quences were identified outside of this region. TheA3 gene locus on equine chromosome 28 is flanked by NPTXR and CBX7, which also flank the humanA3gene locus on human chromosome 22. The CBX6 gene, which is located between humanNPTXRandhA3A, appears to be present in the same location in horses; however, equineCBX6is not yet annotated. Overall, the conservation of gene order between horse chro-mosome 28 and human chrochro-mosome 22 extends fromC22orf28 throughALG12, spanning approximately 15 million bases of the horse chromosome. Therefore, the equineA3gene locus contains six expressed genes and is syntenic to theA3segment of human chromosome 22.
[image:4.585.133.453.67.180.2]Four of the six equine genes encode a single-CDA domain,
FIG. 1. Schematic of the equineAPOBEC3gene locus on chromosome 28. The six equineA3genes are shown between the flanking genes NPTXRandCBX7. The entire region spans 255,301 bp and the schematic is to scale, as indicated by bars. Transcriptional orientation is indicated by arrows; locations of conserved zinc-binding domains are indicated by asterisks. Gene schematics include exons (filled boxes), introns (lines), and untranslated regions (open boxes).⌿, the location of a truncated, EcA3A exon 2-like open reading frame. Gene order, size, and exon/intron boundaries are based on the EquCab1 assembly available at NCBI (accession number NW_001799702).
on November 8, 2019 by guest
http://jvi.asm.org/
and two genes encode double-CDA domains. Phylogenetic analyses indicated the eight equine CDA domains grouped with A3 sequences and segregated by Z1a, Z1b, or Z2 CDA domain designation (15) (Fig. 2). Within the “Z” groups, the CDA domains group by species rather than by gene designa-tion. Using the phylogenetic relationships and the number of CDA domains present per gene, we could group the six genes into twohA3A-like genes, twohA3F-like genes, onehA3C-like gene, and onehA3H-like gene. Although we recognize that it is difficult to assign nomenclature without functional charac-terization, we have designated these genes, listed in the order they occur in the locus,EcA3A1, EcA3A2, EcA3F1,EcA3F2,
EcA3C, andEcA3H(Fig. 1).
The two EcA3Agenes were previously identified asA3 by Conticello et al. (15). Each gene encodes one CDA domain and clusters with human Z1b CDA sequences, characteristics that are shared by hA3A. The first gene in the locus,EcA3A1, contains an atypical CDA sequence (DXEX27PCX2C rather
than HXEX27PCX2C). The aspartate mutation is present in 29
equine EST clones as well as the annotated genome. It is not known how this mutation affects activity, but EcA3A1 was expressed in PBMCs from several horses (data not shown).
TheEcA3F1and EcA3F2genes each contain two Z1a CDA
domains and cluster withA3Fsequences from multiple species. Moreover, the predicted eA3F1 and eA3F2 proteins show ex-tensive sequence homology to hA3F (HsA3F) at the amino acid level (Fig. 3). The transcript forEcA3F1 was amplified from PBMCs stimulated with phorbol 12-myristate 13-acetate, while several single- and double-CDA-domain transcripts for
EcA3F2were amplified from both FEK and equine PBMCs
(data not shown). Single-domainEcA3F2transcripts may rep-resent alternately spliced transcripts and/or PCR artifacts. The
EcA3Cgene contains a single Z1a CDA domain, whileEcA3H
[image:5.585.61.268.67.359.2]contains a single Z2 CDA domain and is thus most similar to
FIG. 2. Phylogenetic analysis of A3 domains. Neighbor-joining tree with bootstrap values based on A3 CDA domains. For proteins con-taining two CDA domains, domains are annotated with “N” for the N-terminal CDA domain or “C” for the C-terminal CDA domain. Domain names start with species: human (Hs), horse (Ec), cow (Bt), sheep (Oa), pig (Ss), cat (Fc), mouse (Mm), and rat (Rn). GenBank accession numbers are given in Materials and Methods.
FIG. 3. Alignment of the predicted amino acid sequences of the full-lengthHsA3F(hA3F),EcA3F1(eA3F1), andEcA3F2(eA3F2) proteins. Critical CDA domain residues are indicated by asterisks. The sequences ofEcA3F1andEcA3F2have been deposited in GenBank under NCBI accession numbers FJ174662 and FJ174663, respectively.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.110.473.466.697.2]hA3C and hA3H, respectively (Fig. 2). Interestingly, EcA3C
and EcA3H are computationally predicted to form a single,
seven-exon, double Z1a/Z2 CDA gene (XM_001501833). This predicted genomic organization and complement of CDA do-mains is most similar to theA3Fgene described in artiodactyl species (33). However, despite repeated attempts, we failed to detect any double-CDA transcripts by RT-PCR (data not shown). Rather, we consistently detected the independent ex-pression of either the 5⬘or the 3⬘half of this predicted double-CDA gene, which was also consistent with the EST clones (Table 1). Therefore, we conclude thatEcA3CandEcA3Hare in fact two separate genes, each with a single-CDA domain. Finally, comparison of the nucleotide sequence identity and amino acid similarity within and among the equineA3genes confirmed the presence of six distinct genes (Table 2). The
EcA3A1 and EcA3A2 genes were closely related, as were
EcA3F1andEcA3F2; however, the percent identity among the
alleles of each gene was higher and did not overlap the percent identity between the different genes. In summary, we identified six expressed genes in the eA3 locus, which is more than currently described for any other nonprimate species. Strik-ingly, the repertoire and organization of the eight equine CDA domains (five Z1a in single- and double-domain genes, two Z1b in single-domain genes, and one Z2 single-domain gene) are most similar to the human CDA organization rather than that of artiodactyl species, cats, or mice (15, 33, 48).
Tissue distribution of e3AF1 and e3AF2.The number and repertoire of the equine A3 genes suggest they may have evolved to play an important role in antiviral defense. Previous work has revealed that A3 proteins that show strong antiret-roviral activity, such as hA3G, generally contain two CDA domains (16), and it was therefore of particular interest to analyze the two equineA3double-CDA genes for breadth of expression and antiviral properties, especially against EIAV. To determine whether EcA3F1 and EcA3F2 transcripts (henceforth termed eA3F1 and eA3F2) were expressed in cells relevant to EIAV replication, a cDNA panel was assembled from tissues and cells that differ in permissiveness for EIAV replication in vivo and in vitro. The panel included seven
so-matic and lymphoid tissues (brain, kidney, liver, lung, bone marrow, lymph node, and spleen), circulating lymphoid cells (PBMCs and monocytes), and in vitro-cultured MDMs, ED cells, and FEK cells. Overall, eA3F1 and eA3F2 showed sim-ilar, variable patterns of mRNA expression in equine cells and tissues (Fig. 4). Abundant levels of each transcript were de-tected in the liver, lung, and lymphoid tissues, including PBMCs and MDMs. Low but detectable levels of mRNA were also present in brain, kidney, and FEK cells and monocytes; however, no amplified products were detected in ED cells. The abundant mRNA expression in spleen cells and MDMs are of particular interest because EIAV is a macrophage-tropic len-tivirus, and the main site of infection and replication in the acute and chronic stages of disease are the spleen and tissue macrophages of the lung, liver, kidney, lymph node, and bone marrow tissues (44, 51, 63). In contrast, very low mRNA levels were detected in circulating monocytes, which are not permis-sive for EIAV replication (43, 63). Field isolates of EIAV do not readily replicate in vitro, but certain isolates have been adapted to fibroblast cell lines, such as ED and FEK. Interest-ingly, low to no levels of eA3F1 and eA3F2 mRNA were present in FEK and ED cells. Collectively, these results dem-onstrate that transcripts encoding the equine double-CDA-domain proteins eA3F1 and eA3F2 are expressed in cells and tissues that are permissive for EIAV replication in vivo and, furthermore, that expression of these genes is not correlated with restriction of EIAV replication in vivo or in vitro.
Functional analyses of e3AF1 and e3AF2. An important attribute of A3 proteins is their ability to edit dC residues to dU on single-stranded DNA templates (71). To test whether eA3F1 and eA3F2 showed this enzymatic activity, we used a previously described (8, 28) DNA mutation assay in bacteria. This assay measures the ability of a protein to mutate theE. coliRNA polymerase B gene (rpoB). Mutations in rpoBare then detected by screening for the frequency of Rifrcolonies.
Expression of hA3A and hA3G has previously been shown to greatly or modestly enhance, respectively, the frequency of rpoBmutations (8, 28).
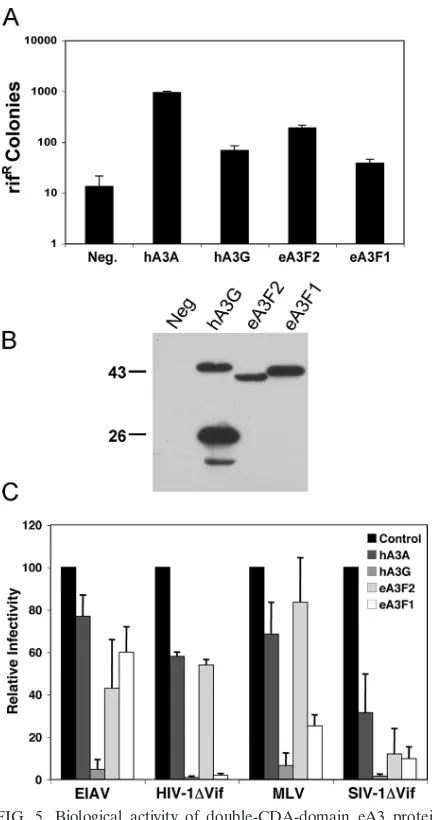
As shown in Fig. 5A, hA3A increased the incidence of Rifr
[image:6.585.42.284.91.169.2]colonies by⬃70-fold when expressed in bacteria, while hA3G increased their incidence by a more modest⬃5-fold. Similarly, expression of eA3F2 increased the number of Rifrcolonies by ⬃14-fold, while eA3F1 had an⬃3-fold enhancing effect. DNA sequence analysis of therpoBgene in the Rifrcolonies derived
[image:6.585.329.510.586.662.2]FIG. 4. Detection of eA3F1 and eA3F2 transcripts in equine cells and tissues. Total RNA isolated from the indicated tissues or cells was amplified by RT-PCR using eA3F1- and eA3F2-specific primers. Am-plified products were visualized following agarose gel electrophoresis and ethidium bromide staining. Specificity of each primer set is shown using peA3F1-HA and peA3F2-HA plasmid templates. -actin was used as an internal control for input RNA.
TABLE 2. Nucleotide identity and amino acid similarity among the equine APOBEC3 genes and allelesa
Gene % nucleotide identity and amino acid similarity of:
EcA3A1 EcA3A2 EcA3F1 EcA3F2 EcA3C EcA3H Allele
EcA3A1 100 85.9 30.3 30.3 41.5 45.7 99.7–100b
EcA3A2 79.0 100 30.0 29.4 39.6 47.7 97.0–100c
EcA3F1 16.2 15.6 100 92.9 46.3 26.4 99.1–100d
EcA3F2 15.9 15.3 85.6 100 45.7 25.2 98.2–100e
EcA3C 30.2 29.0 43.1 42.2 100 34.5 ND
EcA3H 25.9 25.8 12.4 12.4 22.9 100 ND
a
Percent nucleotide identity and amino acid similarity were calculated using sequences from representative clones in the EST database or identified in this study. The percent nucleotide identity is given above the 100% diagonal, and the amino acid similarity is shown in boldface below the diagonal. ND, not deter-mined.
b
Based on 421 bases of exons 1 to 3 from 18 EST clones.
c
Based on 460 bases of exons 1 to 3 from 19 EST clones.
d
Based on 579 bases of exons 2 to 6. Sequences were derived from five
unrelated horses by RT-PCR amplification and cloning usingEcA3F1-specific
primers.
e
Based on 673 bases of exons 2 to 6. Sequences were derived from five
unrelated horses by RT-PCR amplification and cloning usingEcA3F2-specific
primers.
on November 8, 2019 by guest
http://jvi.asm.org/
from the eA3F1- or eA3F2-expressing bacteria showed almost exclusively C-to-T mutations, as predicted (data not shown). We therefore conclude that both eA3F1 and eA3F2 are func-tional CDAs.
We next asked whether eA3F1 or eA3F2 would be able to inhibit the infectivity of HIV-1, simian immunodeficiency virus (SIV), EIAV, or MLV by producing infectious retroviral vec-tor particles based on each of these viruses in the presence of eA3F1 or eA3F2. hA3G, a potent inhibitor of many different retroviruses, and hA3A, a relatively weak inhibitor, were used as controls. For this purpose, we cotransfected 293T cells with a VSV-G expression plasmid (pHIT/G) and a plasmid encod-ing one of these four A3 proteins, together with a plasmid which encodes an HIV-1-based lentiviral reporter virus encod-ing luciferase but lackencod-ing an intact vif gene (pNL-Luc-HXB⌬Vif), or a similar plasmid encoding a⌬Vif version of an African green monkey SIV provirus (pSIV-AGM-Luc⌬Vif). Alternatively, the A3 expression plasmids were cotransfected with a plasmid encoding an MLV-based retroviral vector ex-pressing luciferase (pFB-luc) and an MLV packaging plasmid (pNCS) or a plasmid encoding an EIAV-based lentiviral vector expressing luciferase (pUNC-SIN6.1CLW-1) and an EIAV packaging plasmid (pEV53B). Of note, pEV53B expresses ev-ery known EIAV protein except Env and Tat (53).
As shown in Fig. 5C, hA3G acted as a potent inhibitor of the infectivity of HIV-1⌬Vif, SIVagm⌬Vif, EIAV, and MLV, while hA3A had little effect on viral infectivity, with the excep-tion of SIVagm, which was inhibited by approximately three-fold. eA3F1 inhibited HIV-1⌬Vif infectivity by ⬃25-fold, SIVagm⌬Vif infectivity by⬃10-fold, and MLV infectivity by
⬃3-fold but inhibited EIAV infectivity by ⬍2-fold. Finally, eA3F2 inhibited SIVagm⌬Vif infectivity by ⬃10-fold but re-duced HIV-1⌬Vif, MLV, and EIAV infectivity by ⱕ2-fold (Fig. 5C).
The data presented in Fig. 5C suggest that eA3F1 and eA3F2 are not effective inhibitors of EIAV infectivity but that they are capable of selectively inhibiting other retroviruses. To further confirm this result, we performed a limited dose-re-sponse analysis looking at the abilities of eA3F1, eA3F2, hA3G, and hA3A to inhibit EIAV or HIV-1⌬Vif infectivity (Fig. 6). As may be observed, these data confirmed the data reported in Fig. 5C showing that eA3F1 is a potent inhibitor of HIV-1⌬Vif infectivity, while hA3G is a potent inhibitor of EIAV infectivity. In contrast, neither eA3F1 nor eA3F2 ex-erted a strong inhibitory effect on EIAV.
[image:7.585.53.269.77.487.2]eA3F1 can edit HIV-1 reverse transcripts.Inhibition of ret-roviral infectivity by A3 proteins is usually, but not invariably, correlated with a significant level of editing of retroviral re-verse transcripts (30, 40, 50, 74). Specifically, hA3G and other inhibitory A3 proteins can edit dC residues on the proviral minus strand to dU residues, resulting in misincorporation of A in place of G during copying of the proviral DNA plus strand by the reverse transcriptase (71). As eA3F2 did not exert a strong inhibitory effect on any retrovirus tested (Fig. 5C), we instead asked whether eA3F1 was capable of editing HIV-1 reverse transcripts made by HIV-1⌬Vif virions generated in its presence. As shown in Fig. 7A, eA3F1 in fact induced a level of G-to-A editing that was at least equal to that seen upon coexpression of hA3G with HIV-1⌬Vif. We also noted a sig-nificant increase in C-to-T mutations, which suggests that
FIG. 5. Biological activity of double-CDA-domain eA3 proteins. (A) This assay measures the abilities of A3 proteins to enhance mu-tagenesis levels in bacteria. Plasmids encoding the indicated proteins were introduced into bacteria, and their expression was induced. The level of mutagenesis was then assessed by plating the bacteria on medium containing rifampin and counting the number of Rifrcolonies.
The average of eight experiments with the standard deviation is indi-cated. (B) Western blotting of A3 protein expression in the bacterial strain analyzed in panel A using an HA-tag-specific antibody. The propensity of hA3G to give rise to a truncated, carboxy-terminal form in bacteria has been previously noted (8). Size markers are indicated. (C) 293T cells were cotransfected with 200 ng of the VSV-G expression plasmid pHIT/G, 125 ng of a plasmid encoding the indicated hA3 or eA3 protein, or the parental pcDNA3 plasmid, together with either 2 g of a self-packaging retroviral luciferase expression plasmid (pNL-Luc-HXB⌬Vif or pSIV-AGM-Luc⌬Vif) or 1g each of a retroviral packaging plasmid and a cognate luciferase expression vector (pNCS and pFB-Luc or pEV53B and pUNC-SIN6.1CLW-1). At 44 h post-transfection, released retroviral virions were collected and used to infect naïve 293T cells. A further 24 h later, the infected cells were lysed and luciferase expression levels were determined. Data are pre-sented relative to the control culture, cotransfected with the parental pcDNA3 plasmid, which is set to 100 for each virus. The average of three independent experiments with the standard deviation is indi-cated. Neg., no APOBEC3 protein expressed.
on November 8, 2019 by guest
http://jvi.asm.org/
hA3G and eA3F1 are also able to edit the HIV-1 proviral plus strand, albeit clearly less effectively than the minus strand. In contrast, preliminary experiments have not detected any edit-ing of EIAV reverse transcripts produced from EIAV virions derived from cells expressing eA3F1, although editing of EIAV reverse transcripts by hA3G was readily detected (data not shown).
Previous work from several groups has shown that hA3G prefers to edit C residues located 3⬘to another C, while hA3F prefers to edit C residues located 3⬘to T residues (4, 38, 70). Consistent with the sequence similarity between hA3F and eA3F1 (Fig. 3), we observed that eA3F1 also preferred to edit TC, rather than CC, targets (Fig. 7B).
Virion packaging of eA3F1 and eA3F2.In order to inhibit retroviral infectivity, A3 proteins have to be specifically pack-aged into retroviral virion particles and then exert their inhib-itory effect during the reverse transcription process in newly infected target cells (16). It has been reported that some ret-roviruses that lackvifare able to selectively exclude specific A3 proteins from virion particles and thereby achieve resistance to their inhibitory effects (17, 19). To address whether the poor inhibition of EIAV infectivity exerted by eA3F1 and eA3F2 (Fig. 5C and 6) results from a similar exclusion phenomenon, we produced HIV-1⌬Vif and EIAV virions in the presence of hA3A, hA3G, eA3F1, or eA3F2 and then examined whether these proteins were packaged effectively. We have previously reported that hA3A is not effectively packaged into HIV-1⌬Vif virions (27), so this A3 protein is used here as a control for packaging specificity.
As shown in Fig. 8A, and as expected, hA3A was not pack-aged into HIV-1⌬Vif virions, while packaging of hA3G was
[image:8.585.76.495.67.290.2]FIG. 6. Dose-response analysis of the inhibition of HIV-1 and EIAV infectivity. This experiment was performed as described in the legend to Fig. 4, except that three different levels of each A3 expression plasmid (62 ng, 125 ng, or 250 ng) were analyzed. The upper panel shows the level of each A3 protein expressed in the EIAV-producing 293T cells, as determined by Western blot analysis. The middle panel shows the level of infectivity in each virus sample, given as a percentage of the level of transduced luciferase gene expression seen in the control culture cotransfected with the pcDNA3 parental expression plasmid. The average of three independent experiments with the standard deviation is indicated. The lower two panels show the relative level of production of the EIAV or HIV-1 Gag polyprotein in the producer cells, measured by Western blot analysis. A representative experiment is shown.␣-HA, anti-HA.
FIG. 7. Editing of HIV-1 reverse transcripts by hA3G or eA3F1. Infectious HIV-1 viral particles were produced in 293T cells cotrans-fected with pNL-Luc-HXB⌬Vif and either pcDNA3, phA3G-HA, or peA3F1-HA. These viruses were collected and used to infect naïve 293T cells, and the infected cells were then lysed 24 h later. Total DNA was isolated, and a segment of the virally encoded luciferase gene was amplified by PCR, cloned, and sequenced. (A) These boxes show differences between the predicted luciferase DNA sequence and the observed DNA sequence, given the total number of specific mutations observed in the absence of any exogenous A3 protein (none) or in the presence of hA3G or eA3F1. The number below each box shows the total number of bases sequenced. (B) The G to A mutations compiled in panel A were analyzed to reveal that the sequence context of the dC residue on the proviral minus strand was edited to dU by either hA3G or eA3F1.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.585.43.283.415.588.2]efficient. Interestingly, both eA3F2, which is a weak inhibitor of HIV-1⌬Vif infectivity, and eA3F1, a strong inhibitor (Fig. 4 and 5), appeared to be packaged into HIV-1 virions as effec-tively as hA3G (Fig. 8A). Analysis of A3 protein packaging into EIAV virions gave a closely similar result. Again hA3A failed to package, while the incorporation of hA3G, eA3F1, and eA3F2 was efficient and closely comparable. We therefore conclude that the inability of eA3F1 or eA3F2 to inhibit EIAV infectivity effectively, compared to hA3G, does not reflect a difference in virion incorporation efficiency.
To begin to address whether eA3F1 and eA3F2 were being packaged into retroviral virion cores, the experiments shown in Fig. 8 were repeated in the presence of the detergent Triton X-100, which has been reported to strip away proteins that copurify with HIV-1 virions but that are not located within the virion core (26). However, treatment of virions with this detergent did not reduce the association of hA3G, eA3F1, or eA3F2 with either HIV-1 or EIAV virion particles (data not shown), thus suggesting that these proteins are indeed core associated. The resistance of EIAV to inhibition by eA3F1 and eA3F2 observed in Fig. 5C and 6 must therefore occur via a mechanism that acts subsequent to their incorporation into virions.
One possible mechanism of resistance after packaging, first suggested by Abudu et al. (1) in the case of MLV resistance to inhibition by mA3, is that the packaged eA3F proteins might be cleaved by the viral protease after viral budding. At least in the case of eA3F1, we did in fact occasionally see an eA3F1 cleavage product in EIAV virions that was not present in the producer cells (Fig. 8B, lane 6). However, as cleavage was both far from complete and observed at similar levels in HIV-1 virions (Fig. 8A, lane 6), which are highly susceptible to inhi-bition by eA3F1 (Fig. 5C and 6), protease cleavage seems unlikely to explain the insensitivity of EIAV to eA3F1 inhibi-tion.
[image:9.585.65.260.65.392.2]Previously, we and others have reported that the selective incorporation of A3 proteins into retroviral virions is due to a specific interaction between the packaged A3 protein and the NC domain of the Gag polyprotein (2, 12, 60, 73). As eA3F1 and eA3F2 are apparently able to specifically package into both HIV-1⌬Vif and EIAV virion cores (Fig. 8), we therefore predicted that packaging should correlate with the ability to interact with the EIAV and/or HIV-1 NC protein in vitro. To
[image:9.585.111.473.553.687.2]FIG. 8. Analysis of packaging of eA3F1 and eA3F2 into HIV-1 and EIAV virions. 293T cells were cotransfected with either the HIV-1 proviral vector pNL4-3⌬Vif⌬Env, or the EIAV vectors pEV53B and pUNC-SIN6.1CLW-1, together with phA3A-HA, phA3G-HA, peA3F1-HA, or peA3F2-HA. At 48 h posttransfection, supernatant media were collected and virions were isolated by centrifugation through a sucrose cushion. (A) Western blot analysis of lysed HIV-1 producer cells and HIV-1 virions using an anti-HA (␣-HA) antibody or an anti-HIV-1 p24 (␣-p24) capsid antiserum. (B) Western blot analysis of lysed EIAV producer cells and EIAV virions using an anti-HA antiserum or an anti-EIAV Gag antiserum that recognizes the p26 capsid protein. The mobility of protein size markers is indicated.
FIG. 9. Specific interaction of eA3F1 and eA3F2 with both the HIV-1 and EIAV NC protein. Recombinant bacterial GST, or GST-HIV NC or GST-EIAV NC fusion proteins, were mixed with HA-tagged hA3A, hA3G, eA3F1, or eA3F2 from overexpressing 293T cells, and GST protein complexes were collected by incubation with glutathione-agarose beads. Bound (25% of total) and input (5% of total) proteins were then visualized by Western blot analysis using anti-GST (␣-GST) or anti-HA (␣-HA) antisera as previously described.
on November 8, 2019 by guest
http://jvi.asm.org/
perform this analysis, we incubated recombinant bacterial GST-HIV NC and GST-EIAV NC fusion proteins, or unfused GST, with recombinant mammalian HA-tagged hA3A, hA3G, eA3F1, or eA3F2. As may be observed in Fig. 9, hA3A, which fails to incorporate into either HIV-1 or EIAV virion particles (Fig. 8), also fails to specifically bind either the HIV-1 or EIAV NC protein. In contrast, hA3G, eA3F1, and eA3F2, which are selectively incorporated into both HIV-1 and EIAV virions (Fig. 8), also specifically interact with both GST-HIV NC and GST-EIAV NC but not with unfused GST (Fig. 9). These data therefore suggest that incorporation of eA3F1 and eA3F2 into EIAV virions, like incorporation of hA3G into HIV-1 virions, is mediated by a specific interaction with the NC domain of each Gag polyprotein.
Inhibition of retrotransposon mobility by eA3F1 and eA3F2.
While the A3 proteins first became of interest due to their ability to inhibit the infectivity of exogenous retroviruses, it is now clear that many A3 proteins are also capable of inhibiting retrotransposon mobility (7, 8, 13, 21, 22, 67). To test whether eA3F1 or eA3F2 also shared this activity, we examined their ability to inhibit the mobility of two murine long-terminal-repeat retrotransposons, MusD and IAP, using previously de-scribed retrotransposition indicator constructs (18, 22). These constructs contain aneogene, inserted into the MusD or IAP retrotransposon in the antisense orientation, disrupted by an
intron in the sense orientation. Therefore, Neorcan only occur
if the indicator construct is transcribed and spliced and then reverse transcribed and integrated.
To perform this analysis, we cotransfected HeLa cells with the MusD or IAP retrotransposition indicator constructs to-gether with vectors encoding hA3A, hA3G, eA3F1, or eA3F2. As a control for nonspecific toxicity, we also cotransfected each of these A3 expression plasmids with pcDNA3, which contains an intactneo gene that can confer Neorwithout requiring a
reverse transcription step. As shown in Fig. 10, none of these A3 proteins exerted any nonspecific inhibitory activity on Neor
conferred by pcDNA3. In contrast, and as previously de-scribed, hA3A acts as a potent inhibitor of IAP mobility, re-ducing retrotransposition of IAP by ⬃20-fold, while hA3G exerted a modest⬃2- to 3-fold inhibitory effect on IAP mo-bility (7, 22). Similarly, eA3F2 was observed to inhibit IAP mobility by⬃10-fold, while eA3F1 exerted an⬃5-fold inhibi-tory effect (Fig. 10).
In the case of the MusD retrotransposon, hA3G, again as previously reported, inhibited retrotransposition by⬃10-fold, while hA3A had only a modest inhibitory effect (22). In the case of MusD, eA3F2 proved to be an ineffective inhibitor, while eA3F1 inhibited MusD mobility by⬃10-fold (Fig. 10). We therefore conclude that while eA3F1 and eA3F2 can both function as potent inhibitors of retrotransposon mobility, these two eA3 proteins clearly have different abilities to affect the mobility of distinct retrotransposon species. The molecular bases for these differences are currently unclear.
eA3F1 and eA3F2 show distinct subcellular localizations.
While the majority of the two-CDA-domain A3 proteins, all of which are too large to passively diffuse into the nucleus, are found localized in the cytoplasm, at least one hA3 protein, hA3B, is localized to nuclei and in fact functions as a nucleo-cytoplasmic shuttle protein (8, 67). Analysis of the subcellular localization of the two equine double-CDA-domain proteins revealed that eA3F1, like hA3G and hA3F, localizes to the cytoplasm, while eA3F2, like hA3B, is found predominantly in the nucleus (Fig. 11). While the functional relevance of this difference in subcellular localization is currently unclear, it is interesting to note that both eA3F1 and eA3F2 are able to package into HIV-1 and EIAV virions with comparable
effi-FIG. 10. Analysis of the effect of eA3F1 and eA3F2 on retrotrans-poson mobility. HeLa cells were transfected with either pcDNA3, which contains an intactneogene, or with retrotransposition indicator constructs based on MusD or IAP, which can confer Neoronly after
undergoing retrotransposition. The cells were also cotransfected with pK-based vectors expressing the indicated A3 proteins or with pK itself. At 72 h posttransfection, to allow retrotransposition to occur, the transfected cells were selected for Neor, and resistant colonies were
[image:10.585.60.265.67.275.2]stained and counted 12 days later. (A) These data summarize four independent experiments analyzing the effect of the indicated A3 pro-teins on the mobility of MusD or IAP. The pcDNA3 plasmid repre-sents a control for nonspecific toxicity. Data are given relative to the pK control vector and show the observed standard deviation. (B) The expression of the indicated A3 proteins in the cotransfected HeLa cells was analyzed by Western blot analysis using an anti-HA antiserum. A representative experiment is shown.
FIG. 11. Subcellular localization of eA3F1 and eA3F2 in trans-fected cells. HeLa cells were transtrans-fected with either peA3F1-HA or peA3F2-HA, and the subcellular localization of each protein was de-termined at 44 h posttransfection by immunofluorescence. Nuclei were identified in parallel using Hoechst stain, as previously described (8).
on November 8, 2019 by guest
http://jvi.asm.org/
[image:10.585.335.505.69.204.2]ciency (Fig. 8), even though these are thought to be assembled exclusively in the cytoplasm.
DISCUSSION
Species-specific expansion and divergence of mammalianA3 genes have occurred in response to evolutionary pressure from exogenous and endogenous retroviruses and/or retroelements. The presence of sixA3genes in the equine genome (Fig. 1), including two Z1a/Z1a CDA genes (Fig. 2), is greater than that reported in any other nonprimate species and is nearly equal to that found in the human genome. The variability in the number ofA3genes among mammalian species, ranging from one to seven, results from gene expansion by duplication within a syntenic region (15, 59). An evolutionary history of gene du-plication is also likely for the equineA3genes, sinceeA3A1and
eA3A2are remarkably similar to each other, as areeA3F1and
eA3F2(Fig. 2 and 3; Table 2). It is not known whether the six
equineA3genes have redundant or separate functions. How-ever, the observed differences between eA3F1 and eA3F2 with respect to antiviral activity, subcellular localization, and retro-transposon specificity (Fig. 5C, 6, 10, and 11) indicate that at least some of the equine genes are functionally distinct. Phy-logenetic segregation of CDA domains into Z1a, Z1b, or Z2 types has facilitated comparative analyses of the repertoire of A3 genes within and among different species (15, 33, 52). Based on the complement of Z domains, it is evident that the usage and organization of the equine CDA domains are most similar to that seen in humans (Fig. 2). Like hA3F, the equine double-CDA-domain proteins eA3F1 and eA3F2 comprise two Z1a domains. In contrast, a Z1a/Z2 configuration is found in all of the double-CDA-domain proteins of nonprimate spe-cies characterized to date, including artiodactyls (A3F), cats (A3CH), and mice (A3) (15, 33, 48). Horses are not evolution-arily closer to humans than these other species; therefore, the similar complement of double-CDA domains may have arisen from similar selective pressures during the evolution of A3 genes.
The studies reported here demonstrate that eA3F1 has po-tent antiviral activity against HIV-1 and MLV but only modest activity against EIAV (Fig. 5C and 6). This suggests that EIAV has evolved a mechanism to resist or inhibit the intrinsic anti-viral activity of eA3F1. Retroviruses that lackvifuse various strategies to resist the antiviral activity of A3 proteins, and most, if not all, inhibit incorporation of cognate A3 proteins into virions (16). A possible exception to this generalization has recently been reported for MLV, which appears to package the cognate mA3 protein into MLV virion particles yet is apparently less susceptible to inhibition by virion-incorporated mA3 than by equivalent levels of virion-incorporated hA3G (9, 57). However, other groups have suggested that MLV virions also package hA3G somewhat more efficiently than mA3 (1, 20, 34).
In this paper, we demonstrate that EIAV does not block the expression of eA3F1 or eA3F2 in transfected cells (Fig. 6C and 8) or prevent EIAV NC binding or packaging of eA3F1 or eA3F2 into virions (Fig. 8 and 9). These data suggest that EIAV may employ a novel mechanism to escape the antiviral activities of eA3F1 and eA3F2 that act at a step after virion incorporation (57). This resistance does not result from any
lack of biological activity on the part of the eA3 proteins, as eA3F1 at least is clearly a potent inhibitor of HIV-1 and SIV infectivity and effectively edits HIV-1 reverse transcripts after virion incorporation (Fig. 5C, 6, and 7). Moreover, both eA3F1 and eA3F2 are active CDAs (Fig. 5A). The ability of EIAV to avoid inhibition by eA3F1 and eA3F2 at an undefined step after virion incorporation presumably explains the ability of EIAV to replicate in vivo in tissues that, as measured by mRNA expression levels, appear to produce substantial levels of these equine intrinsic immunity factors (Fig. 4).
ACKNOWLEDGMENTS
R.L.T. was supported by NIH grant T32 AI007025. Work at WSU was partially supported by grants from the Schindler Equine Research Funds and Washington State Equine Research Funds. Work in the laboratory of B.R.C. was supported by NIH grant R01-AI065301.
We thank Steven Goff (Columbia University), Nathaniel Landau (NYU), Thierry Heidmann (Institute Gustave Roussy), Fred Fuller (NCSU), Reuben Harris (University of Minnesota), and John C. Olsen (UNC) for reagents used in this research. The rabbit polyclonal HIV-1SF2 p24 antiserum reagent was obtained through the NIH AIDS
Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. We thank Sue Pritchard and Yousuf Jafarey for excellent tech-nical assistance.
REFERENCES
1.Abudu, A., A. Takaori-Kondo, T. Izumi, K. Shirakawa, M. Kobayashi, A. Sasada, K. Fukunaga, and T. Uchiyama.2006. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr. Biol.
16:1565–1570.
2.Alce, T. M., and W. Popik.2004. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein.
J. Biol. Chem.279:3408 51 -34086.
3.Belshan, M., G. S. Park, P. Bilodeau, C. M. Stoltzfus, and S. Carpenter.
2000. Binding of equine infectious anemia virus rev to an exon splicing enhancer mediates alternative splicing and nuclear export of viral mRNAs.
Mol. Cell. Biol.20:3550–3557.
4.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim.2004. Cytidine deamination of retroviral DNA by diverse
APOBEC proteins. Curr. Biol.14:1392–1396.
5.Bogerd, H. P., and B. R. Cullen. 2008. Single-stranded RNA facilitates
nucleocapsid: APOBEC3G complex formation. RNA14:1228–1236.
6.Bogerd, H. P., B. P. Doehle, H. L. Wiegand, and B. R. Cullen.2004. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad.
Sci. USA101:3770–3774.
7.Bogerd, H. P., H. L. Wiegand, B. P. Doehle, K. K. Lueders, and B. R. Cullen.
2006. APOBEC3A and APOBEC3B are potent inhibitors of
LTR-retro-transposon function in human cells. Nucleic Acids Res.34:89–95.
8.Bogerd, H. P., H. L. Wiegand, A. E. Hulme, J. L. Garcia-Perez, K. S. O’Shea, J. V. Moran, and B. R. Cullen.2006. Cellular inhibitors of long interspersed
element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. USA103:8780–
8785.
9.Browne, E. P., and D. R. Littman. 2008. Species-specific restriction of
Apobec3-mediated hypermutation. J. Virol.82:1305–1313.
10.Carroll, R., L. Martarano, and D. Derse.1991. Identification of lentivirus Tat functional domains through generation of equine infectious anemia
virus/human immunodeficiency virus type 1 tat gene chimeras. J. Virol.
65:3460–3467.
11.Carvalho, M., and D. Derse.1991. Mutational analysis of the equine
infec-tious anemia virus Tat-responsive element. J. Virol.65:3468–3474.
12.Cen, S., F. Guo, M. Niu, J. Saadatmand, J. Deflassieux, and L. Kleiman.
2004. The interaction between HIV-1 Gag and APOBEC3G. J. Biol. Chem.
279:33177–33184.
13.Chen, H., C. E. Lilley, Q. Yu, D. V. Lee, J. Chou, I. Narvaiza, N. R. Landau, and M. D. Weitzman.2006. APOBEC3A is a potent inhibitor of
adeno-associated virus and retrotransposons. Curr. Biol.16:480–485.
14.Conticello, S. G., R. S. Harris, and M. S. Neuberger.2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase
APOBEC3G. Curr. Biol.13:2009–2013.
15.Conticello, S. G., C. J. Thomas, S. K. Petersen-Mahrt, and M. S. Neuberger.
2005. Evolution of the AID/APOBEC family of polynucleotide
(deoxy)cyti-dine deaminases. Mol. Biol. Evol.22:367–377.
16.Cullen, B. R.2006. Role and mechanism of action of the APOBEC3 family
of antiretroviral resistance factors. J. Virol.80:1067–1076.