JOURNAL OF VIROLOGY, Nov. 1993,P. 6365-6378 0022-538X/93/116365-14$02.00/0
Copyright © 1993, American Society for Microbiology
Alternative Splicing of
Human
Immunodeficiency
Virus
Type 1
mRNA
Modulates Viral Protein Expression,
Replication, and Infectivity
DAMIAN F. J. PURCELL* AND MALCOLM A. MARTIN Laboratory ofMoleclularMicrobiology, NationalInstituteof Allergy andInfectious Diseases, Bethesda, Maryland 20892 Received 27April 1993/Accepted21 July 1993
Multiple RNA splicing sites exist within human immunodeficiencyvirus type 1 (HIV-1) genomic RNA, and
these sites enable the synthesis ofmanymRNAsfor each ofseveralviral proteins. We evaluated the biological significance of the alternatively spliced mRNA species during productive HIV-1 infectionsof peripheral blood lymphocytes and human T-celllinestodetermine the potential role of alternative RNA splicing inthe regulation of HIV-1 replication and infection. First,weusedasemiquantitative polymerase chain reaction of cDNAs that were radiolabeled for gel analysis todetermine the relative abundance of the diverse arrayofalternatively
spliced HIV-1 mRNAs. The predominant rev, tat, vpr, and env RNAs contained a minimum of noncoding
sequence,but thepredominant nef mRNAswereincompletely spliced and invariably included noncodingexons.
Second, the effect of altered RNA processingwasmeasuredfollowingmutagenesis of the major5' splice donor
and severalcryptic, constitutive, and competing 3' spliceacceptormotifs of
HIV-1NL4-3.
Mutations that ablated constitutive splice sites led tothe activation ofnewcryptic sites; some of these preserved biological function.Mutations that ablated competing spliceacceptorsitescaused marked alterations inthe pool ofvirus-derived mRNAs and, in some instances, in virus infectivity and/or the profile of virus proteins.The redundant RNA splicing signals in the HIV-1genomeand alternativelyspliced mRNAs providesamechanism forregulating the
relative proportions of HIV-1 proteins and, insome cases, viral infectivity.
Eucaryotic cells control their metabolic activities by regulat-ing gene promoter activity and the processing of RNA and protein. In thesame waythat the study of viralpromoters has servedasaparadigm for the promotersof their host
mamma-lian cells, the investigation of viral RNA processing in
mam-malian cells has provided an insight into various mechanisms
usedtoregulate the steady-state level ofaspecific mRNA. The
complexnature of theprocessing of human immunodeficiency virus type 1 (HIV-1) RNAs provides an important model for
human RNA processing pathways. All retroviruses require RNA splicing to remove upstream gag and pol coding
se-quencesfrom theenvmRNA. Inaddition, HIV-1 generatesa
distinctly complex pattern of spliced RNA to encode the essential regulatory proteins, Tat and Rev, as well asseveral other proteins (Vif, Vpr, and Nef) needed for successful replication in vivo (3, 15, 18, 26, 32, 35, 36, 43, 47, 52). The HIV-1 Rev protein binds viral RNAspecies that contain the Rev-responsive element (RRE), located in the env gene,
thereby promoting the export, and possibly the stability and translation, of partially spliced and unspliced RNAs from the nucleus into thecytoplasmfor its translationand/or packaging
intoprogenyvirions(2, 6, 7, 9, 12-14, 19, 20, 22, 23, 28, 33, 36,
38). The Rev-RRE system alleviates theparadoxical require-mentfor bothspliced and unsplicedHIV-1 RNA for successful virus replication. Rev protein also regulates the temporal changefrommultiply spliced HIV-1 RNAstopartially spliced
orunspliced RNAsduring productive virus infection (27, 29). Thesplicingof HIV-1 RNA isextremely complexbecause of the presence of both constitutive and alternatively used 5' RNA splice donor (SD) and 3' splice acceptor (SA) motifs. Numerous weak SA motifs, located toward the center of the
*Corresponding author.
genomic RNA, are competing points of ligation for splicing,
and their alternate selection usually determines the protein encodedby themature RNA (3, 15, 18, 35, 43, 47). However,
someof these mRNAsare multicistronic, encodingmorethan oneprotein (15, 48, 49). Increased diversity of spliced mRNAs for several HIV-1 proteins results from the alternative casset-ting of two noncoding exons into a proportion of transcripts
(15, 43, 47). In addition, the useofseveral cryptic SA and SD
sitesmayleadtothe synthesis of novel chimeric viral proteins (5, 15, 46, 47). The varieduse of these diverse splicing signals
results in thesynthesis of several setsofstructurally different
RNAsthatserveasalternative templates for the translation of
thesame protein, includingthe viralenvelope, regulatory,and
accessory proteins. Because this complex pattern of RNA
expression is maintained among many HIV-1 isolates of
di-verse origins (42, 43, 50), it is likely that this complexity is
critical for the successfulcompletion of the HIV-1 infectious cycle and not simply an inherent redundancy in viral RNA processing.
The major advantage of examining regulated splicing of a
self-replicating entitysuchasHIV-1 is that suchinvestigations
can usewholecells, rather than in vitro splicing extracts, and the biological consequences can be readily correlated with RNA and protein expression as well aswith virus infectivity. Whilemixedgroups of mRNAs encodemost HIV-1 proteins, it isunclear whether different cell typesusealternativesplicing to regulate HIV-1 RNA expression. The principal cell types thatareinfectedby HIV-1, CD4+ lymphocytesand monocyte-derived macrophages, are known to alternatively splice RNA from some cellular genes, depending on the maturation or
activation statusof the cell (e.g., CD45[54, 55])oron the cell type (e.g., CD46 [44]). Given the wide sequence diversity of HIV-l strains (37), it is likely that sequence differences will
6365
Vol. 67, No. 11
on November 9, 2019 by guest
http://jvi.asm.org/
6366 PURCELL AND MARTIN
affect the
splicing
motifs of different virus isolates in view of what is known in other systems(17,
34).
We
exploited
theself-replicating capacity
of HIV-1 toexamine the role of alternative RNA
splicing
in theregulationofvirus
replication
andinfectivity
andto evaluate the relativeimportance
of the different RNAsencoding
HIV-1proteins.
First,
we used asemiquantitative
polymerase
chain reaction(PCR) protocol
thatpreserved
the relativeproportions
of theHIV-1 RNA
species
in the 1.8- or 4.0-kb class ofpoly(A)+RNAtoevaluatethe
steady-state
levelsof viralmRNAsduringproductive
viral infection.Second,
we introduced mutations into several SD and SA motifs of the HIV-1proviral
clonepNL4-3
to assess their effectsonthecomposition
and relative abundance ofalternatively
spliced
mRNAsduring
virusrepli-cation and infection. HIV-1
splice
site mutantspermitted
anexamination of the
biological significance
of thelarge
redun-dantpool
ofspliced
mRNAs and thepotential
roleof alterna-tive RNAsplicing
in theregulation
of HIV-1during
tissue culture infections.MATERIALSANDMETHODS
Construction of
proviral
mutants. TheHIV-1proviral
mo-lecularclone
pNL4-3
wasconstructed from the NY5 andLAV(LAI)
HIV-1 isolates(1).
A PCR-basedmutagenesis protocol
that used a
mutagenic oligonucleotide
and a secondprimer
positioned
nearaconvenient restrictionendonuclease sitewasusedtogenerateaPCR
product containing
the mutation. Thisproduct
wasgel purified
and usedas amegaprimer
withathirdoligonucleotide positioned
near a second convenient restric-tion site togenerate DNAcontaining
the mutatedsplice
site and thetworestriction sites(31).
Theseproducts
werecloned into thepCR1000
vector(Invitrogen,
SanDiego,
Calif.),screened
by
restrictionmapping,
andthen cloned back into the HIV-1provirus by using
the selected restriction sites. Theoligonucleotides
used,
with mutated nucleotides underlined, were as follows: forSD1,
Odp.008
(5'-TGGCGTACTCTGC
AGTCGCCGCC-3')
withOdp.002
(5'-CTCTGGTAACTAG
AGATCCCTCAG-3')
and thenOdp.007
(5'-CTCATCTGGC
CTGGTGCAATAAGG-3');
forSA4b, Odp.023 (5'-AGGAG
ATGCTCAAGGC7lITYlGTCATG-3'),
and for SA5, Odp.025(5'-GTCTCCGCTTlCTTCGAGCCATAGG-3'),
each withOdp.021 (5'-GAATTGGGTGTCGACATAGCAG-3')
andthen
Odp.030
(5'-TTGFLT7AYTATTATlTTCCAAATTGTTC
TC-3');
forSA6,
Odp.028
(5'-GTGTTAGYTTATCTTG
CACTGATTTGAAG-3')
withOdp.030
and then Odp.021;and for
SA7a,
Odp.033 (5'-CTATAGTGAATTCAGTTAG
GCAGGGAT-3'),
forSA7a+7b, Odp.035
(5'-CTATAGTGAATTCAGTlTlTCGCAGGGATATT-3'),
and for SA7,7a,7b,
Odp.037
(5'-CTATAGTGAATTCAGFT[TCGCAGGGA
TATTCACCATTATCGT'Tl7CGTACCCACCTCCCCTATA
GTGAA T AGAGTTAG G
CAGGGATAT-3'),
each withOdp.032 (5'-CCGCAGATCGTCCCAGATAAG-3')
andthenOdp.031
(5'-AGTAGAGCAAAATGGAATGCCAC-3').
Splice
site mutantproviruses
were sequenced to verify the presence of the desiredchange
aswell as additional changes thatmight
have been introduced during the PCR procedure. Some mutations(ASA4b
Tat G->S,ASA5
Tat R->S, ASA6Env
K--S,
ASA7a Env R->S,ASA7b
Env R->S, and ASA7Env
Q->R)
changed
the codon at that splice site. Otherchanges
were asfollows:pNLASD1,
756A-4T(asilent change in thepackaging
signal);
pNLASA4b,
6002C-*T
(Tat A-V, RevL->F);
pNLASA5,
6143 A--G (Env E->G); pNLASA6,6695T->C
(Env
F->L);
andpNLASA7a+7b,
7361 T->C (EnvF--L),
8069 C->A(Env
L--M),
8107G->T
(EnvW->C), and 8321 T->C(Env
S--P).
Several other proviral splice sitemutants were sought; the resultant plasmids proved to be unstable, however, precluding their functional
analysis.
Cellculture,transfections,andinfections. HeLa
cells,
main-tained in Dulbecco's modified Eagle's mediumsupplemented
with 10% fetal calf serum
(FCS),
were obtained from the American Type Culture Collection. CEM(12D7)
cells,
main-tained in RPMI 1640 medium with 10% FCS, were obtained fromMicrobiological Associates(Gaithersburg,
Md.),
aswereperipheral blood mononuclear cells (PBMC), which were
stimulated with phytohemagglutinin
(0.25
,ug/ml;
Wellcome Diagnostics, Dartford, UnitedKingdom)for 96 h and grown in RPMI 1640with 10% FCS and 10% interleukin-2(Pharmacia
Diagnostics, Fairfield,N.J.).HeLacells(5 x
105)
in T25flaskswere cotransfected bythe calcium phosphate
coprecipitation
technique(57)with20
,ug
ofproviral plasmid
DNAand 0.5,ug
ofahuman growth hormone reporter plasmid, pXGH5
(10);
transfection efficiencywasdeterminedbya
radioimmunoassay
for human growth hormone
(Nichols
Institute, San Juan Capistrano, Calif.). Virus production was monitored with anassay for reverse transcriptase (RT) activity, using
[32P]TTP
incorporation withanoligo(dT)*poly(A) template (59). Cells (2 x 105) were infected with 105 cpm of RT activity of an
HIV-1 inoculum(approximately equivalent to a multiplicity of infection of0.002) in1ml ofRPMI 1640for2hat37°Cbefore addition of 4 ml ofRPMI 1640containing 10%FCS. The cells were fed with RPMI 1640 containing 10% FCS at 2-day
intervals, and aliquots of the medium were assayed for RT
activity.
Isolationof HIV-1mRNA,preparationofcDNA,and
semi-quantitative PCR for spliced HIV-1 mRNA. Total cell RNA was harvested by extraction with RNAzol (TelTest Inc., Friendswood,Tex.) 16or36haftertransfection of HeLa cells or immediatelyprior tothe peak ofRTproductionfollowing
infection of approximately 5 x 106 infected human PBMC; poly(A)+ RNA was selected by the Micro-Fast track
oli-go(dT)-cellulose method (Invitrogen). Forfirst-strand cDNA synthesis, either poly(A)+ RNA or in vitro-transcribed RNA
(seebelow) wasdenatured in 15 mM MeHgOH in the
pres-ence of 130 mM f-mercaptoethanol and 0.5 ,ug of random hexamers at 94°C for 2 min before reverse transcription (two
times) for 1 h with murineleukemia virus RT, using a cDNA cycle kit (Invitrogen). Semiquantitative PCR analysis of cDNA from the 1.8-kbclass of RNA was performed with oligonucle-otide primers Odp.045 (5'-CTGAGCCTGGGAGCTCTCTG
GC-3'; positions 477 to 499) and Odp.032 (5'-CCGCAG ATCGTCCCAGATAAG-3'; positions 8477 to 8498); for the 4.0-kb class of RNA, primers Odp.045 and Odp.070
(5'-ACTATTGCTATTATTATTGCTACTAC-3'; positions 6094
to 6115) were used. Twenty cycles of PCR were performed with 1 Uof Amplitaq (Perkin Elmer Cetus, Norwalk, Conn.), 2 ,lI of first-strand cDNA, 0.2 mM each dATP, dCTP, dGTP, and dTTP, and 1 ,uM each primer in 10 mM Tris-HCI (pH 8.3)-50 mM KCl-1.5 mM MgCl2-O.001% (wt/vol) gelatin by first incubating the mixtures for 5 min at 94°C and then
subjectingthem to thermocycling(94°C for 1.5
min,
55°Cfor 1min,72°C for 2.5 min) before finally incubating them at
72°C
for7min. Ifthe DNA concentration in a reaction mixture was less than 10ng/,l, as assessed byagarose gel electrophoresis,
aliquotswere sequentially reamplified in steps of five cycles after dilution 1:4 with fresh reaction mix to maintain linear amplification. Amplification products (100 ng) were radiola-beledby performing a single round of PCR as before but with addition of 10 ,uCi of[32P]dCTPandsubsequently analyzed by electrophoresis on a 6% polyacrylamide-urea gel at sufficient
current tomaintain a temperature of
65°C;
bands were visu-alizedby autoradiography or quantified with a Fujix BAS 2000J.VlIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
RNA SPLICING MUTANTS OF HIV-1 6367
Bio-image analyzer. An MspI digest of pBR322 was end labeled and used as size markers. Controls used in PCR experiments included cDNA from mock-transfected and
-in-fectedcells and poly(A)+orin vitro-transcribedRNA(below)
that was not incubated with RT during the cDNA synthesis
reaction.
Analysis of PCR products and cloning of HIV-1 cDNAs. Bands were excised from the PCR gel, eluted in 0.5 M
ammoniumacetate-10 mMmagnesium acetate-I mM
EDTA-0.1% sodium dodecyl sulfate (SDS) for 16 h at4°C, and then
precipitated with ethanolbefore rcamplificationwith thesame
primers. Afterit wasconfirmed that the PCRproductmigrated
as a singleband, residual primerswere removed with a Magic
PCR Prep column (Promega, Madison,Wis.), and theproduct
was directly sequenced by using end-labeled nested
oligonu-cleotideprimers in thedsDNACycleSequencing System(Life
Technologies, Inc., Gaithersburg, Md.). Purified PCR bands werealsoclonedintothepCRIIvector(Invitrogen)sothatthe
sense strand was downstream of the T7 promoter sequence, and the identities of clones were confirmed by sequencing. HIV-l cDNA cloneswere linearizedwith SpeI, extracted with phenol and chloroform (50%, vol/vol), and precipitated with
ethanol, and RNA was transcribed in vitro by usingT7 RNA
polymerase (Promega). The DNA template was removed
by
digestion (twice) with RNase-free DNase (Promega),and the RNA produced wasdirectly quantified by spectrophotometry
or density scanning of bands following agarose gel electro-phoresis. Selected in vitro-transcribedRNAswere diluted and then mixed in known proportion as controls for subsequent
cDNA synthesis and PCR amplification.
Analysis of HIV-1 proteins. Lysates were prepared from
transfected HeLacells(5 x 107cells perml) in0.5% Nonidet
P-40 in 10 mM Tris-HCI (pH 7.4)-150 mM NaCI-I mM EDTA-1 mMphenylmethylsulfonylfluoride.Differentamounts of the cell lysate, standardized on the basis of human
growth
hormone activity,wereseparated on SDS-5 to 20%
polyacryl-amide gradient gels, electroblotted onto Immobilon-P mem-branes (Millipore, Bedford, Mass.), and blocked with 5%
powdered skim milk in
phosphate-buffered
saline. HIV-1 proteins were detected by using serum(1:1,000)
from an HIV-1-seropositiveindividual, orrabbitserum to a NefN-ter-minal peptide (1:100) (21) or to
purified
NL4-3gpl60/120
(1:100) (58), and visualized with
'251-labeled
protein
Aby
autoradiography or
phosphoimaging
analysis.
Alternatively,
cells were
biosynthetically
labeled with[35S]methionine-cys-teine (Tran
35S-label; ICN, Irvine,
Calif.)
as describedprevi-ously (59) and
immunoprecipitated
with rabbit serum to Revproduced inEscherichiacoli
(1:100)
(gift
from D. M. D'Agosti-noand G. N. Pavlakis). Levels of Revfunctionalactivity
weremeasuredasdescribed
previously
(24) by
cotransfecting
0.5[Jg
of the pDM128 Rev reporterplasmid
with 20 jig ofproviral
plasmid and 0.5
p.g
ofpXGH5
humangrowth
hormone reporterplasmidandthenassaying
forchloramphenicol
acetyl-transferase
(CAT)
activity
on cell extractsthatwerestandard-ized for growth hormone expression.
RESULTS
The selection of different
competing
SA sites inprimary
HIV-1 transcriptsleads tothe alternativeexoncomposition
offully
processed
mRNA(Fig.
1).
Forexample,
functional Tatprotein, a transactivatorofHIV-I
transcription
(8,
35,40,
51),
isexpressedfromthetwotypesof mRNA
using
SA4(Fig.
IA):
RNAs containing exon
4E,
which is continuous from SA4 tothepoly(A) addition site
(one-exon
tat),
ortranscripts
joining
exons 4 and 7 (two-exon
tat)
(Fig.
IC)
(45).
Two othercompeting
SAsites,SA4a and SA4b(Fig. IA),
mapping
fewer than200bases downstream fromSA4,
give
risetoexons4aand4b,
which arespliced
to exon 7 to generate mRNA for Rev(Fig.
IC).
Anothercompeting splice
site,
SA5(Fig.
lA),
is used for theexpression
ofexons 5and SE(Fig.
IC);
RNAspecies
that contain exon 5E encode
envelope
gp16O,
and thosesplicing
exon 5 to exon 7 encode Nefprotein (15, 47).
Thetranslation initiation sites ofmRNAs
using
SA4a, SA4b, andSA5have poor ribosome
binding
capacities
andthereforehave thepotential
to be multicistronic: mRNAscontaining
exonSE,
4aE, or 4bE encodeVpu
andgpl6O
envelope
proteins,whereas
transcripts joining
exon 4aor4bwith exon 7 encode both Rev and Nef(15,
47, 48,
53).
This increases thecoding
potential
of several HIV-1 mRNAs and thecomplexity
ofHIV-1 mRNA
pool.
Furthercomplexity
results from theinterchangeable
incorporation
oftwononcoding
exons, exons 2and 3(Fig.
IC),
into thespliced
RNAspeciesutilizing
anyof the downstream SA motifs.PCR
protocols
using primers
that promoteamplification
oflimited
subgroups
of RNA have been usedinconjunction
with selectivehybridization probes
to map SA6,SD5,
SA7a,
andSA7b
(Fig. lA)
withinenv,thusgenerating
frameshifting
exons6,
7a,
and 7b(Fig.
IC).
These exons maygenerate
novelchimeric Tat-Env-Rev
(Tev
orTnv),
Rev-X-Tat, and Tat-Envfusion
proteins
following
transfection-infectionby
somederiv-atives ofthe HIV-I1Alstrain
(5,
15, 46,
47).
These aremerely
cryptic
sites in theHIV-1NL4-3
genome(see
below),
andtheir roleduring
HIV-1replication
is notclear(16).
Semiquantitative
PCRanalysis
ofspliced
HIV-1 mRNA.Since Northern
(RNA)
blotanalysis
ofHIV-1 RNA does notdistinguish
thefull array ofalternatively
spliced
RNAsencod-ing
thesameviralprotein,
weusedasensitivesemiquantitative
PCR assay and
urea-acrylamide gel
analysis
thatdiscriminatedbetween RNA species
differing
in sizeby
asingle
nucleotide.Twosuchassayswerecarriedout.Thefirst
selectively
detected the smallermultiply
spliced
1.8-kb RNAs that useSD4
andSA7, SA7a,
orSA7b(nef,
rev',
tat,orvpr) by
using theOdp.045
and
Odp.032
primers(Fig. lD);
the secondspecifically
ana-lyzed
thelarger
4.0-kb RNAspecies,
which containexonsthatextend
beyond SD4
into the envreading
frame(vpu/env,
one-exontat, vpr, and
viJf),
by
using
theOdp.045
andOdp.070
primers
(Fig. IE).
Representative gels,
depicting
the PCRproducts
that resulted from random hexamerpriming
ofthemultiple
species
of HIV-1 RNApresent
in infected PBMC priorto thepeak
ofRTproduction,
areshown inFig.
1D
andE. Each of these
cDNA
bands was excised from thegel,
reamplified,
anddirectly
sequenced.
Severalrepresentative
cDNA clones of each band were also introduced into the
pCRII
vector, which contains a T7promoter
adjacent
to thecloning
site,
and alsosequenced.
To ascertain whether the relativequantity
ofthePCR-amplified
cDNAbands visualizedon the
gel
faithfully
represented
the relative levels of thevarious RNA
species
isolated from infectedcells,
plus-sense
HIV-1 RNAwas
synthesized
fromeight
differentcDNAclones invitroby
using
T7RNApolymerase, directly
quantified,
andthen mixed in known
proportions.
Included among these RNAs was one,designated cryptic,
from a cDNA clonecontaining
exons1,
2x, 5,
and7,
where exon 2xreadsthrough
SD2
toacryptic
SDatposition
5059 invif.
The RNAmixtureswere then used as
templates
for cDNAsynthesis
and PCRamplification,
and the -P-labeled PCRproducts generated
were
analyzed
by
phosphoimaging (Fig.
2).
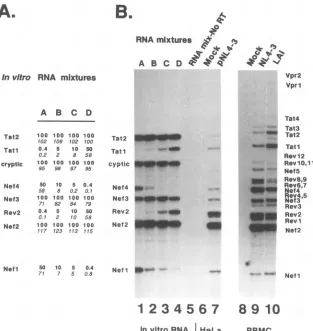
Wefoundthat theproportion
ofradioactivity
measured for each cDNA bandclosely
matched theproportion
ofRNAaddedto themixtureprior
to cDNAsynthesis
(Fig. 2A).
Thus, each cDNA reverse VOL.67, 1993on November 9, 2019 by guest
http://jvi.asm.org/
6368 PURCELL AND MARTIN
A.
Splice junctions
- Splice
fL HJ donors
-SI): I
B. Open reading frames
C'DC7O'
Ln i"in 4' mG, C;i)
Spike CIDO1%IIt-I~Oil IILn 1101,
>acptnjrs tiy t
-S: 2 3 4-ih: 6:
2 3 4
I'c L'Iev ' -'
7
-L
tE=
,-LLIn.-L
NT'PPU'
CO CO
CS} Cc 0
..i.J
CiD
(D C
CD G
_I c
iAAA,^.i.
71)
-8
7 ..
tat21tcv.3
n
,
rvXrC.
Exons 02 *3 -5 06M41) M4a - 4
-4
-- 3a
M.
ml
-
7_ _ 7a
I 2 J7b
- S~~E
4hE 4aE
4cE 4E
3E 2E
D.
PCR for
1.8 kb class of RNA
P('GRGelI RNAname Exoncontent lBand size
- Vpr 2 1/2J3a/ 7 1101 [it
VprI 11/3a/7 1051 nt
Tat4
Tat 3
I'aat2
_doo..It Tat I
Rev 12
Rev 11
Rev 11)
Rev 8
Revr7 Ref 6 Nef4
Re
Reesr4
Nef3 Rev3 Rev 2 Rev I N-ef 2
Nsef I
2/ /4/-7
113/ 7
I1,4/7 4/
1 / 3/ 4t 7
/2/3/4i /7
/2/3/4 /7
3 4c7
13 4a 7
3/4b 7 / /4a 7
I/12i4b 7/'7
/ /7
44il7
/4 /7
/ 5/
788rit 73>8Tlt
714 rIt
664 iit 629 nI 61 ntri
605lit
589Tit 5-79rii
93rin
555 nit 537n nt
5 15ior
487nt
480rit 464jit
-t -- Vif 2
_Vpr
4Vpr3 Tat 8 Tat 7
Tfat6
TI'at5 Env 16 Env 15 - / Env 14 Ern 13
/{nv 12
Env 11 Env 1(I _lp - .Env 8
W-f HlEiv7
lEnv6
. Envrh
En\ 4
Env I
/7 396Iot
E.
PCR for
4.0
kb
class
of
RNA
PCURCGel RNAname Exoncontent Band size
I/ 2E
I1/2 / 3E I/3E
I/2/3/4E 3/4E /2/4E /4E /2/3 /4cE I/2 /3 4aE
I/2/3 / 4bE
I/ 3/SE
/3!4cE
I/3!4aE I/3/4bE I1214cR /3/5E I/2/4aE I12/4bE
I/2/5E
/4cE
I/4aF
I/4bE I/5E
1474nt
1047nt
997ot
734 nt 684 nt 660 nt
610nt
575nt 557 nt 551 nt 535 nt
525nt 507nt 501 nt 501 nt 485 nt 483nt
477 nt 461 nt
451 nt 433nt
426nt 410nt
J. VIROL.
I
on November 9, 2019 by guest
http://jvi.asm.org/
RNA SPLICING MUTANTS OF HIV-1 6369
transcribed from the in vitro RNAwas amplified with equal
efficiency withuseof the Odp.045-Odp.032 primerpair.
PCRanalysisof the 1.8-kb classof mRNAwasperformedon
samples from HeLa cells transfected with HIV-1 proviral
clones or from human PBMC infected with cell-free HIV-1
(Fig. 2B). RNA directed bythe pNL4-3 cloned provirusDNA
intransfected HeLa cellsorby
HIV-lNL4-3
ininfectedhumanT lymphocytes yielded thesamepatternof bands innumerous
independent experiments irrespective of whether RNA was
synthesized followingtransfectionorinfection.Asimilar
band-ingpatternwasalso observed withRNApreparedfrom PBMC
infected with HIV-1LAI (Fig. 2B, lane 10). The results
pre-sented in Fig. 2 demonstrate that HIV-1 RNA is spliced
equivalently in transfectedHeLacellsandinfected PBMC and thatthesplicingpatternissimilar for closely related strainsof
HIV-1 suchas NL4-3and LAI.
These control PCR amplifications also illustrate two other
important points: (i) for each RNA mixture, the relative
proportion of the different amplified RNA species, as
deter-mined experimentally, closely approximated theoriginal
pro-portion added, and (ii) only PCR productsderiving from input RNAwere detected on the gel, indicating that only RNAs
spliced by genuine processing pathways gave bands in this
analysis. AbnormalcDNA species,potentially arising by
tem-plate jumping during reverse transcription of RNA, did not
appear asbands on the final autoradiogram. Thus, the PCR
protocol used here accurately reflects the relative quantityof
oneHIV-1 RNAspeciescompared with the others amplified in
thesamereaction.
Identification ofa newSA site forrevandvpu/envRNAs. Our
PCR analyses ofviral RNA synthesized in transfected HeLa cellsand infected PBMCindicatedthepresenceofapreviously
unreported competing SA site, designated SA4c, thatwasused
togeneratethreenovelrevmRNAs,rev3, rev6, and rev9(Fig.
1D), andthreenovelvpulenvmRNAs, env4, env9, and envl6 (Fig. 1E). TheSA4csite is 18 nucleotides (nt)upstreamfrom SA4a and isconservedamongmost,butnotall,HIV-1 isolates
(Fig. 3).TheSA4csplice site exists inHIV-lHXB2,
HIV-1LAI,
HIV-lJR-CsF,
andHIV-lBA-L,thestrains used in earlier studiesmapping thesplicing motifsofHIV-1, althoughitsuse wasnot noted(3, 15, 18, 35, 43, 47).Sinceno newtranslation initiation sites are introduced into mRNAs using SA4c, transcripts containingexons4c and 4cE would stillencodetypicalHIV-1 Revor Env protein. However, structural changes introduced into these mRNAs may affect their translatability. An ATG codonexists in theHIV-1NL43betweenSA4c and SA4a butis present in acontext unfavorable for efficient translation(30).
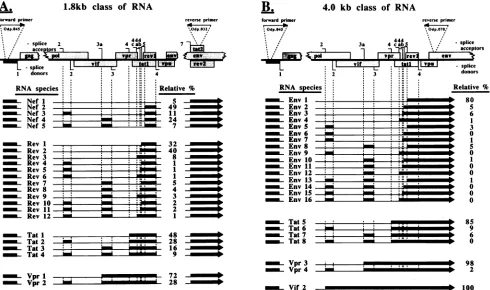
Relative abundance of the alternatively spliced HIV-1 mRNAs. Since PCR amplificationofHIV-1 cDNA preserved the relative proportion of the various alternatively spliced forms of HIV-1 RNA, we used phosphoimaging analysis to
directly determine the relative abundance of each of these
RNAs in a spreading
HIV-1NL4-3
infection of PBMC (Fig. 2and4).Within the1.8-kb class of HIV-1mRNAs(Fig. 4A), nef,
rev,tat,andvprspeciesexisted inaratio of 56:34:9:1;within the
4.0-kb class (Fig. 4B),env,tat, vpr,and vif species existedina
ratio of 92:5:2:1. Of the nef mRNAs, nef2, which includes
noncodingexon 5flanked bySA5 and SD4,wasthe
predom-inant type, comprising 49% of all nefRNA and 28% of all
1.8-kbRNAs.nefRNAscontaining noncodingexon3(nef4)or
2 (nef3) or both 2 and 3 (nef5) were present in decreasing amounts. Nefl RNA, which lacks a noncoding exon, was the
leastabundant nefmRNA. Itisunclear whether the
nontrans-lated RNAfromexon5 contributestonef function,asthenef gene has not been evaluated as an RNA element, only as
protein.
Incontrast, thepredominant spliced mRNAsfor the other
HIV-1 proteins lacked noncoding exons. Only 20% of rev
mRNA includes anoncodingexon, and theuse ofnoncoding exon 3 (rev7, rev8, and rev9) or both exons 2 and 3 (revl0,
revll, andrevl2) predominatesover useofexon2alone(rev4,
revS, and rev6). In addition, revl and rev2 mRNAs, utilizing
SA4a and SA4b, respectively, occur approximately fivefold morefrequently than rev3 mRNA, which usesthe previously
unreportedSA4c. Both theone-andtwo-coding-exonforms of tat mRNA (1.8- and 4.0-kb class RNAs, respectively) infre-quentlyusenoncodingexons2and 3(tatlandtat5). However,
when a tat mRNA contained a noncoding exon, exon 2 was
predominantly used (tat2andtat6).Therelativeproportion of
theone-andtwo-exonformsoftatorvprmRNA couldnotbe
determined here, since the assays for the 1.8- and 4.0-kb mRNAsaremerelysemiquantitativeamongthespecies
repre-sented in each PCR assay.
EightypercentofenvmRNAsuseSA5 (exon SE);however,
12% ofenvRNAs(env2, env3,andenv4)utilize the upstream
SA4a, SA4b,orSA4c(exon4aE, 4bE,or4cE)whennoncoding
exons 2 and 3wereexcluded. Noncoding exons 2and 3were
usedonlyatlow(<5%) frequencyinenvmRNA andmostlyin
conjunction with SA5 (envS, env8, and envl3). This study clearly identifies mRNA species containing both noncoding
exons2and3 in thesametranscript.Thisfindingcontrastswith
previous analyses of RNA directed by the HIV-lHXB2 strain that identified these as mutually exclusive exons (15, 18, 47)
but is in agreement witharecentreportthatcharacterizedviral
FIG. 1. Identification by RT-PCR of alternatively spliced HIV-1 mRNAscontaining a variety ofexons that arise from the existence of numeroussplicejunctionsencodedintheHIV-1 genome.(A) Mapshowingthelocations of the SD and SA sites in thepNL4-3proviral molecular cloneofHIV-1,witheachpositionshown innucleotidesfromthestartof the 5'longterminal repeat(LTR).The SD and SA sitesarenumbered asdoneby Schwartzetal.(47). (B)Organizationofthe HIV-1 genome.Openboxes show locations of theopenreadingframes that encode the HIVproteins. (C)ThedifferentHIV-1 exonsgeneratedfrom the useof different combinations of SA and SD motifs during RNA splicingare shownasbarsandnumberedasdonebyMuesingetal.(35).Exonsrepresented bygraybarswerenot found inanyof the RNAspecies examined inthesestudies.Exons1,2,3,and 5 donotencodeHIV-1protein,andthose numbered withanEreadthroughSD4 into theenvgene. (D) The various HIV-1 mRNA species falling into the 1.8-kb size range in Northern blot analyses were distinguished by acrylamide gel analysis of
PCR-amplifiedcDNA,usingprimersOdp.045(positions477to499)andOdp.032 (positions8477to8498).Arepresentativegel from PCRanalysis
of RNA frompNL4-3-infectedPBMC is shown. Thedistinct HIV-1RNAspecieswereidentifiedbydirect PCRsequencingof excised bands and bycloning,and theidentity,exoncontent,and size ofthe PCRproductareshown.The mRNAspecieshave been namedaccording to theprincipal
protein that they encode andbytheirsize,with thesmallestRNAas1. Faint bandswerevisibleonlongerexposuresof the autoradiogram.rev mRNAsarebicistronicand alsoencodeNef(15, 48,49).(E) Representativeacrylamide gelfrom PCRanalysis of HIV-1 mRNA species falling into the4.0-kb size range frompNL4-3-infectedPBMC,usingprimers Odp.045andOdp.070 (positions6094 to6115). The identity of each RNA species, determinedbydirectsequenceanalysisofexcisedbands,is shown with theexoncontent and size of the PCR product. RNA species not matched to abandonthegelweredifficulttodetectexceptonverylong gelexposuresandwerenotquantified above the background level in
phosphoimaginganalysis.AllenvmRNAsaremulticistronic and also encodeVpuand Nef(48,49).
VOL.67, 1993
on November 9, 2019 by guest
http://jvi.asm.org/
6370 PURCELL AND MARTIN
B.
100 100 100 100 102 106 102 100 0.4 5 10 50
0.2 2 8 58
100 100 100 100 95 98 97 95
50 10 5 0.4 58 8 0.2 0.1 100 100 100 100
71 82 84 79 0.4 5 10 50 0.1 2 10 58 100 100 100 100 117 123 113 115
Tat2
Tat1 cyptic
Nef4
Nef3 Rev2 Nef2
4~
RNAmixtures r g, *
ABCD4tt4*~t
q9¼~
A B C D + ?q #h
Vpr2
Vpr1
Tat4 Tat3
P-'-
- Tat2Tati
Rev12
-nquie
....
Rev1O,11''Nef5
RevB,9
Net3 Rev3
_,n,. NRev2
_ _ = = ~~~~~~~~~RevI
-~~ ~~~~~Nef~~~~~2
Nef1 50 10 5 0.4
71 7 5 0.8 Nef 1 *r
N
A..- Netl
1
234567
Invitro RNA HeLa
89
10
[image:6.612.144.457.77.408.2]PBMC
FIG. 2. The relativeproportionsofalternativelyspliced HIV-1 mRNAswereaccuratelydeterminedbyRT-PCR assay. Thesemiquantitative
capacityof the RT-PCR assay for the 1.8-kb class ofspliced HIV-1 mRNAwasevaluatedby mixing predetermined concentrations of RNA transcribed in vitro fromHIV-1cDNAclones with T7 RNApolymerase(A)andthenperformingreversetranscription,PCRamplification,labeling
with[32P]dCTP,andgelanalysis (B).Proportionsof each RNA added intofourmixtures,Athrough D,areshowninpanelAwith theproportion
of cDNAexperimentallyamplifiedbyPCR(panelB, lanes1to4)and determinedbyphosphoimaging analysis (showninitalics inpanelA). (B)
Lane 5showsapoolof RNA mixtures AthroughDthatweretreatedinparallelbutwithout theadditionof RTduringcDNAsynthesis.RNA from HeLa cellstransfected withreporterhumangrowthhormoneonly(lane6)andwithpNL4-3(lane7)orwith RNAfromPBMC(lane8)and PBMC infected with
HIV-lNL,43
(lane9) orHIV-lLAI (lane 10)wasexamined in thesamesetofreactionsasthe control. Theidentityof each amplified speciesis shownontheright (seeFig. 1D).S
A
ACCAATTGCTATTGTAAAAAGTGTTGCTTTCATTGCCA
----C---A---T
----C--- --_-_
4-SA 4a PTTGTTTCATGACAAPACF
---CA
---
__---A---G---CA--- -
---CA---CA ----
--SA 4b CCTI AC
G----__
"I__
__-
4-Rev SA
start 5 GCATCTCCTT}{GCAC
_________ _____
_________ ____ _
_________ ____ _
---_-_
-AT- -F---C----CA-G----__
G---C--- ---C---
-A---__G---A--F---________________ _---A---G-- __---
Si
-A----G---C--- ----
-C--T-A-AC---__G---AT-G ---C--- -G---A-A--G--__G---J_ _ _ _ __
FIG. 3. LocationofSA4c,a newSAsite forrevandvpu/envmRNAs.Shown is alignment of nucleotidesequencesof severalHIV-1 isolates surroundingthe newSA site forrevandvpu/envmRNAs, SA4c(adaptedfrom Myersetal. [37]).
A.
In vitro RNA mixtures
A B C D
Tat2 Tatl
cryptic
Net4
Nef3 Rev2
Nef2
NL4-3
LAI HXB2 SF2
MN
NY5
ADA BAL
SF162
ELI
MAL
GAAGA
J. VIROL.
4-SA 4c
Z
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.86.513.545.702.2]RNA SPLICING MUTANTS OF HIV-1 6371
A.
forwardprimer
Odp.045:
II
- splice 2
,* acceptors
I M_IU k}
-111illor"Giv.splc.e- ! N.-4a
_~-splice
1 donors
RNA species -Nef
1-=mL.- Nef 2 mmm-- Nef3 mom- Nef4 miii- Nef 5 Rev 1
Rev2 i.
Rev 3 ' Rev 4
-,--Rev5
-Rev6 -Rev 7 Rev 8 Rev 9 Rev 10
-,_i-Rev 11 Rev 12
-1.8kb class of RNA
reverseprimer :Odp.032.
I II
3a 4ca7
I
n7.,
1I"O*2
2Vlli
32 3
- Tat1 - Tat 2 - Tat3 - Tat4
:L I. L.!.,
I.... 4
:: Relative %
;;;i5 -j~
....- Af
49
11 24 7
-,-,-- 32 -j
.... 40 8
1
1-_Li~
44_ -33
.-.. 2
-:_..
2_ -1
48
28
16 9
- Vpr1
- Vpr2 -.n- 7228 -.
B.
4.0 kb class of RNAforwardprimer reverse primer
Odp.1145 Odp.070.'
2 3. 4cab -splice
acceptors 1 , '2 . pl3 vpr Irevi4 I to do s
~~L Ll w~~~~~it 1 Lrz| splice
I
|2|~~~~~~
3 4 donorsRNA species o. Env1
-.-Env 2
-Env 3
Env 4 Env -Env6
in-Env7
-. Env8 Env 9 - Env 10 - Env 11 - Env 12.-- Env
13.-in- Env 14 - Env 15 - Env 16
- Tat5 - Tat6 - Tat7 - Tat 8
Relative % 80
5 _6
mo-_ 1
_o 5
_ 1
_io 1 _ _ _NO O
85 9 6
0
- Vpr 3 - Vpr4 - Vif2
98
2
[image:7.612.72.562.86.376.2]p 100 FIG. 4. Structure and relative abundance of each alternatively spliced HIV-1 mRNA. (A) Splice site usageofthe 1.8-kbclassofHIV-1 mRNA amplified by PCR of randomly primed cDNA, using primers Odp.045 and Odp.032. (B) Splice site usage of the 4.0-kb class of HIV-1 mRNA amplifiedby PCR of randomly primed cDNA, using primers Odp.045 and Odp.070. The solid boxes raised above the line represent the regions ofretained RNA. Shown at the right of each panel is the relative proportion of each mRNA species quantified by phosphoimaging from pNL4-3 virus infections of PBMC, using semiquantitative PCR analysis for the 1.8- and 4.0-kb classes of RNA. In general, values less than 1 were not measured at levels significantly above background and were recorded as zero; however, the existence of these cDNA species was evident on long gelexposures.
RNAininfected MT-2cells (50).Noevidencewasfound for a
vif
RNA among the 1.8-kb species that spliced SD4 to SA7(vifl).
Splicesite mutants ofpNL4-3.During HIV-1 mRNA splic-ing, the cellular spliceosome cleaves at GT and AG dinucle-otides within the SD and SA motifs, respectively. Several of these highly conserved dinucleotide motifs present in the pNL4-3 proviral DNA clone of HIV-1 were replaced with different dinucleotides to block RNA splicing, using a PCR-based strategy (Fig. 5). Seven site-directed provirus mutants weregenerated by inactivating (i) the constitutivelyusedmajor splice donor,pNLASD1; (ii)thecompeting splice acceptorfor
thefirstcodingexonofrev,pNLASA4b; (iii)thecompetitively
selectedmajorspliceacceptorforenvandvpu,pNLASA5; (iv)
the cryptic splice acceptor within env purportedly used to generate theTevprotein (5, 46, 47), pNLASA6; (v) SA7a,the
most5'of twoconservedcrypticSA sequencesmapping34 and 28bases upstreamfrom thesecondcodingexon oftatandrev,
designated pNLASA7a; (vi) both the SA7a and SA7b cryptic sites, pNLASA7a+7b; or (vii) both of these in combination withSA7, the constitutive SA forthesecondcodingexonoftat
and rev, pNLASA7+7a+7b. Some of these mutations also
altered an amino acid codon(s) overlapping the
splice
site(Materials and Methods). The integrityofallmutant
proviral
clones was confirmed by nucleotide sequence
analysis
of the reconstructed regions.Consensus motif
Mutantname pNLASD1
pNLASA4b
pNLASA5 pNLASA6 pNLASA7a pNLASA7a+7b pNLASA7+7a+7b
CAG(1I AAGT
A IG
3' Spilice Acceptor
TTTTTTTTTTTTNC,,2 CCCCCCCCCCCCNTC G
CTGCAGAGTACGCCA
'1
TGACAAAAGCCTTGAG
41
CATCTCCTATGGClPCG
4i TGTGTTAGTTTAIC11T TTCTATAGTGAATI(CA
4,i "I TTCTATAGTGAAT1PCAGTT1PCG
'1 'I I
AAT1CAGTTTCGCAGGGATATTCACCATTATCGTTTCGPA FIG. 5. Basesubstitutions introduced into SD and SA motifs of the pNL4-3proviralclone ofHIV-1.Theconsensusmotifs of mammalian SD and SA sites are shown at the top, with the essential two
dinucleotides of the motif shown in outlined font. These dinucleotides were changed in the pNL4-3 proviral clone to the bases shown in outlined font so as to inactivate several SD and SA sites. Arrows indicate thepointofcleavageand ligationof RNAduringsplicing.
t 'W'st-7- I
VOL.67, 1993
5' Splice Donor
!t
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.330.564.490.658.2]6372 PURCELL AND MARTIN
A.
9
eB.
gpl60/120o Z gp160/120
p55
...,>.. ~~~~~~~~~~~~~~~..~ ~ p55
p24 ..IE...-...
p17 _ _
36 hrs
p24 p17
72hrs
1
2 3
4 56
78 9
0
1 2
FIG. 6. Some HIV-1 splicingmutants direct the synthesis ofan altered profile of HIV-1 protein. Western blot analysis of HIV-1 protein detected with patient serum from HeLa cell lysates was performed 48 h(A) and 72 h(B) after cotransfection of 0.5 pLgofa growth hormone reporter plasmid with 20 pLg ofwild-type pNL4-3 (lane 1), with RNA splice mutant provirus pNLtASD1 (lane 2), pNLASA4b (lane 3), pNLASA5 (lane 4), pNLASA6 (lane 5), pNLASA7a (lane 6), pNLASA7a+7b (lane 7),orpNLASA7+7a+7b (lane8),orwith reporterplasmidalone(lanes9 and0).The volume of celllysatewasstandardizedfor transfectionefficiency accordingtothe determination of humangrowthhormone in the culturesupernatants.
Protein synthesis bysplicingmutantsofHIV-1. Inthe first groupofexperiments, theeffects of thesplicesite mutationson
viral protein production were assessed by Western blotting
(immunoblotting) lysates from transfectedHeLacells, usingan
AIDSpatient'sserum.AsshowninFig.6, each of themutants
except pNLASA7+7a+7b directed the synthesis of the same
complement ofHIV-1 proteinsasdidwild-type pNL4-3,with thefollowing exceptions. Mutation ofSD1 caused a marked decreasein thequantity of viral proteins accumulatingin HeLa
cellsduring the first 48 h despite transfection efficiency
equiv-alent tothatof the wild type,as measuredby coexpression of
ahumangrowth hormone expression plasmid (Fig. 6A, lanes1
and 2). By 72 h after transfection, however, HIV-1 protein accumulation directedbymutantplasmidpNLASD1was
com-parabletothewild-type level (Fig. 6B, lanes1and2). Mutation ofSA4b,used for theproductionofrevandenvmRNAs, ledto
consistent and substantial elevations ofgpl60/120 levels
com-pared with the wild type (Fig. 6A, lane 3). In contrast, the mutation of SA5 markedly reduced, but did not eliminate,
gpl60/120production (Fig. 6A, lane 4). Mutation of SA6 (for
tevmRNA)and the SA7aorSA7a+7bcryptic splice sites (for the secondcodingexonoftatandrev)had little if any effect on
HIV-1protein synthesis (Fig. 6A, lanes 5 to 7), indicating that these three SA sites (and exons 6, 7a, and 7b) play no significant role for HIV-1 protein synthesis in transfected HeLa cells. In contrast, mutant
pNLzASA7+7a+7b,
which contains a triple splice site mutation including SA7, the in-frame acceptor used constitutively for the second codingexonoftatand rev, directs the synthesis of a markedly altered protein profile (Fig. 6A, lane 8), similar to that previously reported for Rev-deficient HIV-1 mutants: minimal levels of
p55,p24, or pl7gagprotein and no detectable gpl60/120 (12, 14, 20, 33) accompanied by a novel and abundant 20-kDa protein that reacted with Tat antiserum (data not shown). No increased accumulation of HIV-1structural proteins was noted
at later times (72 or 96 h after transfection) despite the
HeLa transfection ct
%99 99>D 9999>¢COrC5>rc>rtA 2;c,~~~~~-P7SstvX=sS7?<z,
622bpI 527bpi
404bp
1 2
3 4 5 6
7
8 910
FIG. 7. HIV-1provirusesmutatedatcryptic splicesitessynthesize awild-typeprofileof RNA.SemiquantitativeRT-PCRanalysisof the 1.8-kb class HIV-1 RNAfrom HeLacells transfectedwith 0.5pLgof reporterplasmid alone(lane 2)orcotransfectedwith 20,utgofpNL4-3
(lane 3) or HIV-1 RNA splicing mutant pNLASD1 (lane 4),
pNLASA4b (lane 5), pNLASA5 (lane 6), pNLASA6 (lane 7),
pNLASA7a (lane 8),pNLASA7a+7b (lane 9),orpNLASA7+7a+7b (lane 10).AnMspI digest of pBR322is shownontheleft(lane 1)as a sizemarker.
expression of levels of total cellular HIV-1 RNA similar to
wild-type levels by Northern blot analysis (not shown). These
results suggest that mutant pNLASA7+7a+7b failed to
ex-press functional Rev and resulted in reduced amounts of Rev-dependent cytoplasmic mRNAs encoding Gag and Env
proteins.
mRNAanalysis from cryptic splice site mutants of HIV-1.
When the 1.8-kb class of RNAfrom HeLa cells transfected with HIV-1 proviralDNAclonescontaining splice site
muta-tions was analyzed, it was clear that several expressedthesame
RNApattern as didwild-typepNL4-3 (Fig. 7). For example, mutants pNLASA6, pNLASA7a, and pNLASA7a+7b were indistinguishable from the wild type (Fig. 7, lanes 7 to 9), indicating that the SA6, SA7a, and SA7b sites are not used with significant frequency by HIV-1NL43, even though it is virtually identical to HIV-lHXB2, the strain in which these splice siteswereoriginally described. Furthermore, no cDNAs
usingthe SA6, SA7a, or SA7bsplice site were ever detected amongthePCR-amplifiedbands that were directly sequenced
fromcellstransfected or infected with derivatives ofHIVLAIor
HIV-lN"-3,
and no clone generated from these PCR-ampli-fiedcDNAscould beshown to utilize these splice sites. Thus, the SA6, SA7a, and SA7b cryptic sites are used extremely rarely, if at all, and may be active only in theHIV-1LAI
derivativeHIV-lHxB2.
Wefound that careful selection and testing of PCR primers
was required, in conjunction with the use of poly(A)+ RNA andnonspecific cDNA priming, to avoid the amplification of aberrant cDNAs. Our semiquantitative PCR assays depended
onequal use of all the HIV-1 RNA species in either the 1.8- or 4.0-kb mRNA classes as competing templates for amplifica-tion. Otherprotocols may selectively amplify transcripts using SA6, SA7a, and SA7b (47): reports characterizing the
se-J. VIROL.
"I
n'.),
cp
.+ V %,:.' 1118 ll.v
..
..:
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.348.515.80.297.2] [image:8.612.54.293.82.246.2]RNA SPLICING MUTANTS OF HIV-1 6373
A.
e
B. Q9~,'o
Vpr3-
-Tat4
Tat3.
Tat2-Tati- _ _ _
Nef5- Rev6-
Nel4- Revl,-Net2- F
Nett-__
Tat7-
Tat6-Tat5- __
Envg-Env8-..
Env5-
Env4- Env3-Env2- _
Envi- -__
1 2 3 4 5 1 2 3 4 5
FIG. 8. An altered profile of spliced RNA is synthesized byHIV-1
provirus mutated at constitutive orcompeting splice sites. Shown is
semiquantitativeRT-PCR analysis of the1.8-kb class HIV-1 RNA (A)
and the 4.4- to 5.5-kb class of HIV-1 RNA (B) from HeLa cells
transfected with 20 p.g of pNL4-3 (lane 1) or HIV-1 RNA splicing
mutantpNLASD1 (lane 2),pNLA&SA4b (lane 3),pNLA&SA5 (lane 4),
orpNLASA7+7a+7b(lane 5).
quence of cDNA clones around the SA7 splice site (15) or
using hybridization analyses of cDNAs (50) also failed to
detectusageof SA7aorSA7b.
Analysis of mRNA from HIV-1 with mutated constitutive and competitive splice sites. The cDNAs amplified from cells
transfected withmutantpNLASD1 had apatternverysimilar to that of the wild type but with a slightly slower
electro-phoretic mobility (compare lanes 3 and 4 in Fig. 7). Direct
sequence analysis of these PCR bands as well as several
individual cDNA clones indicated that spliced RNA from
pNLASD1 usedaGTdinucleotide4ntdownstream from the
mutated majorSD (constitutiveSD1) siteas acrypticSDsite
in this HIV-1 proviral DNA. The HIV-1 proteins translated
fromtheseslightly largermRNAswereindistinguishablefrom thewild type (Fig. 6), but the rate of splicing of these viral
transcripts may be slower, perhaps explaining the delay in
protein expression previouslyobserved. This cryptic SD
(incor-rectly annotated as the major SD in the alignment of this
region byMyers et al. [37]) is strongly conserved among all
HIV-1 isolates.BecausethecrypticSD1sitemaybeused only
whenthegenuine siteis inactivated, it is likely that thestrong
conservation of this alternative splice site results from addi-tional selective pressures. Nevertheless, its existence greatly
reduces thepossible loss of virus infectivity due to a
sponta-neous mutation affecting the major SD that would otherwise
blocktheproduction of functional splicedmRNAs.
ThecDNAsamplified from the remaining mutantsdiffered from the pNL4-3 wild-type pattern (Fig. 7 and 8). For the provirus mutant inactivating the constitutive SA7 for the
secondcoding exon oftat andrev, pNLASA7+7a+7b, every
cDNA bandclearly differedinsizecompared with the wildtype
followingamplificationofthe 1.8-kbspecies ofHIV-1 mRNA
(Fig. 8A, lane 4); the overall pattern was similar to the
wild-typepattern except that each band had a faster
electro-phoretic mobility. A direct sequence analysis of these PCR bands and sequencing of individual pNLASA7+7a+7b cDNA clones indicated that all used an AG dinucleotide situated 20 nt downstream from SA7 as the alternative SA site; this resulted in cDNAs that were 20 bases shorter than the wild type. The activation of this cryptic downstream SA site, however, precluded the generation of mRNA capable of encoding a functional Rev protein and resulted in an immu-noblot devoid of HIV-1 structural proteins (Fig. 6A, lane 8). It should be noted that all of the wild-type 1.8-kb spliced RNAs were transcribed as truncated species by mutant pNLA SA7+7a+7b except for the nefl RNA, which was not detected on the autoradiogram shown in Fig. 8A. The cDNA bands obtained for pNLASA7+7a+7b after PCR for the 4.0-kb mRNA were identical to those of wild-type virus, indicating that RNA splicing to SA and SD sites upstream of SA7 was not affected by the absence of functional two-exon Rev protein. This finding demonstrates that Rev is not required for splice site selection in HIV-1 RNAs; however, protein expression from RNAs containing an RRE clearly requires functional Rev protein.
Mutation of each of two competing SA sites, SA4b and SA5, caused a different usage frequency for neighboring SA sites but not the activation of any cryptic sites. The first competing SA mutant, pNLASA4b, failed to generate bands for revl (Fig. 8A, lane 2), rev4, rev7, andrevlO (evident after long gel exposures; not shown) following PCR with primers for the 1.8-kb mRNA species and for env2 (Fig. 8B, lane 2), env6, envlO, andenvl4
(evident after long gel exposures; not shown) after PCR with primers for the 4.0-kb mRNA. The failure to detect these bands was consistent with the absence of RNAs splicing to the mutagenized SA4b site. All other cDNAs amplified from pNLASA4b were identical to wild-type cDNAs. The cDNA pattern associated with the second competing SA mutant, pNLASA5, lacked several predominant nef species (nef2, nef3, nef4, andnef5)after PCR for the 1.8-kb mRNA (Fig. 8A, lane 3), as well as the major env species (envl, env5, env8, and envl3) after PCR for the 4.0-kb mRNA (Fig. 8B, lane 3), reflecting the absence of RNAs splicing to the mutagenized SA5 site. In addition, there was a compensatory increase in the use of SA4, SA4a, SA4b, and SA4c, resulting in increased levels ofrevl, rev2, rev3, tatl, tat2, tat3, tat4, tat5, tat6, tat7, env2, env3, and env4 mRNAs.
Translational consequences of altered RNA profile for com-petitive splice site mutants. Since the mRNAs using the competing SA4a, SA4b, SA4c, andSA5 splice sites are multi-cistronic, potentially coding several proteins from each mRNA (15, 48, 49), we examined the effect of altered proportions of HIV-1 mRNAs on protein expression (Fig. 9). Mutant
pNLAiSA4b
directed the synthesis of a significantly increased level of envelopegpl60/120and Nef but decreased amounts of Revcompared with wild-type provirus (Fig. 9A to C, lanes 3). Because Rev protein plays a prominent role in regulating the level of gpl60/120, we evaluated Rev functional activity by measuring theRev-dependent rescue of CAT activity following cotransfection ofpNLASA4b with the pDM128 Rev reporter plasmid (Fig. 9E and F) (24). Again, Rev activity directed bypNLASA4b, as measured in this assay, was lower than that directed bywild-type pNL4-3. Thus, the observed elevation of
gpl60/120wasnot due to any increase in Rev activity. Signif-icantly, the expression of both Rev-dependent gpl60/120 and Rev-independent Nef proteins were increased in similar pro-portions. These proteins share SA5 as the predominant com-peting SA for theirmRNAs. Thus, the increased synthesis of
gpl60/120 and Nef from the pNLASA4b mutant most likely reflects the increased use of SA5 (and SA4c) and results in
VOL.67, 1993
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.90.277.76.295.2]6374 PURCELL AND MARTIN
A.
'.
* gpl60/120
1 2 3 4
B.
,15 ey.
as* t,.. ,...
__ Nef
1 2 3 4
C.
*.- Re
r)- 2 3
E. Mock pNL4-3
0 5 1
0.5 0.5 0.5
pNL.NSA4b
5
0.5 0.5
pNL.\SA5
5 1 Ltg provirus
0.5 0.5 LtgpDM128
*
W. ... AcCM SA7CM
0 14.0 13.7 3.1 0.4 26.2 11.6 %conversion
FIG. 9. Translational consequences ofalternativeusageofcompeting splicesitesbymutantHIV-1proviruses.Westernblotanalysiswithrabbit serum togpI60/120(A)orNef(B)andimmunoprecipitationwithrabbitserum toRevfrom HeLacelllysatesprepared48 h after cotransfection with 0.5 ,ug of humangrowthhormone reporterplasmidalone(lane 1)orwith 20 ,ug ofpNL4-3(lane 2),pNLASA4b (lane 3),orpNLA&SA5 (lane
4)(C). Lysatevolumeswerestandardizedaccordingtohumangrowthhormonedetermination. Functional Revactivitywasmeasured withuseof the pDM128 Rev-dependent reporter plasmid of Hopeet al. (24) (D) by measuringthe amountofCAT activity rescueddue to Rev-RRE interactionaftercotransfectingthe indicated amountsofplasmids pDM128andpNL4-3, pNLASA4b, pNLASA5with0.5jigof humangrowth
hormone reporterplasmid(E).The percentageofchloramphenicol(CM)convertedtotheacetylatedforms(AcCM)is shownatthe bottom.SV40,
simianvirus40 promoterandenhancer; LTR, long terminalrepeat.
increased synthesis of both proteins. This occurs despite the loss ofmRNA species using SA4b which encode Rev. Since increaseduseof SA4c does not preventthe reduction ofRev
expression, if islikelythat multicistronic mRNAs usingSA4c
arelessefficient for Revexpression than for Neforgpl60/120
expression.
HeLa cells transfected with pNLASA5 express very low levels ofgpl60/120and Nefproteinsbut elevated levels ofRev
compared with wild-type pNL4-3 (Fig. 9A to C, lanes 4).
Elevated Rev activity was also measured in the assay of Rev-dependent rescue of CATactivity, showing that the low level ofgpl60/120 expression didnotresult from anydeficiency
in Rev function. The low-level expression ofgpl60/120most
likely results from the inefficient translation of the multicis-tronic env2, env3, and env4 mRNAs, which are present at
relatively increased levels in cells transfected with pNLASA5 compared with the wild-type pNL4-3 (Fig.8B, lanes 1 and3). This result indicates that the multicistronic env mRNAs are markedly less competent forgpl60/120expression than is envl.
pNLASA5 alsofails to synthesize the major nef RNAs (nef2, through nef5; Fig. 4), although the levels of the nefl cDNA species, which results from splicing of SD1 directly to consti-tutive SA7,were equivalent in thepNLASA5- and wild-type-transfected cells (Fig. 8A). This low expression of Nef protein indicates that the increased amount of bicistronic
revinef
andenvinefmRNAs encoded by mutant pNLASA5 fails to com-pensate for the loss of the predominant monocistronic nef mRNAs.The elevated levels of Rev protein observed
demon-strate that the bicistronic
revinef
andenvinef
mRNAs encodeRevwith significantly higher efficiency than Nef.
Infectivity of splicing mutants of HIV-1. The ability of the
HIV-1 splice site mutants to generate progeny virions was assessed by measuring the RT activity released into the
medium following cotransfection of HeLa cells with proviral
and human growth hormone DNAs (Fig. 10A). All of the
splice sitemutantsexceptpNLASA7a+7b generated less par-ticle-associated RT than did the wild-type plasmid pNL4-3;
pNLASA7+7a+7b andpNLASD1 produced only 2and 15% of viral progeny, respectively, comparedwith the wild type.
Theinfectivity of the splice site mutants was evaluated by
inoculating CEM (12D7) cells with equal amounts of virus harvested from transfected HeLa cell supernatants as deter-minedby RTassay (for wild-type pNL4-3, themultiplicity of infectionwas approximately 0.002) (Fig. lOB). Spreading in-fectionwasestablishedbyoneof thetwoproviruses containing
amutated constitutive site. Mutant pNLASD1 was infectious butexhibiteddelayedreplication kinetics compared with wild-typepNL4-3,reflectingthedelayed kinetics of protein synthe-sisbythismutant (Fig. lOB).LowerefficiencyofRNAsplicing from the cryptic donor activated in pNLASD1 is very likely responsible for the delayed replication and infectivity kinetics.
Mutant
pNLASA7+7a+7b,
which lacks the constitutive SA7site,couldnot infect CEM(12D7) cells, reflecting its inability
toproduceRev and the HIV-1 structuralproteins.
Mutationsaffecting competingSAsitesin HIV-1 had
differ-enteffectsonviralinfectivity. MutantpNLASA4b,whichfails
togenerate severalHIV-1mRNAs(revl, rev4, rev7, and revlO andenv2, env6,envlO, and envl4)and consistently expressed elevated levels ofgpl60/120,exhibited growth kinetics similar
tothe wild-typevirus kinetics (Fig. lOB). In this case, altered
splicing of viral mRNA had no effect on viral infectivity. Mutant pNLASA5, which lacks the processing site for the
major envl mRNAspecies, was not infectious despite being abletosynthesize low levels ofgpl60/120in transfected HeLa cells by utilizing alternative SAs (Fig. 6A, lane 4; Fig. 9A). Thesecontrasting results demonstrate that alternative splicing
tocompetingSAsitescanaffectgpl60/120production, leading
tosynthesisof either fullyinfectious or defective virion.
D.
Revdependent reporterplasmid pDM128 SD4
SV40 CAT RRE
* *.
s ,
*
@ .
J.VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:10.612.139.478.78.288.2]RNA SPLICING MUTANTS OF HIV-1 6375
A.
20000
- 15000
jk.
E
a,6
10000 50000
Bo
40000
'a
0. ie
30000
-200000
10000
DISCUSSION
0
* pNL4-3wildtype * pNLASDI O pNLASA4b
0 pNLASA5 O pNLASA6 13 pN1ASA7a a pNLASA7a+7b M pNLASA7+7a+7b
* Mock
pNL4-3wildtype pNLA pNLASA4b
pNLASAS
pNLA pNLA pNLASA7a+7b
pNLASA7+7a+7b
Mock 0
--+
-0---20
Days post-infection
30
FIG. 10. Efficiency ofvirion production and infectivity of HIV-1 splicing mutants. (A) Particle production by HIV-1 RNA splicing
mutantswasmeasuredby usingthe accumulation of RT activity40h aftercotransfectingHeLa cells with20 jig ofmutantprovirus plasmids and 0.5 ,ug of human growth hormone reporter plasmid. The RT activitywasstandardized fortransfectionefficiency by using the ratio of human growth hormonecomparedwithwild-type pNL4-3 virus. (B) Kinetics ofinfection of HIV-1 RNAsplicingmutantsfollowing trans-fer ofcell-free virionobtained from transfectionsupernatants(105cpm
fromtheRTassay)toCEM(12D7)cells.
The infectivity of mutants affecting the cryptic splice sites
was moresubtle andcouldnotbepredictedfrom theirability
to generate virus particles in the transfection experiments
shown in Fig. 1OA. Mutant pNLASA7a was infectious, but
peakvirus productionwasdelayed4 to5 days comparedwith the wild type. Mutant pNLASA7a+7b, which directed large
amountsofprogenyvirionproduction followingtransfection of HeLacells,was notinfectious. MutantpNLASA6wasalsonot
infectious, despite directing the synthesis of the wild-type complement and quantities of protein and spliced mRNAs
(Fig. 6 and 7). These latter results could indicate that the
crypticSA6 and SA7b sitesparticipateinsomeotheraspectof RNA processing (e.g., folding or branch point formation). Alternatively, amino acid changes introduced into the
enve-lope protein as a result of mutagenesis
'(see
Materials andMethods) mayeliminate virus infectivity, possibly by affecting gpl60 processing as suggested by reduced amounts ofgpl20
observed inFig. 6A. Inaprevious study,asimilar mutation of SA6 in theHIV-lHXB2isolate resultedinthe loss ofinfectivity,
whereas SD5 mutants,also defectivefor Tevexpression,hada
wild-type phenotype (16).
Relativeproportions of alternatively spliced HIV-1mRNAs.
Various regulatory mechanisms control the expression and function of HIV-1 during a cycle of virus infection. The
regulation of RNAprocessing is one such prominent
mecha-nism,andthe balancedsplicing of genomic length RNA into a
complexset of alternative RNA transcripts is required for the
synthesis of several viral proteins essential for replication. Several transcripts are capable of expressing each of the regulatory and accessory HIV-1 proteins, and most of these
transcripts have the potential to encode two or more proteins with different efficiencies. To evaluate the importance of the
complex group of mRNAs synthesized during infection by
HIV-1, wefirstrigorouslydetermined the identities and rela-tivequantitiesof viralmRNAsresulting from transfection and infection experiments. Our analysis shows that some RNA
speciesaresynthesizedinpreference to others. Generally, the
most highly spliced forms of RNA that exclude noncoding
exons aremostcommonexceptinthe case of nef, in which case the inclusion of the 68-nt noncoding exon 5 is favored. A
previouslyunrecognized SA site forrev-and env/vpu-encoding
mRNAs (designated SA4c)wasidentified among a cluster of
competing SA sites in the tat coding sequence. This site was selected at fivefold-lower frequency than the SA4a or SA4b site for both rev and env mRNAs in PBMC infected with
HIV-lNL-3
orHIV-lLAI.
The SA4csite is used by many strains of HIV-1 and is thepredominant SA used for rev mRNA bysomeHIV-1 strains(42).Theaddition of thenewSA4c site to the centralcompeting SA sites(SA4,SA4a, SA4b, SA4c, and
SA5)
determined that 16 alternative mRNAs may encodegpl60/120. However,mostofthese existatverylow levels, and the most common env mRNAs either used SA5 or excluded both noncoding exons 2 and 3. The shortest possible env
transcript (envl)accountsfor 80% of allenvRNAdirected by
HIV-'NL4-3.
Competing SA site usage determines gpl60/120 levels and
virus infectivity. Changing the balanced usage of competing
splicesites caused alterations in theproportionsof both RNA andprotein species and,in somecases, viralinfectivity.HIV-1 mutants containing changes affecting SA4b gave rise to an
increasedproportion of themRNAspecies using neighboring
SA4a, SA4c,
and SA5. This caused elevated expression ofenvelope
gpl60/120but no loss of virus infectivity. Increasedsynthesis
of HIV-1 env mRNA using SA5, which moreeffi-ciently yields gpl60/120, is likely to explain the increase in
gpl60/120.Mutation of theclosely adjacent SA5,themajorSA forenvmRNA,resulted in increased useofSA4a, SA4b, and SA4c butwas
accompanied
byamarked reduction in boththeexpression of envelope gp160/120 and virus infectivity. The reduced levels ofgpl60/120werenotassociated with reduced
Rev activity, and Gag protein production was not altered.
These results demonstrate that the level of expression of
envelope proteins
can bedramatically
alteredby
forcingdifferent
splicing
patternsonHIV-lNLA-3
through
thedispro-portionate
usage of theseemingly
redundant SA sites in thisregion
of the viral genome.Two determinants could control the selection of the
com-peting
SA sites if this alternativesplicing
mechanismweretooperate invivo.
First,
the different sequence structure of thecompeting
SA motifs(Fig. 3)
orthe branchpoint
structure(s)in individual HIV-1 strains could alter thebalance of the SA usage. Thelocation of the branch
point(s)
for thecompeting
HIV-1 SA sites is unknown. We have confirmed that HIV-1
strains with different sequences have different
splicing
patterns in a survey of various HIV-1 isolatesexhibiting
variableVOL.67, 1993