Copyright © 1998, American Society for Microbiology
Increased Induction of Apoptosis by a Sendai Virus Mutant Is
Associated with Attenuation of Mouse Pathogenicity
MASAE ITOH,
1* HAK HOTTA,
1ANDMORIO HOMMA
2Department of Microbiology, Kobe University School of Medicine, Chuo-ku, Kobe 650,
1and Faculty of Home
Economics, Kobe Women’s University, Suma-ku, Kobe 654,
2Japan
Received 2 September 1997/Accepted 12 December 1997
An avirulent mutant of Sendai virus, Ohita-MVC11 (MVC11), was generated from a highly virulent field
strain, Ohita-M1 (M1), through successive passages in LLC-MK
2cell cultures (M. Itoh, Y. Isegawa, H. Hotta,
and M. Homma, J. Gen. Virol. 78:3207-3215, 1997). In LLC-MK
2cells, MVC11 induced a high degree of
apoptotic cell death that was demonstrated by chromatin condensation of the nucleus and DNA fragmentation,
and production of MVC11 declined markedly after prolonged culture. On the other hand, M1 did not induce
prominent apoptosis and maintained high virus titers. In primary mouse pulmonary epithelial cell cultures,
M1 replicated rather slowly to reach maximum level of virus production at 3 days postinfection, and high levels
of virus production were maintained thereafter without causing apoptosis. In contrast, MVC11, which
pro-duced 20 times more progeny virus than M1 at 1 day postinfection, inpro-duced a high degree of apoptotic cell
death before the virus replication cycle was completed. Accordingly, the production of progeny virus was
strongly inhibited thereafter. In the lungs of mice infected with MVC11, virus antigens and signals of DNA
fragmentation detected by the in situ terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling
technique colocalized in bronchial epithelial cells, clearly demonstrating that infection by MVC11 triggered
apoptosis in vivo as well as in vitro. These results suggest the possibility that induction of apoptosis by MVC11
plays an important role in attenuation of mouse pathogenicity by restricting progeny virus production in the
lung. The C protein was shown to have the capacity to induce apoptosis, and the increased level of the C protein
in MVC11-infected cells was considered to account partly, if not entirely, for the induction of apoptosis.
Sendai virus (SeV), a member of the paramyxoviruses, is
also called murine parainfluenza virus type 1 and often causes
outbreaks of lethal pneumonia in mouse colonies.
Experimen-tal infection in mice has been studied as a model for
respira-tory viral infection. Although there are SeV strains differing
remarkably in pathogenicity to mice, the determinants of their
pneumovirulence are largely unknown. In our previous work,
we demonstrated that the pathogenicity of SeV closely
corre-lates with virus replication in the mouse lung (35). We then
showed that susceptibility of the fusion (F) envelope
glycopro-tein to trypsin and to the activating proteases in the mouse
respiratory tract determines the efficiency of virus replication,
and therefore the virulence, by supporting multiple-cycle
rep-lication through cleavage and activation of the F protein (16).
Kato et al. (20) reported that an SeV mutant lacking the V
protein replicated less efficiently in the lungs of mice and was
strongly attenuated. However, the mechanism by which the
lack of the V protein leads to the decreased production of
progeny virus is not known.
When infected with a virus, the host attempts to suppress
virus replication in infected cells and viral spread to
neighbor-ing cells by means of host defense mechanisms such as
induc-tion of immune responses, interferon producinduc-tion, and suicidal
cell death (apoptosis). By turning on the switch for apoptosis
before the virus has completed the replication cycle, the host
cells prevent the virus from producing progeny virions that
infect neighboring cells. This idea is supported by recent
find-ings that many viruses, such as poxviruses (2, 13), adenovirus
(30, 31), Epstein-Barr virus (9–11), hepatitis C virus (7, 32),
[image:1.612.348.507.382.647.2]* Corresponding author. Mailing address: Department of
Microbi-ology, Kobe University School of Medicine, 7-5-1 Kusunoki-cho,
Chuo-ku, Kobe 650, Japan. Phone: 81-78-341-7451, ext. 3302. Fax:
81-78-351-6347. E-mail: masae@med.kobe-u.ac.jp.
FIG. 1. Progeny virus production in and viability of LLC-MK2cells infected
with M1 or MVC11. LLC-MK2cells infected with M1 (a) or MVC11 (b) at an
MOI of 10 were incubated in MEM supplemented with 8% FBS in the absence of trypsin at 38°C. Culture medium was taken and cells were refed with fresh medium every 6 or 12 h after infection as indicated. Virus released into the culture medium within 6 or 12 h before the sampling was assayed after activation with trypsin (15). Another set of LLC-MK2cells infected with M1 or MVC11 as
described above was collected every 24 h, and viability of the cells was deter-mined by the trypan blue dye exclusion test.
2927
on November 9, 2019 by guest
http://jvi.asm.org/
and baculovirus (3), contain genes whose function appears to
interfere with the apoptotic process, presumably to allow cell
survival and continued virus replication (36).
Recently, we reported the characterization of an avirulent
mutant of SeV, the Ohita-MVC11 (MVC11) strain, which was
derived from a highly virulent field strain, the Ohita-M1 (M1)
strain, and possessed two amino acid mutations, one in the C
protein and the other in the L protein (17). MVC11 exhibited
strongly suppressed virus replication in mouse lungs and had
almost entirely lost pathogenicity to mice. In this work, we
studied replication of M1 and MVC11 in cultured mouse
pul-monary epithelial cells, as well as in LLC-MK
2cells, in order to
elucidate the mechanism of restricted replication of MVC11 in
mouse lungs. We found that whereas M1 maintained to
pro-duce progeny virus for a long period of time without killing the
host cells, MVC11 induced apoptotic cell death that interfered
with the following progeny virus production. We propose a
hypothesis that the increased capacity of MVC11 to induce
apoptosis may play an important role in attenuation of
viru-lence through restricting virus spread in mouse lungs.
MATERIALS AND METHODS
Viruses, cells, and antibodies.The M1 strain of SeV, a fresh isolate from an outbreak of SeV in experimental animal facilities, and the MVC11 strain, a laboratory-adapted mutant of M1 obtained through passaging in rhesus monkey (LLC-MK2) cells, were described elsewhere (17). Infective virus titers were
determined as described previously and expressed as PFU or cell-infecting units (CIU) (19). CIU were essentially equivalent to PFU. LLC-MK2cells were grown
in Eagle’s minimum essential medium (MEM) supplemented with 8% fetal bovine serum (FBS). Polyclonal anti-SeV antibodies were made by immunizing rabbits with purified SeV strain Fushimi. Polyclonal anti-C guinea pig serum (29) was a kind gift from K. Iwasaki (Tokyo Metropolitan Institute of Medical Sci-ence).
Primary culture of mouse pulmonary epithelial cells.Tracheotomy was per-formed on 6-week-old male ICR/CRJ (CD-1) mice under ether anesthesia, and 1 ml of protease type X (2 mg/ml; Sigma) was infused into the lung with a syringe. After incubation for 10 min at room temperature, the lung was taken and minced in phosphate-buffered saline (PBS). Blocks of the tissues were removed by filtration through four layers of sterilized gauze, and single cells were collected by
FIG. 2. Apoptosis induced by SeV. (a) Condensation of chromatin. LLC-MK2cells infected with M1 or MVC11 at an MOI of 10 were fixed with ethanol
at 48 h p.i. and stained with Hoechst 33342. (b) DNA fragmentation. DNAs were extracted from M1- or MVC11-infected LLC-MK2cells (MOI of 10) at 24, 36,
[image:2.612.50.287.69.442.2]and 48 h p.i. and electrophoresed in a 1% agarose gel.
FIG. 3. Replication of M1 or MVC11 in primary cultures of mouse pulmo-nary epithelial cells. (a) Progeny virus production. Primary cultures of mouse pulmonary epithelial cells were infected with M1 (F) or MVC11 (E) at an MOI
of 10 and incubated in Dulbecco’s MEM supplemented with 10% FBS in the absence of trypsin. Culture medium was harvested and cells were refed with fresh medium at the indicated days after infection, and virus titers were determined as PFU per milliliter. (b) SeV-specific proteins. On the indicated days after infec-tion, lysates prepared from cells infected with M1 or MVC11 as described for panel a were subjected to Western blot analysis to detect virus-specific proteins with anti-SeV polyclonal rabbit antiserum.
on November 9, 2019 by guest
http://jvi.asm.org/
centrifugation, suspended in Dulbecco’s MEM supplemented with 10% FBS, and cultivated at 38°C.
Determination of apoptotic cell death.Occurrence of apoptosis was deter-mined by the following two methods.
For detection of chromatin condensation of the nuclei, cells grown on coverslips were fixed with ethanol for 20 min at room temperature and stained with 10mg of Hoechst 33342 per ml in PBS for 10 min at room temperature.
For detection of DNA fragmentation, cells (53105) were lysed in 0.5% Triton
X-100–10 mM Tris-HCl (pH 7.5)–10 mM EDTA, and nuclei were removed by centrifugation at 10,0003g for 5 min at 4°C. The supernatants were treated with
50mg of proteinase K per ml and 10mg of RNase A per ml for 1 h at 37°C, and DNAs were extracted with phenol-chloroform and precipitated with ethanol. The pellets were dissolved in Tris-EDTA (pH 7.5) and separated by electro-phoresis in 1% agarose gels.
Western blot analysis.Cells were lysed by treatment with 0.5% Triton X-100 in 10 mM Tris-HCl (pH 7.5) for 15 min on ice, and nuclei were removed by centrifugation at 10,0003g for 5 min at 4°C. Supernatants were subjected to
sodium dodecyl sulfate-polyacrylamide gel electrophoresis (8 or 16% acrylamide for detection of the C protein) under reducing conditions, and the proteins were electrotransferred onto nitrocellulose membranes. After blocking with 3% skim milk in PBS, the membranes were incubated with SeV rabbit anti-serum and subsequently with peroxidase-conjugated goat anti-rabbit immu-noglobulin G (IgG). To detect the C protein, the membranes were treated with anti-C guinea pig antiserum and then with peroxidase-conjugated goat anti-guinea pig IgG. Virus-specific proteins were visualized with an ECL chemiluminescence kit (Amersham) according to the manufacturer’s instruc-tions.
In situ terminal end-labeling.The terminal deoxynucleotidyl transferase-me-diated dUTP nick end-labeling (TUNEL) technique was used to label DNA
strand breaks in apoptotic cells. Paraffin-embedded lung sections were deparaf-finized with absolute and 95, 75, and 50% ethanol solutions and then washed with PBS. After endogenous peroxidase was inactivated in 3% hydrogen peroxide, slide preparations were treated with 50mg of proteinase K per ml for 30 min at room temperature. Subsequent steps were performed with a kit (Apop Tag; Oncor), according to the manufacturer’s instructions, to end label the frag-mented DNA with digoxigenin-11-dUTP and peroxidase-conjugated antidigoxi-genin antibody. Color development was achieved with diaminobenzidine as the substrate.
Double labeling of tissue sections for detection of SeV antigens and TUNEL signals.The TUNEL reaction was completed as far as the wash steps after the labeling reaction. The sections were then incubated sequentially with anti-SeV rabbit antiserum, biotin-conjugated goat anti-rabbit IgG, and alkaline phos-phatase-conjugated streptavidin. Finally, color reactions were performed with diaminobenzidine to detect the peroxidase-labeled DNA fragmentation and then with 5-bromo-4-chloro-3-indolyl phosphate–nitroblue tetrazolium to detect al-kaline phosphatase-labeled SeV antigens.
Expression of the C protein.Sau3AI-BglII fragments (nucleotides [nt] 107 to
812) of cDNAs of the P genes of M1 and MVC11, both containing the start codon for the C protein (nt 114) but lacking those for the P (nt 104) and the C9
(nt 81) proteins, were inserted into the unique BamHI site of the pSG5 mam-malian expression vector (8). EcoRI linker-ligated NP cDNA (nt 26 to 1674) derived from M1 was inserted into the EcoRI site of pSG5. The resulting plasmids, pSG-CM, pSG-CMVC, and pSG-NP, which express the C protein of M1 (C170F), that of MVC11 (C170S), and the NP protein, respectively,
[image:3.612.55.550.68.412.2]under the control of the simian virus 40 early promoter, were introduced into cells by calcium phosphate-mediated transfection or by using Lipofectin (GIBCO BRL). We also used pSV2-C, which expresses the C protein of SeV strain Z (kindly supplied by H. Taira, Faculty of Agriculture, Iwate Univer-sity).
FIG. 4. Chromatin condensation in primary cultures of mouse pulmonary epithelial cells infected with SeV. Primary cultures of mouse pulmonary epithelial cells were infected with either M1 (a to d) or MVC11 (e to h). At 2 or 6 days p.i., cells were fixed with ethanol and subjected to immunofluorescence staining with anti-SeV rabbit antiserum (aSeV) as the first antibody and fluorescein isothiocyanate-conjugated goat anti-rabbit IgG as the second antibody for detection of virus antigens (a, c, e, and g), followed by staining with Hoechst 33342 for detection of chromatin condensation (b, d, f, and h). The arrows in panel d show the nuclei of dividing cells infected with M1, and the arrowheads in panels f and h depict chromatin condensation in the nuclei.
on November 9, 2019 by guest
http://jvi.asm.org/
RESULTS
Detection of apoptosis in SeV-infected LLC-MK
2cells.
We
previously showed that progeny virus production of MVC11 in
LLC-MK
2cells within 24 h postinfection (p.i.) was higher than
that of M1 through increased mRNA synthesis (17). We also
reported that, although it was higher than that of M1 within 1
day p.i., replication of MVC11 in mouse lungs was strongly
restricted at 2 days, p.i. and thereafter (17). In the present
study, therefore, we first examined M1 and MVC11 virus
rep-lication in LLC-MK
2cells for a longer period of time. The
culture media of M1- and MVC11-infected LLC-MK
2cells
were replaced every 6 h (0 to 48 h p.i.) or every 12 h (48 to 96 h
p.i.), and the virus titers were assayed. M1-infected LLC-MK
2cells continued to release high titers of progeny virus until 96 h
p.i., with approximately 90% of infected cells being alive (Fig.
1a). All the living cells were confirmed to be infected with M1
by immunofluorescence analysis with anti-SeV antiserum (data
not shown). Since the cells were infected at a multiplicity of
infection (MOI) of 10 and cultivation was performed in the
absence of trypsin, multiple-cycle replication would not take
place. Thus, a high degree of virus replication was maintained
in M1-infected LLC-MK
2cells throughout the observation
pe-riod without killing the host cells. On the other hand,
MVC11-infected LLC-MK
2cells underwent injury and died.
Accord-ingly, MVC11 progeny virus production diminished rapidly
after 24 h p.i. (Fig. 1b).
Since SeV was reported to induce apoptosis (38), we
exam-ined whether the strong cytopathic effect of MVC11 was
caused by apoptosis. Staining of nuclei with Hoechst 33342
clearly demonstrated condensed chromatin in
MVC11-in-fected, but not M1-inMVC11-in-fected, LLC-MK
2cells (Fig. 2a). Also,
strong DNA fragmentation was detected at 36 h p.i. and later
in MVC11-infected cells, whereas slight DNA fragmentation
was observed in M1-infected cells only at 48 h p.i. (Fig. 2b).
Restricted production of progeny virus as a result of
apo-ptosis in MVC11-infected mouse pulmonary epithelial cells.
MVC11 has almost completely lost lethality against mice, with
its replication being extremely suppressed in the mouse lungs
(17). To get information on the mechanism(s) of suppression
of progeny virus production, we investigated whether MVC11
could trigger apoptosis to interrupt virus production in mouse
pulmonary epithelial cells, the target cells of SeV in vivo, as
observed with LLC-MK
2cells. In a primary culture of mouse
pulmonary epithelial cells, M1 virus growth took place more
slowly than in LLC-MK
2cells and reached a maximum titer at
[image:4.612.122.482.68.429.2]3 days p.i., which was maintained at least until 11 days p.i. (Fig.
3a). On the other hand, virus titers in MVC11-infected cells
reached a maximum level at 1 day p.i., and diminished rapidly
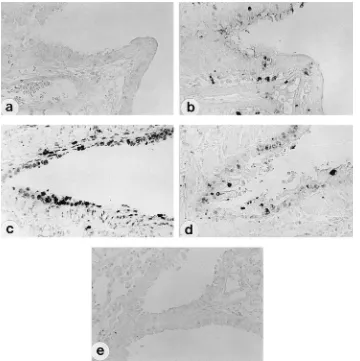
FIG. 5. Detection of apoptosis in lungs of mice infected with SeV. Three-week-old male ICR mice were inoculated intranasally with 1.253105CIU of either M1
(a and b) or MVC11 (c and d) in 25ml of PBS or with 25ml of PBS as a control (e). Tissue sections from 2 days p.i. (a, c, and e) and 6 days p.i. (b and d) were labeled by the TUNEL reaction for detection of DNA fragmentation.
on November 9, 2019 by guest
http://jvi.asm.org/
thereafter. Figure 3b shows synthesis of SeV-specific proteins
in the primary culture of epithelial cells. In accordance with the
time course of virus production (Fig. 3a), synthesis of viral
proteins in M1-infected mouse pulmonary epithelial cells took
place relatively slowly, reaching a maximum level at 3 days p.i.,
which was maintained throughout the observation period. On
the other hand, protein synthesis in MVC11-infected cells
oc-curred more rapidly to a maximum level at 1 day p.i. and was
strongly suppressed thereafter.
SeV-infected mouse pulmonary epithelial cells were
exam-ined for apoptosis by staining the nuclei with Hoechst 33342.
The nuclei of M1-infected cells appeared to be intact at both 2
and 6 days p.i. (Fig. 4b and d). Some of the M1-infected cells
were even found to proliferate (Fig. 4d), suggesting that M1
caused persistent infection without killing the host cells. On
the other hand, the nuclei of MVC11-infected cells
demon-strated condensed chromatin at both 2 and 6 days p.i. (Fig. 4f
and h), with large numbers of cells having detached from the
bottom of the plastic dish on day 6. These results indicate that
MVC11 could induce strong apoptosis of the infected cells,
which interrupted the following synthesis of viral proteins and
progeny virus production. On the other hand, M1 possessed a
very limited capacity, if any, to trigger apoptosis, which allowed
M1 to replicate for a prolonged period of time.
Induction of apoptosis in the lungs of mice infected with
MVC11.
To examine whether MVC11 could induce apoptotic
cell death in mouse lungs in vivo, lung sections obtained from
M1- and MVC11-infected mice were stained for TUNEL
sig-nals. TUNEL signals were detected only slightly in the lungs of
mice infected with M1 at 2 days p.i. (Fig. 5a). Although M1
infection spread over the lung, even to the alveoli, and
pro-duced high titers of virus on day 6 (17), only a few TUNEL
signal-positive cells were detected (Fig. 5b). On the other
hand, nuclei of bronchial epithelial cells in MVC11-infected
mice were strongly stained by TUNEL at 2 days p.i. (Fig. 5c).
On day 6, the number of cells with TUNEL-positive nuclei
decreased and the intensity of the signals diminished (Fig. 5d),
which was associated with both the elimination of MVC11
antigen-positive cells and the sharp decline of virus titers in the
lung (17).
Lung sections prepared from the mice described above were
then dually stained for SeV antigens and DNA fragmentation
to confirm that the cells dying by apoptosis were infected with
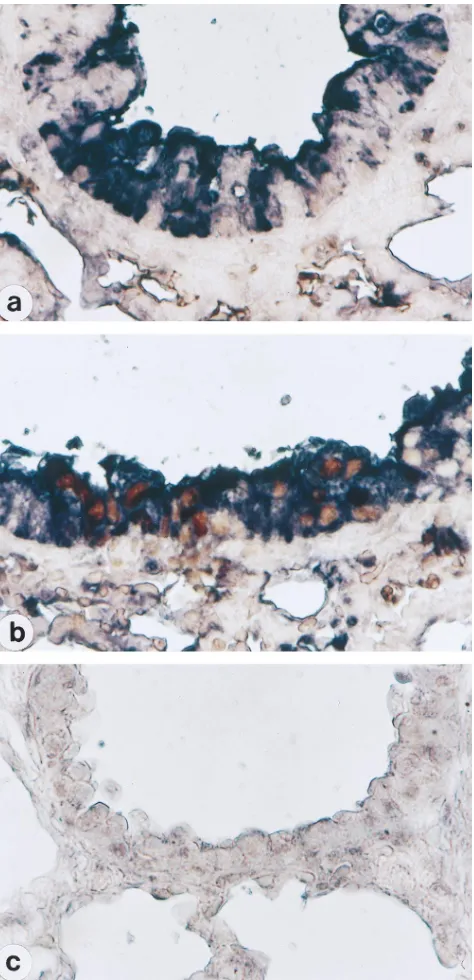
MVC11. Although bronchial epithelial cells of mice inoculated
with M1 were stained dark purple, which verified the infection
by SeV, brown TUNEL signals were not detected in those cells
(Fig. 6a). In MVC11-inoculated mice, bronchial epithelial cells
with DNA fragmentation (brown) were shown to be positive
for SeV antigens (purple) (Fig. 6b). The lungs of control mice
inoculated with PBS alone demonstrated neither SeV antigens
nor TUNEL signals (Fig. 6c). These results clearly
demon-strate that MVC11 triggered apoptosis as a result of virus
infection, while M1 did not.
Induction of apoptosis by the C protein.
MVC11 possesses
two amino acid mutations; one is in the C protein at position
170 (Phe
3
Ser), and the other is in the L protein at position
2050 (Glu
3
Ala) (17). We tested possible involvement of the C
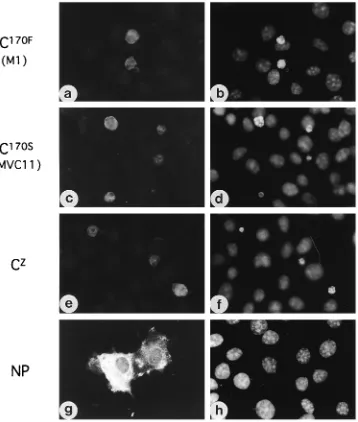
protein in the induction of apoptosis. As demonstrated in Fig.
7, COS-7 cells transiently expressing the C protein exhibited
condensation of chromatin. There was no apparent difference
in apoptosis-inducing capacity among the C proteins of strains
M1 (C
170F) (Fig. 7b), MVC11 (C
170S) (Fig. 7d), and Z (Fig. 7f).
It was unlikely that the apoptosis was induced simply by
over-expression of a protein, since strong over-expression of the NP
protein did not induce apoptosis in the cells (Fig. 7h).
Induc-tion of apoptosis by C
170Fand C
170S, but not by the NP protein,
was confirmed also in HeLa and L929 cells (data not shown).
Since the C proteins of M1 and MVC11 induced apoptosis
equally, we then examined the synthesis of the C protein in
M1- and MVC11-infected cells. In M1-infected LLC-MK
2cells, the C protein accumulated gradually until 96 h p.i. (Fig.
8a). In MVC11-infected LLC-MK
2cells, on the other hand,
synthesis of the C protein took place more rapidly and to a
larger extent than with M1 until 24 h p.i. but decreased rapidly
thereafter. In mouse pulmonary epithelial cells, the C protein
of M1 was not detected throughout the course of infection,
prob-FIG. 6. Colocalization of viral antigens and TUNEL signals in SeV-infected mouse lungs. Tissue sections were prepared on day 2 from the same mice as in Fig. 5 inoculated with M1 (a), MVC11 (b), or PBS (c). Double labeling was performed with anti-SeV antiserum (purple) and the TUNEL reaction (brown) to demonstrate colocalization of DNA fragmentation and SeV antigens.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.53.290.68.558.2]ably due to the limited number of cells, whereas that of MVC11
was detected at 1 day p.i. and diminished thereafter (Fig. 8b).
DISCUSSION
Apoptosis is a built-in cell suicide program required for
normal embryonic development, tissue homeostasis, and
sev-eral immunological processes. Infection of cultured cells with a
wide variety of viruses, including herpesviruses (14, 18, 21, 22,
25), parvoviruses (27), retroviruses (5, 23, 26, 28, 33),
paramyxoviruses (4), myxoviruses (6, 12, 34), alphaviruses (24,
39), and picornaviruses (37), results in activation of the
apo-ptosis pathway. It has been shown that virus-induced activation
of programmed cell death in certain cell populations, such as
neurons and immune cells, may be directly associated with viral
pathogenicity (1). In such cases, virulent strains cause
apopto-sis more strongly than avirulent strains. On the other hand,
data have accumulated that host cells trigger apoptosis when
infected with viruses, which interferes with virus production,
offering an important host defense mechanism to combat virus
infection (36). Suppression of virus production by apoptosis
was reported with some viruses, such as poliovirus (37) and
vaccinia virus (13).
In this study, we demonstrated that MVC11, an avirulent
mutant of SeV derived from the virulent wild-type isolate M1,
induced apoptosis in mouse pulmonary epithelial cells within 2
days p.i. (Fig. 4f). As shown in the one-step growth experiment
(Fig. 3a), replication of SeV appears to take place relatively
slowly in primary cultures of mouse epithelial cells, and
prob-ably in vivo as well, reaching a maximum titer at 3 days p.i.
Apoptosis triggered by MVC11 therefore could have caused
cell death before the virus replication cycle was completed and,
as a result, the following synthesis of virus proteins and
pro-FIG. 7. Induction of apoptosis by the C protein. The C proteins of M1 (C170F), MVC11 (C170S), and Z (CZ) and the NP protein of M1 were expressed transiently
in COS-7 cells. At 60 h after transfection, COS-7 cells transfected with pSG-CM (a and b), pSG-CMVC (c and d), or pSV2-C (e and f) were stained with anti-C guinea pig serum and fluorescein isothiocyanate-conjugated goat anti-guinea pig IgG (a, c, and e), and cells transfected with pSG-NP were stained with anti-SeV polyclonal rabbit antiserum and fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (g). The cells were then stained with Hoechst 33342 to detect chromatin condensation (b, d, f, and h). Cells positive for C170F, C170S, or CZexhibit chromatin condensation and/or nuclear fragmentation, whereas NP-positive cells do not.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.123.481.70.492.2]duction of progeny virus were strongly suppressed.
Consider-ing that about 10
4mouse pulmonary epithelial cells were used
for the virus growth experiment of Fig. 3a, a single
MVC11-infected cell produced approximately 100 virus particles on the
first day of infection and much less thereafter. On the other
hand, an M1-infected cell released 1,000 virus particles every
day throughout the cultivation period without undergoing
ap-optosis. Therefore, it is clear that apoptosis induced by MVC11
inhibited virus production in the culture. The same concept
could be applied to the in vivo experiments, where apoptosis
interfered with the replication and spread of MVC11 in mouse
lungs. Thus, it is likely that induction of apoptosis by MVC11
plays an important role in attenuation of the mouse
pathoge-nicity of the virus.
There is increasing evidence that many viruses encode
teins that interact with the cellular pathways regulating
pro-grammed death (36). However, the molecular mechanism by
which SeV infection activates the death pathway is unknown.
Tropea et al. (38) reported that alpha interferon had no effect
on SeV-induced apoptosis. We demonstrated in the present
study that the C protein of SeV induces apoptosis when
tran-siently expressed in COS-7 cells (Fig. 7) and in HeLa and L929
cells (data not shown). To our knowledge, this is the first study
that pinpoints an SeV protein as an apoptosis-inducing
pro-tein.
Despite the marked difference between M1 and MVC11 in
the capacity to induce apoptosis through viral infection, the C
protein of M1 (C
170F) induced apoptosis to practically the
same extent as the C protein of MVC11 (C
170S) in
transient-expression experiments (Fig. 7). A possible explanation for this
discrepancy is that apoptosis is triggered by the increased level
of C protein expression in MVC11-infected cells: the amounts
of the C protein in MVC11-infected cells in the early stage of
infection (12 to 24 h p.i. in LLC-MK
2cells and 1 day p.i. in
mouse pulmonary epithelial cells) were significantly larger
than those in M1-infected cells throughout the infection (Fig.
8). Another possibility that should also be taken into
consid-eration is that another SeV protein(s) is involved in the
induc-tion of apoptosis. Further analysis to elucidate the mechanism
of SeV-induced apoptosis is in progress.
ACKNOWLEDGMENTS
We thank K. Iwasaki and H. Taira for their kind gifts of anti-C
guinea pig antiserum and pSV2-C, respectively.
This work was supported in part by a Grant-in-Aid for Scientific
Research from the Ministry of Education, Science, Sports and Culture,
by a Research Program for Slow Virus Infection grant from the
Min-istry of Health and Welfare, Japan, and by a research grant from
Yakult Co., Ltd.
REFERENCES
1. Auwaerter, P. G., H. Kaneshima, J. M. McCune, G. Wiegand, and D. E.
Griffin.1996. Measles virus infection of thymic epithelium in the SCID-hu mouse leads to thymocyte apoptosis. J. Virol. 70:3734–3740.
2. Brooks, M. A., A. N. Ali, P. C. Turner, and R. W. Moyer. 1995. A rabbitpox virus serpin gene controls host range by inhibiting apoptosis in restrictive cells. J. Virol. 69:7688–7698.
3. Clem, R. J., M. Fechheimer, and L. K. Miller. 1991. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254:1388–1390. 4. Esolen, L. M., S. W. Park, J. M. Hardwick, and D. E. Griffin. 1995. Apoptosis as a cause of death in measles virus-infected cells. J. Virol. 69:3955–3958. 5. Estaquier, J., T. Idziorek, F. De Bels, F. Barre-Sinoussi, B. Hurtrel, A. M.
Aubertin, A. Venet, M. Mehtali, E. Muchmore, P. Michel, Y. Mouton, M. Girard, and J. C. Ameisen.1994. Programmed cell death and AIDS: signif-icance of T-cell apoptosis in pathogenic and nonpathogenic primate lentivi-ral infections. Proc. Natl. Acad. Sci. USA 91:9431–9435.
6. Fesq, H., M. Bacher, M. Nain, and D. Gemsa. 1994. Programmed cell death (apoptosis) in human monocytes infected by influenza A virus. Immunobi-ology 190:175–182.
7. Fujita, T., S. Ishido, S. Muramatsu, M. Itoh, and H. Hotta. 1996. Suppres-sion of actinomycin D-induced apoptosis by the NS3 protein of hepatitis C virus. Biochem. Biophys. Res. Commun. 229:825–831.
8. Green, S., I. Issemann, and E. Sheer. 1988. A versatile in vivo and in vitro eukaryotic expression vector for protein engineering. Nucleic Acids Res.
16:369.
9. Gregory, C. D., C. Dive, S. Henderson, C. A. Smith, G. T. Williams, J.
Gordon, and A. B. Rickinson.1991. Activation of Epstein-Barr virus latent genes protects human B cells from death by apoptosis. Nature (London)
349:612–614.
10. Henderson, S., M. Rowe, C. Gregory, D. Croom-Carter, F. Wang, R.
Long-necker, E. Kieff, and A. Rickinson.1991. Induction of bcl-2 expression by Epstein-Barr virus latent membrane protein 1 protects infected B cells from programmed cell death. Cell 65:1107–1115.
11. Henderson, S., D. Huen, M. Rowe, C. Dawson, G. Johnson, and A.
Rickin-son.1993. Epstein-Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death. Proc. Natl. Acad. Sci. USA 90:8479–8483.
12. Hinshaw, V. S., C. W. Olsen, N. Dybdahl-Sissoko, and D. Evans. 1994. Apoptosis: a mechanism of cell killing by influenza A and B viruses. J. Virol.
68:3667–3673.
13. Ink, B. S., C. S. Gilbert, and G. I. Evan. 1995. Delay of vaccinia virus-induced apoptosis in nonpermissive Chinese hamster ovary cells by the cowpox virus CHOhr and adenovirus E1B 19K genes. J. Virol. 69:661–668.
14. Ishii, H. H., and G. C. Gobe. 1993. Epstein-Barr virus infection is associated with increased apoptosis in untreated and phorbol ester-treated human Burkitt’s lymphoma (AW-Ramos) cells. Biochem. Biophys. Res. Commun.
192:1415–1423.
[image:7.612.119.472.68.238.2]15. Itoh, M., and M. Homma. 1987. Single amino acid substitution of Sendai FIG. 8. Synthesis of the C protein in M1- and MVC11-infected cells. At the indicated times after infection, lysates prepared from LLC-MK2cells (a) or mouse
pulmonary epithelial cells (b) infected with M1 or MVC11 (MOI of 10) were subjected to Western blot analysis with anti-C polyclonal guinea pig antiserum to detect the C protein. For panel b, the same cell lysates as for Fig. 3b were used.
on November 9, 2019 by guest
http://jvi.asm.org/
virus at the cleavage site of the fusion protein confers trypsin resistance. J. Gen. Virol. 68:2939–2944.
16. Itoh, M., D.-M. Tan, T. Hayashi, Y. Mochizuki, and M. Homma. 1990. Pneumopathogenicity of a Sendai virus protease-activation mutant, TCs, which is sensitive to trypsin and chymotrypsin. J. Virol. 64:5660–5664. 17. Itoh, M., Y. Isegawa, H. Hotta, and M. Homma. 1997. Isolation of an
avirulent mutant of Sendai virus with two amino acid mutations from a highly virulent field strain through adaptation to LLC-MK2cells. J. Gen. Virol.
78:3207–3215.
18. Johnson, P. A., A. Miyanohara, F. Levine, T. Cahill, and T. Friedmann. 1992. Cytotoxicity of a replication-defective mutant of herpes simplex virus type 1. J. Virol. 66:2952–2965.
19. Kashiwazaki, H., M. Homma, and N. Ishida. 1965. Assay of Sendai virus by immunofluorescence and hemadsorbed cell-counting procedures. Proc. Soc. Exp. Biol. Med. 120:134–138.
20. Kato, A., K. Kiyotani, Y. Sakai, T. Yoshida, and Y. Nagai. 1997. Importance of the V protein for determining in vitro phenotypes and in vivo pathoge-nicity of Sendai virus. EMBO J. 16:578–587.
21. Kawanishi, M. 1993. Epstein-Barr virus induces fragmentation of chromo-somal DNA during lytic infection. J. Virol. 67:7654–7658.
22. Koga, Y., K. Tanaka, Y. Lu, M. Oh-Tsu, M. Sasaki, G. Kimura, and K.
Nomoto.1994. Priming of immature thymocyte to CD3-mediated apoptosis by infection with murine cytomegalovirus. J. Virol. 68:4322–4328. 23. Laurent-Crawford, A. G., B. Krust, S. Muller, Y. Riviere, M.-A. Rey-Cuille,
J.-M. Bechet, L. Montagnier, and A. G. Hovanessian.1991. The cytopathic effect of HIV is associated with apoptosis. Virology 185:829–839. 24. Levine, B., Q. Huang, J. T. Isaacs, J. C. Reed, D. E. Griffin, and J. M.
Hardwick.1993. Conversion of lytic to persistent alphavirus infection by the bcl-2 cellular oncogene. Nature (London) 361:739–742.
25. Lewis, J., S. L. Wesselingh, D. E. Griffin, and J. M. Hardwick. 1996. Alpha-virus-induced apoptosis in mouse brains correlates with neurovirulence. J. Virol. 70:1828–1835.
26. Meyaard, L., S. A. Otto, R. R. Jonker, M. J. Mijnster, R. P. M. Keet, and F.
Miedema.1992. Programmed death of T cells in HIV-1 infection. Science
257:217–219.
27. Morey, A. L., D. J. Ferguson, and K. A. Fleming. 1993. Ultrastructural features of fetal erythroid precursors infected with parvovirus B19 in vitro: evidence of cell death by apoptosis. J. Pathol. 169:213–220.
28. Ohno, K., Y. Okamoto, T. Miyazawa, T. Mikami, T. Watari, R. Goitsuka, H.
Tsujimoto, and A. Hasegawa.1994. Induction of apoptosis in a T lympho-blastoid cell line infected with feline immunodeficiency virus. Arch. Virol.
135:153–158.
29. Omata-Yamada, T., K. Hagiwara, K. Katoh, H. Yamada, and K. Iwasaki. 1988. Purification of the Sendai virus nonstructural C protein expressed in E.
coli, and preparation of antiserum against C protein. Arch. Virol. 103:61–72.
30. Pilder, S., J. Logan, and T. Shenk. 1984. Deletion of the gene encoding the adenovirus 5 early region 1B 21,000-molecular-weight polypeptide leads to degradation of viral and host cell DNA. J. Virol. 52:664–671.
31. Rao, L., M. Debbas, P. Sabbatini, D. Hockenbery, S. Korsmeyer, and E.
White.1992. The adenovirus E1A proteins induce apoptosis, which is inhib-ited by the E1B 19-kDa and Bcl-2 proteins. Proc. Natl. Acad. Sci. USA
89:7742–7746.
32. Ray, R. B., K. Meyer, and R. Ray. 1996. Suppression of apoptotic cell death by hepatitis C virus core protein. Virology 226:176–182.
33. Rey-Cuille, M.-A., J. Galabru, A. Laurent-Crawford, B. Krust, L.
Montag-nier, and A. G. Hovanessian.1994. HIV-2 EHO isolate has a divergent envelope gene and induces single cell killing by apoptosis. Virology 202:471– 476.
34. Takizawa, T., S. Matsukawa, Y. Higuchi, S. Nakamura, Y. Nakanishi, and R.
Fukuda.1993. Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. J. Gen. Virol. 74:2347–2355. 35. Tashiro, M., and M. Homma. 1983. Pneumotropism of Sendai virus in
relation to protease-mediated activation in mouse lungs. Infect. Immun.
39:879–888.
36. Teodoro, J. G., and P. E. Branton. 1997. Regulation of apoptosis by viral gene products. J. Virol. 71:1739–1746.
37. Tolskaya, E. A., L. I. Romanova, M. S. Kolesnikova, T. A. Ivannikova, E. A.
Smirnova, N. T. Raikhlin, and V. I. Agol. 1995. Apoptosis-inducing and apoptosis-preventing functions of poliovirus. J. Virol. 69:1181–1189. 38. Tropea, F., L. Troiano, D. Monti, E. Lovato, W. Malorni, G. Rainaldi, P.
Mattana, G. Viscomi, M. C. Ingletti, M. Portolani, C. Cermelli, A. Cossa-rizza, and C. Franceschi.1995. Sendai virus and herpes virus type 1 induce apoptosis in human peripheral blood mononuclear cells. Exp. Cell Res.
218:63–70.
39. Ubol, S., P. C. Tucker, D. E. Griffin, and J. M. Hardwick. 1994. Neuroviru-lent strains of alphavirus induce apoptosis in bcl-2-expressing cells: role of a single amino acid change in the E2 glycoprotein. Proc. Natl. Acad. Sci. USA
91:5202–5206.