Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Integrin-Using Rotaviruses Bind
␣
2

1 Integrin
␣
2 I Domain via VP4
DGE Sequence and Recognize
␣
X

2 and
␣
V

3 by Using
VP7 during Cell Entry
Kate L. Graham,
1Peter Halasz,
1Yan Tan,
1Marilyn J. Hewish,
1Yoshikazu Takada,
2Erich R. Mackow,
3Martyn K. Robinson,
4and Barbara S. Coulson
1*
Department of Microbiology and Immunology, The University of Melbourne, Victoria 3010, Australia1; Department of
Cell Biology, Division of Vascular Biology, The Scripps Research Institute, La Jolla, California 923072; The
Department of Medicine and Department of Molecular Genetics and Microbiology, Stony Brook University, Stony Brook, and Northport VAMC, Northport, New York 117943; and Celltech
Group plc., Slough SL1 4EN, United Kingdom4
Received 8 April 2003/Accepted 25 June 2003
Integrins␣21,␣X2, and␣V3 have been implicated in rotavirus cell attachment and entry. The virus
spike protein VP4 contains the␣21 ligand sequence DGE at amino acid positions 308 to 310, and the outer
capsid protein VP7 contains the␣X2 ligand sequence GPR. To determine the viral proteins and sequences
involved and to define the roles of␣21,␣X2, and␣V3, we analyzed the ability of rotaviruses and their
reassortants to use these integrins for cell binding and infection and the effect of peptides DGEA and GPRP on these events. Many laboratory-adapted human, monkey, and bovine viruses used integrins, whereas all porcine viruses were integrin independent. The integrin-using rotavirus strains each interacted with all three integrins. Integrin usage related to VP4 serotype independently of sialic acid usage. Analysis of rotavirus reassortants and assays of virus binding and infectivity in integrin-transfected cells showed that VP4 bound
␣21, and VP7 interacted with␣X2 and␣V3 at a postbinding stage. DGEA inhibited rotavirus binding to
␣21 and infectivity, whereas GPRP binding to␣X2 inhibited infectivity but not binding. The truncated VP5*
subunit of VP4, expressed as a glutathioneS-transferase fusion protein, bound the expressed␣2 I domain.
Alanine mutagenesis of D308 and G309 in VP5* eliminated VP5* binding to the ␣2 I domain. In a novel
process, integrin-using viruses bind the␣2 I domain of␣21 via DGE in VP4 and interact with␣X2 (via
GPR) and␣V3 by using VP7 to facilitate cell entry and infection.
Rotaviruses are leading causes of acute gastroenteritis in human infants and young animals. Their restricted tropism suggests that very specific virus-host cell interactions are nec-essary to establish infection. The viral spike protein VP4, the major cell attachment protein, is cleaved by trypsin for en-hanced infectivity into two subunits, VP5* (60 kDa) and VP8* (28 kDa). VP4 and the major outer capsid protein VP7 inde-pendently define serotype specificities. The inner capsid pro-tein VP6 contains group-specific antigenic determinants. Re-assortant rotaviruses containing combinations of the 11 double-stranded RNA genes from two parental viruses can be generated (27) which occasionally show unexpected pheno-types due to VP4-VP7 interactions (43).
Integrins␣21,␣X2, and␣41 were implicated in group A rotavirus cell attachment and entry (11, 23), and ␣V3 was proposed to mediate rotavirus cell entry (20). Integrin usage by rotaviruses was discovered when VP5* was shown to contain the type I collagen-derived, ␣21 ligand sequence DGE at amino acids 308 to 310, and the fibrinogen-derived␣X2 in-tegrin ligand sequence GPR was identified in VP7 at amino acids 253 to 255 (11). Peptides containing these viral integrin ligand sequences (GPRP and RDGEE), monoclonal
antibod-ies (MAbs) to␣21 and MAbs to2, inhibited the infection of monolayers of MA104 and Caco-2 cells by simian rotavirus SA11 and/or human rotavirus RV-5 by 30 to 90% in additive fashion (9, 11). Infectivity blockade in MA104 cell monolayers by anti-␣2 MAbs has now been reported for a range of human and animal rotaviruses (7). Infection of MA104 cell monolay-ers by the rhesus monkey rotavirus RRV and the human ro-tavirus Wa also are inhibited by anti-␣X2 MAbs (20).
Glycoconjugates have been proposed as rotavirus receptors (26, 45). Terminal sialic acids (SA) can be used by a few animal strains, including SA11 and RRV (8), but are not essential for infection (55). Infection by the majority of rotaviruses is inde-pendent of terminal SA, but it might involve subterminal SA (14, 22). SA11 binding to ␣21 expressed on K562 cells as a result of transfection specifically resulted in increased infectiv-ity, which was inhibited by cellular treatment with an anti-␣2 MAb (23). Cellular receptors for viruses bind virus via virus-specific proteins, and this binding leads to productive cellular infection. Thus, these data show that SA11 can use␣21 as a receptor (23). Collagen type I binds the inserted (I) domain of
␣2 (29). As anti-␣2 I domain MAbs blocked rotavirus binding to ␣21 (23), this domain could be important for rotavirus binding to␣21. It has been proposed that rotavirus may bind to SA via multiple low-affinity contacts prior to more specific interactions that determine host and cell tropism (16) and that these specific interactions could be with integrins (23).
Binding of MA104 cells in suspension by an RRV mutant
* Corresponding author. Mailing address: Department of Microbi-ology and ImmunMicrobi-ology, Gate II, The University of Melbourne, Mel-bourne, Victoria 3010, Australia. Phone: 38344-8823. Fax: 61-39347-1540. E-mail: [email protected].
9969
on November 8, 2019 by guest
http://jvi.asm.org/
that was no longer dependent on SA for infectivity (nar) was inhibited by an anti-␣2 MAb and expressed VP5* containing DGE, whereas nar binding was not affected by blockade with mutated VP5* containing D308A (55). However, binding of SA11, RRV, and other rotaviruses to␣21 on suspended cells could not be demonstrated (7, 55). This led to the alternative conclusion by the authors that␣21 is not a rotavirus receptor, but it may function in later entry events (7). However, their data could be reconciled with the original report that ␣21 binds SA11 if rotavirus binding to␣21 on MA104 cells is not detectable when the binding assay is performed with sus-pended cells.
We designed experiments to further establish and clarify the roles of ␣21,␣X2, and␣V3 in rotavirus cell attachment and entry by using MA104 cell monolayers and integrin-trans-fected K562 cells. We also aimed to locate the viral proteins and domains required for integrin recognition. In this study, the extent of usage of␣21,␣X2, and␣V3 for cell binding and infection by a large range of laboratory-adapted rotavi-ruses from several animal species has been established for the first time. Integrin-independent rotaviruses, which do not use these three integrins for cell attachment and entry, also were identified for the first time. We demonstrate, for a much greater range of integrin-using rotaviruses than reported pre-viously, that virus-cell binding specifically involves␣21. Our studies show that the inability of others to detect virus-␣21 binding was related to assay of binding to suspended cells rather than to monolayers. We determined for the first time that integrin usage relates to VP4 serotype and that this is independent of the relation of SA usage to VP4 serotype. In this study the viral proteins required for integrin recognition have been identified by using reassortant mapping and inhibi-tion of virus-cell binding and infecinhibi-tion by integrin ligands (col-lagen, fibrinogen) and integrin ligand peptides DGEA and GPRP. This positively identified the rotavirus protein that binds␣21 as VP4 and the protein interacting with␣X2 and
␣V3 as VP7. VP7 has not previously been suggested to in-teract with␣V3. In novel experiments the DGE sequence in VP4 was shown to be specifically involved in binding of inte-grin-using viruses to␣21 by DGEA peptide blockade of virus binding to␣2-transfected K562 cells. Finally, we have demon-strated binding of VP4 to␣21 at the protein level for the first time and have determined that the␣2 I domain is sufficient for binding, as RRV VP5* (expressed as a glutathione S -trans-ferase [GST] fusion protein) bound to expressed␣2 I domain protein. As we showed that this binding requires VP5* D308 and/or G309, either one or both of these VP5* residues are critical for rotavirus binding to␣21.
MATERIALS AND METHODS
Cell lines and viruses.MA104 cells were obtained from the National Institutes of Health, Bethesda, Md. The derivation of the integrin-transfected K562 cells used in this study has been described previously (23, 42). The maintenance of K562 and MA104 monkey kidney cells and K562 cells transfected with cDNA encoding␣2 (␣2-K562),␣3 (␣3-K562),␣X2 (␣X2-K562),␣L2 (␣L2-K562), ␣M2 (␣M2-K562), and empty vector (PBJ-K562) and monitoring of their cell surface integrin expression by flow cytometry were carried out as before (23, 42). Flow cytometric data are expressed as the relative linear median fluorescence intensity (MFI), defined as the median fluorescence intensity with test MAb divided by the median fluorescence intensity with isotype-matched control MAb. An MFI ofⱖ1.20 was considered positive (34). The origins of the rotaviruses
used in this study and their cultivation in MA104 cells have been described previously (10, 27, 31–33, 39, 40).
Antibodies, proteins, and peptides.MAbs AK7 (␣2I), 8A2 (1), MHM23 (2), MEM-148 (2), and MOPC21 were obtained as previously described (2, 3). Non-I domain anti-␣2 MAb HAS3 (3, 29) and anti-major histocompatibility complex class I MAb W6/32 were provided by F. Watt, Imperial Cancer Re-search Fund, London, England, and by A. Brooks, The University of Melbourne, respectively. W6/32 (10 to 40 g/ml) bound 100% of MA104 cells by flow cytometry, with an MFI of 20⫾1. MAbs P4H9-G11 (2), LM609 (␣V3), and CH-1 (GST) were purchased from Chemicon. MAb P4H9-G11 at 20g/ml bound 100% of MA104 cells by flow cytometry with an MFI of 33⫾1, and LM609 also binds all MA104 cells (20). Anti-VP5* MAb 2G4 (47) was provided by M. Estes, Baylor College of Medicine, Waco, Tex. Anti-VP6 MAb RVA and anti-VP7 MAb RV-4:1 were produced as described previously (12). Human placental type I collagen, human plasma fibrinogen, chicken egg ovalbumin, and bovine serum albumin (BSA) were used according to the manufacturer’s proto-col (Sigma). Peptides (ⱖ90% pure by high-performance liquid chromatography) were purchased from Auspep (DGEA, GPRP) and Mimotopes (GHRP) or were provided by D. Jackson and W. Zeng of The University of Melbourne.
Virus binding and infectivity assays.Assays of infectious rotavirus binding to transfected and parental K562 cells and MAb inhibition of virus binding were carried out as described previously (23). For assays of infectious rotavirus bind-ing to MA104 cells, confluent cell monolayers (5⫻105cells; seeded 3 to 4 days
previously) in 24-well trays were washed and then incubated with MAbs for 2 h at 37°C. In the presence of MAb, cells were cooled on ice for 20 min. Virus infectivity was activated with 10g of porcine trypsin (Sigma)/ml for 20 min at 37°C (34). Activated virus, which had been cooled to 4°C, at a multiplicity of infection (MOI) of 3.5 was allowed to attach to cells on ice for 1 h. This MOI was chosen because insufficient infectious virions bound to most cell lines for the infectivity counts to be reliable at lower MOI (ⱕ1). An MOI of 3.5 to 5 gave the best discrimination between virus binding to integrin-transfected K562 cells over parental cells. At MOI of 10 to 20 there was an increased level of infectious virus binding to parental cells that did not result in productive infection, so the additional binding at high MOI appears to be nonspecific (B. Coulson and M. Hewish, unpublished observations). In MA104 cells an MOI of 3.5 also gave reproducible data. Cell-bound virus titers were determined by indirect immuno-fluorescent staining of infected MA104 cells as described previously (23). For assay of MAb inhibition of virus binding to suspended MA104 cells, cells were placed in suspension by treatment with phosphate-buffered saline containing 0.75 mM EDTA, and then the assay was completed as for K562 cells (23), with gentle rocking of cell suspensions to prevent adhesion. In some experiments MA104 cells in suspension were cooled to 4°C and then were treated with anti-integrin MAb for 2 h at 4°C rather than at 37°C. For assays of peptide inhibition of virus-cell binding, prior to cell cooling and virus addition as above cells were incubated with peptides diluted as described previously (11) at pH 7.5 for 1 h at 37°C.
Peptide, protein, and MAb blockades of virus infectivity at an MOI of 0.02 by using MA104 cells seeded 3 to 4 days previously into 96-well plates (5⫻105cells)
were measured by fluorescence focus reduction assay as described previously (11, 23) and are expressed as mean percentages⫾standard deviations. Anti-integrin MAb and peptide inhibition of rotavirus infectivity is maximal in the MOI range 0.01 to 0.1, and individual virus-infected MA104 cells are most readily distin-guished when the infectivity assay is carried out at an MOI of 0.02 (B. Coulson and M. Hewish, unpublished). On graphs, error bars represent standard devia-tions.
Plasmids, mutagenesis, and production of purified, expressed protein. Pro-duction of GST-␣2 I domain fusion protein (GST-␣2I) and GST have been described previously (29). Following thrombin protease cleavage of GST-␣2I to produce␣2I, carried out according to the manufacturer’s protocol (Amersham Biosciences), free GST was removed from␣2I by using glutathione-agarose beads (Sigma). A trace of GST-␣2I remained in␣2I after thrombin cleavage. Cleavage of GST-␣2I did not affect␣2I function, as both cleaved and uncleaved proteins bound the anti-␣2 I domain MAb AK7 similarly. A pET-6HIS plasmid containing truncated VP5* (amino acids 248 to 474) was used to generate the Asp308Ala, Gly309Ala mutant of VP5* (VP5*D308A/G309A) by site-directed mutagenesis as previously described (17). Viral genes were subcloned into pGEX (Pharmacia) by standard methods. To produce soluble VP5* and VP5*D308A/ G309A,Escherichia coliBL21(DE3) cultures that had been transformed with pGEX-VP5* or pGEX-VP5*D308A/G309A and grown overnight at 20°C were induced with 0.01 mM isopropyl-1-thio--D-galactopyranoside (IPTG) for 6 h at
20°C. Protein was extracted and purified as described previously (29). Copurify-ing, heat shock bacterial chaperonin GroEL (⬃65 kDa) was removed from VP5* and VP5*D308A/G309A by using ATP and partner chaperonin GroES (Sigma),
on November 8, 2019 by guest
http://jvi.asm.org/
as described previously (52). As complete removal of GroEL rendered VP5* insoluble, GroEL was not removed from VP5* for further experiments. The gene carrying murine rotavirus EW VP6 (51) was subcloned into pGEX-4T-1 (Phar-macia) by using theEcoRI/XhoI sites. Expression and purification of GST-VP6 were carried out as for GST-VP5*, except that transformed bacteria were cul-tured at 30°C for 4 h, induced with 0.075 mM IPTG, and culcul-tured for 2 h at 30°C. The identity of all plasmid inserts was verified by DNA sequencing.
EIA. For detection of MAb binding to expressed proteins, immunoplates (96-well; Nunc) were coated for 2 h at 37°C with purified proteins diluted in 50 mM Tris-HCl buffer, pH 7.4, containing 150 mM NaCl, 5 mM CaCl2, 0.1 mM
MgCl2, and 0.1 mM MnCl2(TSC). After being washed with TSC containing
0.05% Tween 20 (Sigma), plates were blocked with TSC containing 2.5% skim milk powder (TSC-S; Diploma) for 30 min at 37°C, reacted with MAbs diluted in TSC-S containing 0.05% Tween 20 (TSC-ST) overnight at 4°C, washed as before, and incubated with sheep anti-mouse immunoglobulin conjugated to horseradish peroxidase (Silenus) diluted in TSC-ST for 1.5 h 37°C. After being washed, color was developed by using the tetramethyl benzidine-H2O2substrate/
indicator system, as described previously (12). For assay of␣2I and control protein binding to viral fusion proteins,␣2I, BSA, or GST diluted in TSC-ST was reacted with blocked, fusion protein-coated plates overnight at 4°C. Washed plates were incubated with MAb AK7 or MOPC21 diluted in TSC-ST for 2 h at 37°C. The enzyme immunoassay (EIA) was completed as described above.
RESULTS
Usage of␣21,␣X2, and␣V3 by rotaviruses.The ability
of MAbs to ␣2, 2, and ␣V3 to inhibit the infectivity of laboratory-adapted rotaviruses was examined in MA104 cells (Table 1). The viruses tested fell into two groups: integrin-using and integrin-independent strains. Integrin-integrin-using rotavi-ruses were defined as those which used ␣21,2, and␣V3 integrins during cell binding and entry, and integrin-indepen-dent rotaviruses were defined as those that did not use these integrins for these functions. Infection by 7 out of 12 human, 2 out of 2 simian, 1 out of 2 bovine, 0 out of 5 porcine, and 0 out of 1 avian rotaviruses was inhibited by all three anti-integrin MAbs but not by control MAb MOPC21. Infection by other virus strains was not affected by any MAb tested. The anti-class I major histocompatibility complex MAb did not affect the infectivity of three integrin-using viruses (RRV, SA11, and Wa) or integrin-independent strain CRW-8. Virus usage of
␣21,2, and␣V3 related to P (VP4) serotype rather than to G (VP7) serotype. Integrins␣21,2, and␣V3 were used by disease-causing rotaviruses of children (Wa, RV-4, RV-5, P, VA70, and K8) of P serotypes 1A and 1B but not by strains from asymptomatically infected neonates (M37, 1076, RV-3, and ST-3) of P serotype 2A. A disease-associated virus (S12/ 85), serologically identical to RV-3 (31), showed the same integrin independence as RV-3, whereas asymptomatic neona-tal virus 116E (P serotype 8) used␣21,2, and␣V3. Infec-tion by bovine rotavirus NCDV (P serotype 7) was integrin dependent, whereas bovine rotavirus UK (P serotype 6) and porcine rotaviruses (P serotypes 2B and 9) did not use␣21,
2, and ␣V3. MAb inhibition levels were similar between viruses and between MAbs, as integrusing viruses were in-hibited by 33%⫾4% by anti-␣2 MAb (n⫽10), 28%⫾6% by anti-2 MAb (n⫽9), and 27%⫾6% by anti-␣V3 MAb (n⫽
7). All integrin-using viruses, except 116E and K8, and all integrin-independent strains, except MDR-13, had DGE in VP5*. 116E, K8, and MDR-13 had DGI, DEE, and NGE, respectively, instead of DGE. All integrin-using viruses had GPR in VP7, as did all integrin-independent strains, except Ty-1, which had DPR. Thus, integrin usage was confined to rotaviruses with D308 in VP5* and GPR in VP7, but
posses-sion of these sequences alone did not determine integrin usage, as the infectivity of many strains with these sequences was integrin independent.
Integrin and terminal SA usage independently related to the P serotype, as all possible combinations of SA and integrin usage were observed.
Viral proteins required for integrin recognition. The viral
[image:3.603.300.540.89.368.2]genes required for integrin usage were determined by using 10 laboratory-generated reassortant rotaviruses. Seven reassor-tants possessed the gene encoding VP4 from an integrin-using virus and the gene encoding VP7 from an integrin-indepen-dent virus or vice versa, two had both VP4 and VP7 from two different integrin-using viruses, and one had both VP4 and VP7 from two different integrin-independent viruses (Table 1). Most of these reassortants have been shown previously to exhibit the expected VP4 and VP7 phenotypes of anti-VP4 and
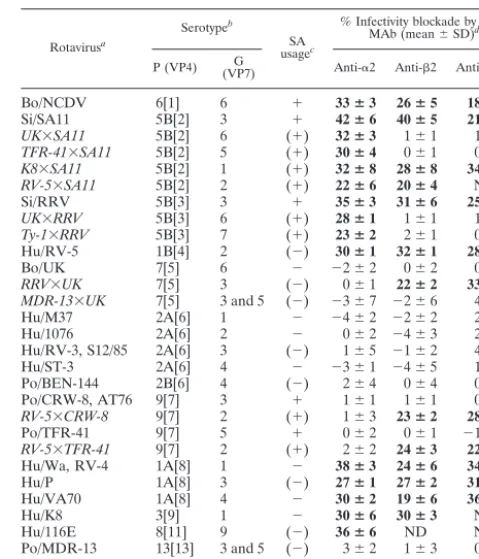
TABLE 1. Usage of MA104 cell␣21,2, and␣V3 integrins for infection by rotaviruses and their reassortants
Rotavirusa
Serotypeb
SA usagec
% Infectivity blockade by given MAb (mean⫾SD)d
P (VP4) (VP7)G Anti-␣2 Anti-2 Anti-␣V3
Bo/NCDV 6[1] 6 ⫹ 33ⴞ3 26ⴞ5 18ⴞ5
Si/SA11 5B[2] 3 ⫹ 42ⴞ6 40ⴞ5 21ⴞ1
UK⫻SA11 5B[2] 6 (⫹) 32ⴞ3 1⫾1 1⫾1
TFR-41⫻SA11 5B[2] 5 (⫹) 30ⴞ4 0⫾1 0⫾1
K8⫻SA11 5B[2] 1 (⫹) 32ⴞ8 28ⴞ8 34ⴞ9
RV-5⫻SA11 5B[2] 2 (⫹) 22ⴞ6 20ⴞ4 ND
Si/RRV 5B[3] 3 ⫹ 35ⴞ3 31ⴞ6 25ⴞ6
UK⫻RRV 5B[3] 6 (⫹) 28ⴞ1 1⫾1 1⫾1
Ty-1⫻RRV 5B[3] 7 (⫹) 23ⴞ2 2⫾1 0⫾1 Hu/RV-5 1B[4] 2 (⫺) 30ⴞ1 32ⴞ1 28ⴞ0
Bo/UK 7[5] 6 ⫺ ⫺2⫾2 0⫾2 0⫾1
RRV⫻UK 7[5] 3 (⫺) 0⫾1 22ⴞ2 33ⴞ7
MDR-13⫻UK 7[5] 3 and 5 (⫺) ⫺3⫾7 ⫺2⫾6 4⫾5
Hu/M37 2A[6] 1 ⫺ ⫺4⫾2 ⫺2⫾2 2⫾1
Hu/1076 2A[6] 2 ⫺ 0⫾2 ⫺4⫾3 2⫾3
Hu/RV-3, S12/85 2A[6] 3 (⫺) 1⫾5 ⫺1⫾2 4⫾7 Hu/ST-3 2A[6] 4 ⫺ ⫺3⫾1 ⫺4⫾5 1⫾3 Po/BEN-144 2B[6] 4 (⫺) 2⫾4 0⫾4 0⫾4 Po/CRW-8, AT76 9[7] 3 ⫹ 1⫾1 1⫾1 0⫾2
RV-5⫻CRW-8 9[7] 2 (⫹) 1⫾3 23ⴞ2 28ⴞ1
Po/TFR-41 9[7] 5 ⫹ 0⫾2 0⫾1 ⫺1⫾3
RV-5⫻TFR-41 9[7] 2 (⫹) 2⫾2 24ⴞ3 22ⴞ1
Hu/Wa, RV-4 1A[8] 1 ⫺ 38ⴞ3 24ⴞ6 34ⴞ5
Hu/P 1A[8] 3 (⫺) 27ⴞ1 27ⴞ2 31ⴞ4
Hu/VA70 1A[8] 4 ⫺ 30ⴞ2 19ⴞ6 36ⴞ2
Hu/K8 3[9] 1 ⫺ 30ⴞ6 30ⴞ3 ND
Hu/116E 8[11] 9 (⫺) 36ⴞ6 ND ND
Po/MDR-13 13[13] 3 and 5 (⫺) 3⫾2 1⫾3 0⫾3
Ty/Ty-1 [17] 7 ⫺ ⫺2⫾3 2⫾3 ⫺1⫾1
aLaboratory-adapted rotaviruses are shown with strain name preceded by
species of origin. Bo, bovine; Si, simian; Hu, human; Po, porcine; Ty, turkey. In laboratory-generated reassortants (shown in italics), VP7 and VP4 originated from the first- and last-named virus, respectively (27, 32, 33, 39, 40). UK⫻SA11, UK⫻RRV, Ty-1⫻RRV, and RRV⫻UK were originally designated as R2/1-2, 28-1, TyRh, and 12-1, respectively.
bSerotypes designations are as previously described (8, 24, 31). P serotype
precedes P genotype, which is shown in square brackets.
cTerminal SA usage (⫹,⫺) determined previously (8) or inferred [(⫹), (⫺)]
from identity of P serotype and species of origin with a tested rotavirus strain.
dData are for MAbs at saturating levels of 20g/ml (anti-␣2 MAb AK7), 40
g/ml (anti-2 MAb MHM23), and 10g/ml (anti-␣V3 MAb LM609). Positive levels of MAb blockade (⬎10%) are in bold. With the exceptions of K8⫻SA11, RV-5⫻SA11, and MDR-13⫻UK, viruses also were tested at four additional serial twofold MAb dilutions, to 1.2 or 2.5g/ml, in 3 to 10 experiments. The percent infectivity blockade of integrin-dependent viruses was MAb dose depen-dent. Integrin-independent viruses showed no blockade at any MAb concentra-tion tested. At 1.2 to 40g/ml, MOPC21 MAb did not affect infectivity of any virus. ND, not done.
on November 8, 2019 by guest
http://jvi.asm.org/
anti-VP7 MAb neutralization (UK⫻SA11, TFR⫻SA11, RV-5⫻SA11, MDR-13⫻UK, RV-5⫻CRW-8, RV-5⫻TFR-41) (33, 39, 40) and of antiserum neutralization, hemagglutination, plaque size, and protease-enhanced plaque formation (UK⫻RRV, RRV⫻UK) (27). Thus, it is likely that other VP4 or VP7 phenotypes of these reassortants, such as receptor binding, would segregate as expected (43). The exception to this is Ty-1⫻RRV, which contains the gene encoding RRV VP4 on the Ty-1 genetic background. The ability of anti-VP5* MAb 2G4 and anti-VP8 MAbs to neutralize Ty-1⫻RRV was reduced compared to that of RRV (32). As shown in Table 1, infection blockade by the anti-␣2 MAb segregated with VP4 originating from an integrin-using rotavirus, whereas blockade by the anti-2 and anti-␣V3 MAbs segregated with VP7 from an integrin-using virus. Integrin usage did not cosegregate with any other rotavirus gene and segregated independently with genes encoding VP4 and VP7 only in reassortants. Interest-ingly, Ty-1⫻RRV infectivity was inhibited to a lesser extent by anti-␣2 MAb (23%) than was RRV infectivity (35%), consis-tent with the reduced neutralization of Ty-1⫻RRV by anti-VP4 MAbs. As expected, other reassortants did not show any VP4-VP7 interaction. The infectivity of integrin-using reassor-tants was inhibited by 28%⫾4% by anti-␣2 MAb (n⫽6), 23%
⫾3% by anti-2 MAb (n⫽5), and 29%⫾5% by anti-␣V3 MAb (n ⫽ 4), so anti-integrin MAb effects were similar be-tween reassortants and laboratory-adapted viruses. Overall, these data show that virus usage of ␣21 maps to VP4, and virus usage of␣X2 and␣v3 is dependent on VP7.
Collagen and fibrinogen inhibited infection of only
integrin-using rotaviruses. Type I collagen and fibrinogen bind␣21
and␣X2 through DGEA and GPRP sequences, respectively (11, 48). Collagen and fibrinogen reduced the infectivity of integrin-using viruses NCDV, SA11, RRV, Wa, RV-4, and K8 by 42%⫾7% and 39%⫾7%, respectively, in MA104 cells but had no effect on integrin-independent strains UK, CRW-8, and MDR-13 (Table 2). These results suggest a role for viral DGE and GPR sequences in mediating virus-integrin interactions.
Rotavirus binding to␣21 depended on VP4, and virus did
not bind␣X2 and␣V3.Anti-integrin antibodies were used
to investigate rotavirus binding to MA104 cells. The titers of RRV bound to suspended MA104 cells after treatment at 37 or 4°C with the anti-␣2 MAb AK7 at 20 g/ml (1.7 ⫻ 105/ml),
control MAb MOPC21 at 20 g/ml (1.9 ⫻ 105/ml), and no
antibody (1.7 ⫻105/ml) were all similar to each other,
con-firming the previous findings that wild-type rotavirus binding to integrins on MA104 cells is not detectable when cells are placed in suspension (7, 55). The binding of integrin-indepen-dent rotavirus CRW-8 to MA104 cells in suspension also was unaffected by the anti-␣2 MAb. However, the anti-␣2 MAb inhibited binding to MA104 cell monolayers of all integrin-using rotaviruses and reassortants containing VP4 proteins derived from integrin-using viruses (Fig. 1). Cell binding of integrin-independent rotaviruses was not affected by the an-ti-␣2 MAb. Cellular treatment with a control MAb (anti-class I at 10 to 40g/ml) did not alter binding of RRV, SA11, Wa, or CRW-8 to cells. An anti-2 MAb (P4H9-G11) at 2.5 to 40
g/ml and an anti-␣V3 MAb (LM609) at 1 to 10 g/ml showed mean percent infectivity blockades of⫺1%⫾2% and
⫺2%⫾2%, respectively, with all the rotaviruses listed in Fig. 1, so it failed to alter virus binding to cells. Levels of SA11 binding to␣X2-K562 cells were indistinguishable from those of SA11 bound to K562, ␣L2-K562, and ␣M2-K562 cells, and an anti-2 MAb (MEM-148) and 2-activating MAbs (KIM 127, KIM 185) had no effect on SA11 binding to these K562 cell lines at 1 to 20g/ml (data not shown). Thus, we confirmed that SA11 is unable to bind␣X2 directly. Collec-tively, these findings indicate that VP4 from an integrin-using rotavirus is necessary for virus binding to␣21 on MA104 cells and suggest that␣X2 and␣V3 participate in virus infection at a postattachment stage.
Recombinant␣21 conferred cell binding by RRV and Wa
but not CRW-8.Previously, SA11 was shown to bind␣21 on
[image:4.603.309.530.67.224.2]␣2-K562 cells (23). The ability of other rotaviruses to bind
FIG. 1. Binding to MA104 cell␣21 by laboratory-adapted rotavi-ruses and their reassortants. Data are for MAb AK7 relative to control MAb MOPC21 at the highest concentration used (20g/ml). Viruses were tested with serial twofold MAb dilutions (1.2 to 20g/ml) in three to five experiments. MOPC21 (1.2 to 40g/ml) treatment gave virus titers that were indistinguishable from titers obtained in the absence of MAb. The percent binding blockade of integrin-dependent viruses was AK7 dose dependent. Integrin-independent viruses showed no blockade at any AK7 concentration tested.
TABLE 2. Inhibition of rotavirus infection of MA104 cells by integrin ligands collagen and fibrinogena
Virus
% Infectivity blockade by given ligand (mean⫾SD)
Type I collagen Fibrinogen
Bo/NCDV 39⫾2 29⫾4
Si/SA11 47⫾14 38⫾5
Si/RRV 45⫾4 45⫾1
Bo/UK ⫺3⫾13 ⫺1⫾8
Po/CRW-8 5⫾7 ⫺7⫾8
Hu/Wa 51⫾3 47⫾5
Hu/RV-4 32⫾8 ND
Hu/K8 35⫾4 34⫾4
Po/MDR-13 ⫺9⫾5 1⫾7
aData are for collagen and fibrinogen at 160g/ml and are expressed relative
to the percent infectivity in the absence of protein. Viruses were tested by using five serial twofold dilutions of each ligand, over the range 10 to 160g/ml, in two to three experiments. The percent infectivity blockade of all integrin-dependent viruses was ligand dose dependent. Integrin-independent viruses UK, CRW-8, and MDR-13 showed no blockade at any ligand concentration tested. Cellular treatment with BSA (SA11, RRV, CRW-8, Wa, RV-4, K8, MDR-13) or ovalbu-min (NCDV, UK) at 10 to 160g/ml did not inhibit infection. Virus nomencla-ture is the same as that used in Table 1.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.603.43.283.89.214.2]␣21 on these cells was investigated. As shown in Fig. 2, titers of RRV and Wa bound to␣2-K562 cells were increased 2.2-fold and 3.2-2.2-fold, respectively, over titers of RRV and Wa bound to PBJ-K562 and␣3-K562 cells. Cellular treatment with anti-␣2 MAb eliminated this binding to␣21, as the titers of RRV or Wa bound to␣2-K562, PBJ-K562, and␣3-K562 cells treated with anti-␣2 MAb were similar to each other and were indistinguishable from titers of RRV or Wa bound to cells treated with control MAb. CRW-8 did not bind␣21 on␣ 2-K562 cells, as the titers of CRW-8 bound to ␣2-K562, PBJ-K562, and␣3-K562 cells treated with control or anti-␣2 MAb were indistinguishable.
Activation of1 increased SA11 binding to␣21 on K562
cells.Activating anti-1 MAbs induce integrins into
high-inte-grin-affinity states for ligand binding that, for example, increase type I collagen binding by ␣2-K562 cells (30). In order to determine if the ligand-binding state of 1 integrins alters rotavirus binding, we assayed SA11 binding to␣2-K562 cells which had been treated with activating anti-1 MAb 8A2 at the concentration (0.25g/ml) previously shown to increase K562 cell binding to fibronectin (18). Activation of 1 increased SA11 binding to␣2-K562 cells by 70% (Fig. 2). This shows that rotavirus binding to␣21 is enhanced by1 integrin activation and may depend on the activation state of1 integrins.
Effect of integrin ligand peptides DGEA and GPRP on
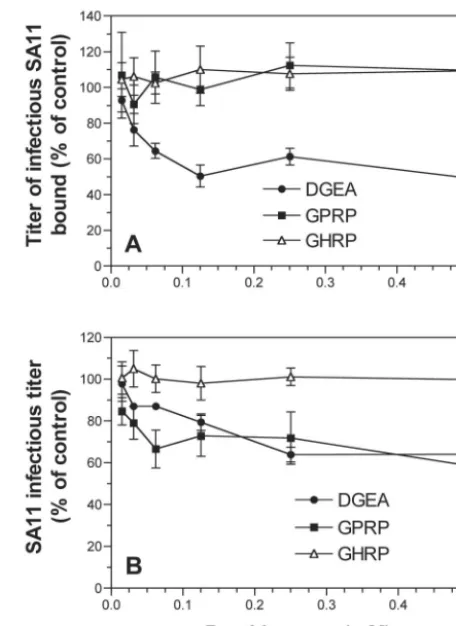
ro-tavirus cell binding and infectivity.Integrin-specific blocking
peptides were used to further define the role of integrins in rotavirus attachment and entry into cells. The collagen-derived peptide DGEA, which blocked type I collagen binding to␣21 (48), inhibited SA11 binding and infection in MA104 cells in a dose-dependent fashion by up to 55% at 0.5 mM (Fig. 3). Significant levels of blockade were detected at 0.03 to 0.06 mM DGEA. Virus binding was inhibited to a greater extent than was infection by DGEA. This could relate to a more efficient blockade of the initial function affected (binding) than of that of the downstream function (infectivity). Like DGEA, the GPRP peptide inhibited SA11 infection of cells by up to 41%. However, GPRP had no effect on virus binding to cells (Fig. 3). Binding and infection by integrin-independent virus CRW-8
were unaffected by DGEA or GPRP (Table 3). In contrast, DGEA and GPRP inhibited binding and infection of integrin-using viruses RRV and Wa at levels similar to those of SA11 (Table 3). These results suggest that the DGE sequence in the VP5* subunit of VP4 may direct rotavirus binding to ␣21. They also implicate the GPR sequence in rotavirus VP7 in the interaction of integrin-using viruses with␣X2 during entry.
DGEA, but not GPRP, inhibited binding by SA11, RRV, and
Wa to recombinant␣21.In order to determine if the peptide
[image:5.603.307.535.69.382.2]effects were integrin specific, levels of peptide inhibition of
FIG. 2. SA11, RRV, and Wa but not CRW-8 bound␣21 on K562 cells, virus-␣21 binding was eliminated by anti-␣2 MAb AK7, and SA11 binding was increased after␣21 activation with anti-1 MAb 8A2. Titers of infectious virus bound in the presence of MAb MOPC21 were indistinguishable from those of virus bound to cells in the absence of antibody. PBJ, K562-PBJ;␣2,␣2-K562;␣3,␣3-K562.
[image:5.603.48.277.70.214.2]FIG. 3. DGEA but not GPRP inhibited SA11 binding, but both DGEA and GPRP inhibited infection. Virus infectivity inhibition ex-periments are shown in panel A, and virus binding inhibition data are shown in panel B. conc., concentration.
TABLE 3. Effect of integrin ligand peptides DGEA and GPRP on RRV, Wa, and CRW-8 binding and infectivity in MA104 cells
Virus
% Blockade by given peptide (mean⫾SD)a
Virus-cell binding Virus infectivity
DGEA GPRP DGEA GPRP
RRV 34⫾1 ⫺2⫾6 34⫾1 42⫾2
CRW-8 1⫾8 1⫾8 ⫺1⫾1 7⫾2
Wa 38⫾1 ⫺2⫾4 27⫾1 31⫾4
aData are for 0.5 mM DGEA and GPRP. Data obtained with negative control
peptide GHRP (0.015 to 0.5 mM) and without peptide were indistinguishable. Viruses were tested by using six serial twofold dilutions of each peptide, over the range of 0.015 to 0.5 mM, in two to four experiments. The blockade of all integrin-dependent viruses was peptide dose dependent. Integrin-independent virus CRW-8 showed no blockade at any peptide concentration tested.
on November 8, 2019 by guest
http://jvi.asm.org/
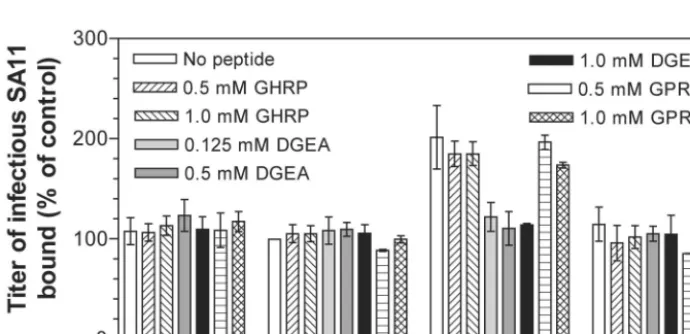
[image:5.603.302.542.600.676.2]virus binding to K562 cells expressing recombinant integrins were measured. DGEA completely inhibited SA11 binding to
␣21 on ␣2-K562 cells, whereas GPRP and GHRP had no effect on SA11 binding to␣21 (Fig. 4). DGEA similarly in-hibited RRV and Wa binding to␣2-K562 cells, and GPRP and GHRP had no effect on binding (Table 4). These peptides had no effect on SA11, RRV, and Wa binding to cells lacking␣21 (K562, PBJ-K562, and ␣3-K562) and did not affect CRW-8 binding to any K562 cell line (Fig. 4, Table 4). Since GPRP did not affect virus binding to␣21, the GPRP inhibition of virus infectivity was not through interaction with␣21. These find-ings indicate that DGEA specifically inhibited binding of inte-grin-using rotaviruses to recombinant ␣21 expressed on the K562 cell surface.
Mutagenesis of VP5* DG residues abolished VP5* binding to
the ␣2 integrin subunit I domain. Integrin-using rotaviruses
contain a conserved DGE sequence within the VP5* subunit of VP4 that could mediate binding to␣21. Within␣21, the I domain of the ␣2 subunit is the most likely region to be rec-ognized by rotavirus, as it is required for collagen and MAb AK7 binding (3, 29). In order to directly test VP5* binding to
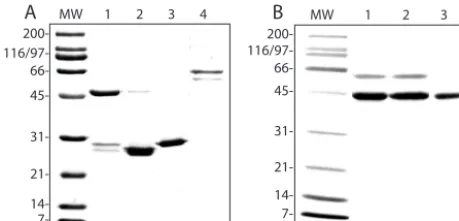
␣21, recombinant-expressed proteins were used in assays of the␣2 I domain binding to VP5*. The DGE motif in VP5* was mutagenized, DG3AA (308 to 309), and was tested as a GST fusion protein for its ability to bind␣2 I domain that had been released from GST by thrombin cleavage. The gel profiles of
purified GST-␣2I (⬃53 kDa), ␣2I (⬃27 kDa), GST (⬃28 kDa), GST-VP5* (⬃45 kDa), GST-VP5*(D308A/G309A) (⬃45 kDa), and GST-VP6 (⬃66 kDa) proteins are shown in Fig. 5. The identities of GST and GST fusion proteins were confirmed by Western blot by using anti-GST MAb CH-1 (data not shown). Expressed integrin and rotaviral proteins were specifically recognized by appropriate function-blocking MAbs in EIA and therefore retained functional epitopes (Fig. 6A and B). Mutagenesis of VP5* D308 and G309 did not affect rec-ognition of expressed VP5* by virus-neutralizing MAb 2G4 (Fig. 6B).
[image:6.603.42.543.92.167.2]To measure␣2 I domain binding, viral proteins coated on EIA plates were reacted with ␣2I or control proteins and binding was detected with I domain-specific or control MAbs (Fig. 6C). GST-VP5* alone bound the ␣2 I domain protein. Control proteins GST and GST-VP6 did not bind␣2I, and no binding of any control protein or control MAb to any viral protein was detected. Mutagenesis of the DG residues within VP5* abolished VP5*-directed binding of the ␣2 I domain (Fig. 6C). This demonstrates that RRV VP5* binding to the␣2 I domain of␣21 is directed by D308 and/or G309 in VP5*. These findings show that the I domain of the ␣2 subunit is sufficient for VP5* binding to ␣21, and the rotavirus VP5* DG sequence is critical for specific rotavirus binding to␣21 integrin.
FIG. 4. DGEA inhibited SA11 binding to␣2-K562 but not to K562, PBJ-K562, and␣3-K562 cells, whereas GPRP had no effect on SA11 binding to these cells. The titers of virus bound to cells are expressed as a percentage of the titers of virus bound to PBJ-K562 cells in the absence of peptide.
TABLE 4. Effect of integrin ligand peptides DGEA and GPRP and control peptide GHRP on RRV, Wa, and CRW-8 binding to␣2-K562, ␣3-K562, and PBJ-K562 cells
Virus
% Virus bound to listed K562 cell line after treatment with given peptide (mean⫾SD)a
GHRP DGEA GPRP
␣2 ␣3 PBJ ␣2 ␣3 PBJ ␣2 ␣3
RRV 182ⴞ24 83⫾6 99⫾6 95⫾10 87⫾3 95⫾2 167ⴞ25 86⫾8
Wa 263ⴞ26 97⫾10 95⫾4 110⫾11 104⫾2 98⫾4 299ⴞ17 97⫾3
CRW-8 112⫾10 103⫾10 102⫾10 104⫾8 105⫾14 104⫾11 100⫾13 101⫾6
aPercent virus bound relative to that bound to PBJ-K562 cells in the absence of peptide. Data are for 0.5 mM peptide. Positive levels of virus binding are in boldface.
GHRP treatment of PBJ-K562 cells gave results similar to those for treatment of␣3-K562 cells.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.603.120.465.527.694.2]DISCUSSION
To date among viruses, rotaviruses are unique in possessing integrin ligand sequences on more than one outer capsid pro-tein (11). Here we present evidence that many disease-causing rotaviruses of humans, monkeys, and cattle can use these se-quences on VP4 and VP7 to employ cellular integrins in a specific, novel process to establish infection in vitro. These viruses bind the␣2 I domain of␣21 integrin by using DGE in the VP5* subunit of VP4 and use VP7 to interact with␣X2 (via GPR) and␣V3 during virus-cell entry.
Specific binding to␣21 and the involvement of the DGE in VP5* in this binding were demonstrated with both permissive and integrin-transfected cells and were shown to lead to infection. A new understanding of the binding of rotaviruses to ␣21 was achieved by our direct demonstration of␣2 I domain binding to RRV VP5* for the first time and the requirement for VP5* D308 and/or G309 for binding. This is consistent with the D308 con-servation we found here in integrin-using rotaviruses, the abro-gation of nar mutant cellular binding by VP5* containing D308A reported previously (55), and the requirement for Asp in the DGEA peptide for inhibition of collagen binding to ␣21 by DGEA (48). It also confirms and extends studies reported else-where, which demonstrated by deletion of the␣2 I domain that this region is necessary for RRV and SA11 binding to human
␣21 expressed on CHO cells (35). As we have shown here that the␣2 I domain is both necessary and sufficient for VP5* binding to occur, structural studies of rotavirus-␣21 interactions can now be focused on VP5* binding to the␣2 I domain. The RRV-based tetravalent rotavirus vaccine has been withdrawn following asso-ciation of its use in children with an increased incidence of intus-susception, so development of therapeutic agents is of particular importance. Our identification of VP5* residues that are critical for rotavirus-␣21 binding and the␣2 I domain as sufficient for binding are essential for development of virus-specific inhibitors of this process.
We showed that many laboratory-adapted rotaviruses, in-cluding RRV, bound␣21 on adherent MA104 cells. In con-trast, RRV did not bind to␣21 on MA104 cells in suspension. This confirms previous reports that RRV (55), SA11, and Wa (7) do not bind␣21 on suspended MA104 cells. Removal of cells from the extracellular matrix produces changed
expres-sion and activation of integrins (6), alterations in activation of signal transduction pathways, and anoikis (46, 50). Thus, a reduction in␣21 expression and activation levels is a likely explanation for the lack of wild-type rotavirus binding to inte-grin on suspended MA104 cells. Supporting this, we found that activation of ␣21 expressed on K562 cells facilitated SA11 binding to␣21. The detection of nar mutant binding to␣21 on suspended cells (48) may have resulted from the high level of dependence on␣21 binding by this virus. Assays of
[image:7.603.49.279.70.180.2]rota-FIG. 5. Gel profiles of Coomassie-stained, expressed proteins. (A) Lanes 1 to 4, GST-␣2I, thrombin-cleaved GST-␣2I, GST, and GST-VP6, respectively. (B) Lanes 1 to 3, GST-VP5*, GST-VP5* (D308A/G309A), and GST-VP5* after GroEL removal (see Materials and Methods), respectively. MW, molecular size standards.
FIG. 6. Expressed␣2 I domain and viral proteins contained func-tional epitopes (A and B), and␣2 I domain binding to GST-VP5* required D308/G309 of VP5* (C). (A and B) Expressed protein (1.25 to 20 g/ml) on the solid phase reacted with MAbs (10 g/ml) to functional (closed symbol) or unrelated (matching open symbol) epitopes in EIA. GST (1.25 to 20g/ml) reacted with test or control MAbs gave optical densities at 450 nm (OD450) ofⱕ0.06. (C) Viral
GST fusion protein or GST (40g/ml) on the solid phase, reacted with ␣2I (2.5 to 80g/ml), was detected with anti-␣2I MAb AK7, negative control MAb HAS3 (non-I domain anti-␣2), or MOPC21 (10g/ml) in EIA. GST fusion protein or GST reacted with GST or BSA (2.5 to 80 g/ml) gave an OD450ofⱕ0.05 by EIA. conc., concentration.
on November 8, 2019 by guest
http://jvi.asm.org/
virus binding to adherent cells should be carried out on cell monolayers to avoid false-negative results.
The␣21 integrin is the cellular receptor for echoviruses 1 and 8. Like rotavirus VP5*, echovirus 1 binds the␣2 I domain. Activation of1 increased binding to␣21 by rotavirus (this study) and collagen (30) but not by echovirus 1 (2). The␣2 cytoplasmic domain is essential for␣21-specific signal trans-duction but not for echovirus 1 infection (2). The increased binding of SA11, but not that of echovirus 1, to activated␣21 suggests that integrin-using rotavirus binding to ␣21 might induce outside-in signaling. Studies of downstream effects of rotavirus-␣21 binding will advance understanding of both ro-tavirus pathogenesis and the DGEA role in collagen-␣21 binding. MAb activation of 1 integrins has been shown to enhance␣51-mediated adenovirus infection (13).
We identified VP7 as the rotavirus protein responsible for virus interaction with␣X2 and␣V3 by analysis of the usage of␣X2 and␣V3 for infection by reassortant rotaviruses and by GPRP peptide blockade of virus infectivity. Previously, the viral protein responsible for␣V3 recognition was not known, and VP7 had not been proposed to fill this role. This mapping of␣X2 and
␣V3 interactions to VP7 will greatly facilitate the identification of the viral regions interacting with␣V3 and will further studies of the roles of␣X2 and␣V3 in rotavirus infection. Rotaviruses did not bind ␣X2 and ␣V3. However, these integrins were shown to be involved in early virus-cell interactions, as proposed previously (11, 20). The GPRP peptide inhibited integrin-using virus infectivity but not binding, and it did not affect virus-␣21 binding, so the GPR sequence in VP7 is likely to be important for virus interaction with␣X2. As integrin usage, including␣X2 usage, by laboratory-adapted rotaviruses is related to the viral VP4 serotype rather than to the VP7 serotype, it appears that the viral VP7 serotype has little influence on␣X2 recognition. This is consistent with the conservation of GPR among all mammalian rotaviruses tested. For 8 to 11 amino acids on either side of the GPR, the sequence is also highly conserved among these rotavi-ruses. There were no conserved VP7 amino acid sequence differ-ences between integrin-using and integrin-independent rotavi-ruses of the same G serotype. However, if conformational change in VP4 or VP7 is needed for virus recognition of ␣X2 (and
␣V3), development of new virus-cell binding assays using con-formationally altered virions may allow detection of virus binding to␣X2 and␣V3 and analysis of the binding specificity.
Infection blockade by MAbs to␣21,␣X2, and␣V3 has been shown to be additive. Anti-␣2 MAb AK7 and anti-2 MAb MHM23 together inhibited SA11 infectivity by 65%, whereas each alone inhibited infectivity by 30 to 42% (11). Similarly, anti-␣2 MAb P1E6 and an anti-␣V3 antibody in-hibited RRV, Wa, and nar infectivity by 55 to 65% when combined but by 20 to 30% separately (20). The combination of the anti-␣2, anti-2, and anti-␣V3 MAbs used in the present study inhibited RV-5 and VA70 infection by 85% of the sum of the blockade by each MAb separately (P. Halasz and B. S. Coulson, unpublished data). Overall, combinations of MAbs to␣21,␣X2, and␣V3 have been shown to reduce integrin-using rotavirus infection by ⬃70%. Additionally, the VP4-derived peptide RDGEE, containing the␣21 ligand se-quence DGE, inhibited SA11 infection of MA104 cells by 90% (11). In this study, we found that SA and members of the integrin family of cell adhesion molecules independently act as
cellular receptors or entry molecules for several rotaviruses (SA11, RRV, NCDV). Analogously, SA and the junctional adhesion molecule (JAM) serve independently as receptors for reovirus, another member of the familyReoviridae(1, 53). The levels of blockade of rotavirus infectivity (⬃70%) mediated by anti-integrin MAbs are very similar to those of the blockade of reovirus binding (⬃70%) mediated by anti-JAM MAbs (1), so the integrin pathway is likely to be as significant for rotavirus cell attachment and entry as the JAM pathway is for these reovirus functions.
Anti-2 reagents and␣V3 MAbs did not affect virus bind-ing to␣21, although these and the anti-␣2 reagents inhibited infectivity. This suggests that VP4 binding to ␣21 precedes the interactions of VP7 with ␣X2 and␣V3. However, the VP4 and VP7 interactions with integrins were not interdepen-dent. Rotavirus binding to␣21 was independent of interac-tions with␣X2 and␣V3 in reassortants, and the effects of anti-integrin reagents on the infectivity of laboratory-adapted rotavirus were additive (11, 20). It can be concluded that virus interaction with␣X2 and␣V3 following binding to␣21 is not essential for infection to occur, and usage of receptors other than␣21 does not preclude virus interaction with␣X2 and␣V3.
Although rotavirus usage of␣21 could be segregated from
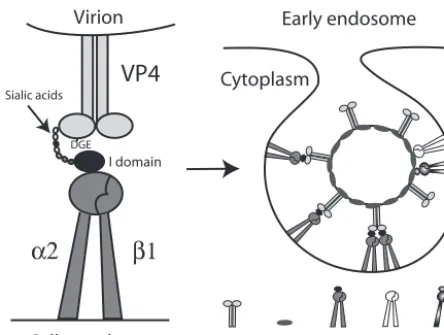
␣X2 and␣V3 usage in reassortants, all laboratory-adapted rotaviruses that bound␣21 also used␣X2 and␣V3. There-fore, multiple integrin usage by rotaviruses, involving both VP4 and VP7, might facilitate virus-cell entry more than partial integrin usage involving only one viral outer capsid protein. This could be by increasing the range of possible alternative pathways for cell attachment and entry and could result in increases in virus infection efficiency or the range of cell types and hosts infected. A model of the role of rotavirus-integrin recognition in early virus-cell interactions is shown in Fig. 7.
We showed for the first time that integrin usage by labora-tory-adapted rotaviruses is related to the viral P serotype. At the viral MOI we used (0.02), not all rotaviruses used integrins for infection. However, it has been reported that all 11 rota-viruses tested could use␣21 for infection at the high MOI of 10 (7). These viruses included OSU and WC3, which are of P types that are identical to those of viruses from the same species that we showed are integrin independent at low MOI. In our hands,⬃80% of MA104 cells were infected by rotavi-ruses at an MOI of 10, so it was necessary to titrate harvested, virus-infected cultures in MA104 cell monolayers to accurately determine MAb blockade of virus infectivity at this high MOI. We found that the infectivity of RRV was inhibited to a similar degree by MAb AK7 (10g/ml) at MOI of 0.02, 3.5, and 10, whereas CRW-8 infectivity was unaltered by MAb AK7 at any of these MOI (P. Halasz and B. S. Coulson, unpublished). This indicates that RRV and CRW-8 retain their respective inte-grin-using and integrin-independent phenotypes over a wide MOI range. The original report that rotavirus strains OSU, WC3, and PA169 (P serotype 11, G serotype 6) use␣21 at an MOI of 10 (7) needs to be reevaluated in the light of our findings. The integrin independence of P serotype 9 porcine rotaviruses found in our study is consistent with the ganglioside binding of P serotype 9 porcine rotavirus OSU (45). Also, the integrin utilization by NCDV and Wa and the integrin inde-pendence of CRW-8 are in agreement with the deinde-pendence of
on November 8, 2019 by guest
http://jvi.asm.org/
NCDV and Wa, but not CRW-8, on protein synthesis for regeneration of receptors (26). The integrin independence of P2A and P2B rotaviruses and the integrin usage by both P1A and P1B rotaviruses is not surprising given the close serological and VP4 sequence relationship between viruses within each P serotype. Although both rotavirus usage of integrins and ter-minal SA binding ability (8) relate to P serotype, integrin usage was independent of SA binding ability. This is consistent with the distinct locations on the virion of the SA-binding region (VP8*), the␣21-binding region (VP5*), and the regions in-volved in␣X2 and␣V3 interactions (VP7).
Although almost all laboratory-adapted rotavirus strains possess DGE sequence in VP4, we showed here that not all these strains use integrins during cell attachment and entry. Changes in VP4 or VP7 conformation resulting from the dif-fering combinations of these proteins between virus strains could affect the ability of rotaviruses to recognize both inte-grins and other receptors. It has been documented by using rotavirus reassortants that VP4-VP7 interactions occur that affect phenotypes, including receptor binding specificity (38), presentation of neutralization epitopes on VP4 or VP7, plaque size, protease-enhanced plaque formation, and hemagglutina-tion (5, 27). More recently it was shown that in some reassor-tants VP4 conformation is subtly altered, leading to expression of unexpected VP4-related phenotypes. It was considered that interactions between heterologous VP4 and VP7 in these re-assortants induced the conformational changes observed in VP4 (43). Thus, conformational changes in VP4 and/or VP7 induced by the interaction of a particular VP4-VP7 combina-tion could lead to loss of accessibility of the DGE sequence in VP4 for␣21 binding.
Many laboratory-adapted, human rotaviruses of P serotypes 1A, 1B, and 3 that cause disease in older infants and young children used integrins, whereas the disease-associated P sero-type 2A virus S12/85 was integrin independent. Laboratory-adapted, human rotaviruses that have been associated with asymptomatic disease in neonates were either integrin inde-pendent (P2A viruses) or integrin deinde-pendent (P8 virus 116E). Overall, we found that many laboratory-adapted, disease-asso-ciated rotaviruses tested used integrins, whereas many of the viruses asymptomatically infecting neonates were P2A and in-tegrin independent. In recent epidemiologic studies, increasing numbers of rotaviruses bearing the P[6] allele (associated with P2A serotype specificity), in conjunction with various G types, have been found in older infants and children with gastroen-teritis worldwide (44, 49). Analysis of a wider range of human rotaviruses for their ability to use integrins, including these new P[6] strains, will be needed to determine if integrin usage by rotaviruses has any relationship to disease severity or tro-pism in humans.
ACKNOWLEDGMENTS
We are grateful to I. H. Holmes for his continuing support, even in his retirement, and we thank him, H. Nagesha, and H. Greenberg for reassortants; H. Greenberg and O. Wijburg for the VP6 plasmid; F. Watt, A. Brooks, M. Estes, and D. Leavesley for MAbs; M. Sandrin for advice on transfection; D. Jackson and W. Zeng for peptides; M. Thomson for some of the studies using RV-4, K8, MDR13, and 116E; and G. Holloway for assistance in sequencing.
This work was supported by Project Grants 980635 and 208900 from the National Health and Medical Research Council (NHMRC) of Australia. B.S.C. is a Senior Research Fellow of the NHMRC.
REFERENCES
1. Barton, E. S., J. C. Forrest, J. L. Connolly, J. D. Chappell, Y. Liu, F. J. Schnell, A. Nusrat, C. A. Parkos, and T. S. Dermody.2001. Junction adhe-sion molecule is a receptor for reovirus. Cell104:441–451.
2. Bergelson, J. M., B. M. Chan, R. W. Finberg, and M. E. Hemler.1993. The integrin VLA-2 binds echovirus 1 and extracellular matrix ligands by differ-ent mechanisms. J. Clin. Investig.92:232–239.
3. Bergelson, J. M., N. F. St. John, S. Kawaguchi, R. Pasqualini, F. Berdichev-sky, M. E. Hemler, and R. W. Finberg.1994. The I domain is essential for echovirus 1 interaction with VLA-2. Cell Adhes. Commun.2:455–464. 4. Chemello, M. E., O. C. Aristimuno, F. Michelangeli, and M. C. Ruiz.2002.
[image:9.603.52.274.67.235.2]Requirement for vacuolar H⫹-ATPase activity and Ca2⫹gradient during entry of rotavirus into MA104 cells. J. Virol.76:13083–13087.
FIG. 7. Model for involvement of␣21,␣X2, and␣V3 in early cellular interactions of integrin-using rotaviruses. Initial cell binding could be via low-affinity binding of VP8* to terminal or subterminal SA (and/or of VP4 to other sugars, such as galactose) on glycolipids or glycoproteins (14, 16, 22, 26), including on integrins such as␣21. Terminal SA binding is not necessary for␣21 recognition, but SA binding could promote conformational change in VP4, exposing the VP5* DGE region, or could bring VP4 into closer proximity to the␣2 I domain. The VP5* binding to the␣2 I domain of the activated, extended form of␣21 via the type I collagen ligand sequence DGE in VP5* could result in integrin clustering and signal transduction, possibly enhanced by the binding of more than one␣21 molecule per multimeric VP4 spike. This binding could facili-tate VP7 interaction with␣X2 and␣V3 (perhaps by further confor-mational change in VP4), produce further integrin clustering, and lead to endocytosis. There is evidence that integrin-using and integrin-indepen-dent rotaviruses can be endocytosed, and activated1 integrins can enter cells by clathrin-mediated endocytosis (4, 41). Adenovirus recognition of ␣V3 leads to both clathrin-mediated endocytosis and macropinocytosis of virus (37). The ability of heat shock cognate protein 70 to inhibit the infectivity of integrin-using rotaviruses (19) is consistent with usage of the clathrin pathway, as this heat shock protein facilitates vesicle uncoating and clathrin recycling to the cell membrane (25). Alternatively, as caveo-lin-1 associates with␣21 and␣V3 (54) and echovirus 1 binding to␣21 results in virus internalization through caveolae (36), rotavirus recognition of␣21 and␣V3 could lead to rotavirus entry through caveolae. It is also possible that rotavirus could be endocytosed via a clathrin- and caveolin-independent route. The ability of methyl--cyclodextrin to in-hibit rotavirus infectivity suggests involvement of cell membrane choles-terol during virus-cell entry (21). Although cholescholes-terol was proposed to be involved as a lipid raft component (21), cholesterol depletion also inhibits endocytosis. As has been proposed previously, fusogenic VP5* domains could then selectively permeabilize the endosomal membrane, lowering the [Ca2⫹] surrounding the virions and leading to virus uncoating (4, 15).
This reduction in [Ca2⫹] also could release bound VP4 and VP7 from
integrins, as Ca2⫹and other divalent cations are necessary for integrin
binding to ligands. A direct entry mechanism for rotavirus (28) appears to be less likely than endocytosis (4, 17) but also would be compatible with integrin usage.
on November 8, 2019 by guest
http://jvi.asm.org/
5. Chen, D., J. W. Burns, M. K. Estes, and R. F. Ramig.1989. Phenotypes of rotavirus reassortants depend upon the recipient genetic background. Proc. Natl. Acad. Sci. USA86:3743–3747.
6. Chen, D., V. Magnuson, S. Hill, C. Arnaud, B. Steffensen, and R. J. Klebe.
1992. Regulation of integrin gene expression by substrate adherence. J. Biol. Chem.267:23502–23506.
7. Ciarlet, M., S. E. Crawford, E. Cheng, S. E. Blutt, D. A. Rice, J. M. Bergelson, and M. K. Estes.2002. VLA-2 (␣21) integrin promotes rotavirus entry into cells but is not necessary for rotavirus attachment. J. Virol.76:1109–1123. 8. Ciarlet, M., J. E. Ludert, M. Iturriza-Gomara, F. Liprandi, J. J. Gray, U.
Desselberger, and M. K. Estes.2002. Initial interaction of rotavirus strains withN-acetylneuraminic (sialic) acid residues on the cell surface correlates with VP4 genotype, not species of origin. J. Virol.76:4087–4095. 9. Coulson, B. S.1997. Effects of workshop monoclonal antibodies on rotavirus
infection of cells, p. 391–393.InT. Kishimoto, H. Kikutani, A. E. G. Kr. von dem Borne, S. M. Goyert, D. Y. Mason, M. Miyasaka, L. Moretta, K. Okumura, S. Shaw, T. A. Springer, K. Sugamura, and H. Zola (ed.), Leu-cocyte typing VI. Garland Publishing, Inc., New York, N.Y.
10. Coulson, B. S., and C. Kirkwood.1991. Relation of VP7 amino acid se-quence to monoclonal antibody neutralization of rotavirus and rotavirus monotype. J. Virol.65:5968–5974.
11. Coulson, B. S., S. L. Londrigan, and D. J. Lee.1997. Rotavirus contains integrin ligand sequences and a disintegrin-like domain that are implicated in virus entry into cells. Proc. Natl. Acad. Sci. USA94:5389–5394. 12. Coulson, B. S., L. E. Unicomb, G. A. Pitson, and R. F. Bishop.1987. Simple
and specific enzyme immunoassay using monoclonal antibodies for serotyp-ing human rotaviruses. J. Clin. Microbiol.25:509–515.
13. Davison, E., R. M. Diaz, I. R. Hart, G. Santis, and J. F. Marshall.1997. Integrin␣51-mediated adenovirus infection is enhanced by the integrin-activating antibody TS2/16. J. Virol.71:6204–6207.
14. Delorme, C., H. Brussow, J. Sidoti, N. Roche, K. A. Karlsson, J. R. Neeser, and S. Teneberg.2001. Glycosphingolipid binding specificities of rotavirus: identification of a sialic acid-binding epitope. J. Virol.75:2276–2287. 15. Denisova, E., W. Dowling, R. LaMonica, R. Shaw, S. Scarlata, F. Ruggeri,
and E. R. Mackow. 1999. Rotavirus capsid protein VP5* permeabilizes membranes. J. Virol.73:3147–3153.
16. Dormitzer, P. R., Z. Y. Sun, O. Blixt, J. C. Paulson, G. Wagner, and S. C. Harrison.2002. Specificity and affinity of sialic acid binding by the rhesus rotavirus VP8* core. J. Virol.76:10512–10517.
17. Dowling, W., E. Denisova, R. LaMonica, and E. R. Mackow.2000. Selective membrane permeabilization by the rotavirus VP5* protein is abrogated by mutations in an internal hydrophobic domain. J. Virol.74:6368–6376. 18. Faull, R. J., J. Wang, D. I. Leavesley, W. Puzon, G. R. Russ, D. Vestweber,
and Y. Takada.1996. A novel activating anti-1 integrin monoclonal anti-body binds to the cysteine-rich repeats in the1 chain. J. Biol. Chem.
271:25099–25106.
19. Guerrero, C. A., D. Bouyssounade, S. Zarate, P. Isa, T. Lopez, R. Espinosa, P. Romero, E. Mendez, S. Lopez, and C. F. Arias.2002. Heat shock cognate protein 70 is involved in rotavirus cell entry. J. Virol.76:4096–4102. 20. Guerrero, C. A., E. Mendez, S. Zarate, P. Isa, S. Lopez, and C. F. Arias.2000.
Integrin alpha(v)beta(3) mediates rotavirus cell entry. Proc. Natl. Acad. Sci. USA97:14644–14649.
21. Guerrero, C. A., S. Zarate, G. Corkidi, S. Lopez, and C. F. Arias.2000. Biochemical characterization of rotavirus receptors in MA104 cells. J. Virol.
74:9362–9371.
22. Guo, C. T., O. Nakagomi, M. Mochizuki, H. Ishida, M. Kiso, Y. Ohta, T. Suzuki, D. Miyamoto, K. I. Hidari, and Y. Suzuki. 1999. Ganglioside GM(1a) on the cell surface is involved in the infection by human rotavirus KUN and MO strains. J. Biochem. (Tokyo)126:683–688.
23. Hewish, M. J., Y. Takada, and B. S. Coulson.2000. Integrins␣21 and␣41 can mediate SA11 rotavirus attachment and entry into cells. J. Virol.74:
228–236.
24. Hoshino, Y., R. W. Jones, and A. Z. Kapikian.1998. Serotypic characteriza-tion of outer capsid spike protein VP4 of vervet monkey rotavirus SA11 strain. Arch. Virol.143:1233–1244.
25. Jiang, R., B. Gao, K. Prasad, L. E. Greene, and E. Eisenberg.2000. Hsc70 chaperones clathrin and primes it to interact with vesicle membranes. J. Biol. Chem.275:8439–8447.
26. Jolly, C. L., B. M. Beisner, E. Ozser, and I. H. Holmes.2001. Non-lytic extraction and characterisation of receptors for multiple strains of rotavirus. Arch. Virol.146:1307–1323.
27. Kalica, A. R., J. Flores, and H. B. Greenberg.1983. Identification of the rotaviral gene that codes for hemagglutination and protease-enhanced plaque formation. Virology125:194–205.
28. Kaljot, K. T., R. D. Shaw, D. H. Rubin, and H. B. Greenberg.1988. Infectious rotavirus enters cells by direct cell membrane penetration, not by endocyto-sis. J. Virol.62:1136–1144.
29. Kamata, T., and Y. Takada.1994. Direct binding of collagen to the I domain of integrin alpha 2 beta 1 (VLA-2, CD49b/CD29) in a divalent cation-independent manner. J. Biol. Chem.269:26006–26010.
30. Kawaguchi, S., and M. E. Hemler.1993. Role of the alpha subunit cytoplas-mic domain in regulation of adhesive activity mediated by the integrin VLA-2. J. Biol. Chem.268:16279–16285.
31. Kirkwood, C. D., R. F. Bishop, and B. S. Coulson.1998. Attachment and growth of human rotaviruses RV-3 and S12/85 in Caco-2 cells depend on VP4. J. Virol.72:9348–9352.
32. Kool, D. A., S. M. Matsui, H. B. Greenberg, and I. H. Holmes.1992. Isolation and characterization of a novel reassortant between avian Ty-1 and simian RRV rotaviruses. J. Virol.66:6836–6839.
33. Lazdins, I., B. S. Coulson, C. Kirkwood, M. Dyall-Smith, P. J. Masendycz, S. Sonza, and I. H. Holmes.1995. Rotavirus antigenicity is affected by the genetic context and glycosylation of VP7. Virology209:80–89.
34. Londrigan, S. L., M. J. Hewish, M. J. Thomson, G. M. Sanders, H. Mustafa, and B. S. Coulson.2000. Growth of rotaviruses in continuous human and monkey cell lines that vary in their expression of integrins. J. Gen. Virol.
81:2203–2213.
35. Londrigan, S. L., K. L. Graham, Y. Takada, P. Halasz, and B. S. Coulson.
2003. Monkey rotavirus binding to␣21 integrin requires the␣2 I domain and is facilitated by the homologous1 subunit. J. Virol.77:9486–9501. 36. Marjomaki, V., V. Pietiainen, H. Matilainen, P. Upla, J. Ivaska, L. Nissinen,
H. Reunanen, P. Huttunen, T. Hyypia, and J. Heino.2002. Internalization of echovirus 1 in caveolae. J. Virol.76:1856–1865.
37. Meier, O., K. Boucke, S. V. Hammer, S. Keller, R. P. Stidwill, S. Hemmi, and U. F. Greber.2002. Adenovirus triggers macropinocytosis and endosomal leak-age together with its clathrin-mediated uptake. J. Cell Biol.158:1119–1131. 38. Mendez, E., C. F. Arias, and S. Lopez.1996. Interactions between the two
surface proteins of rotavirus may alter the receptor-binding specificity of the virus. J. Virol.70:1218–1222.
39. Nagesha, H. S., L. E. Brown, and I. H. Holmes.1989. Neutralizing mono-clonal antibodies against three serotypes of porcine rotavirus. J. Virol.63:
3545–3549.
40. Nagesha, H. S., and I. H. Holmes.1991. VP4 relationships between porcine and other rotavirus serotypes. Arch. Virol.116:107–118.
41. Ng, T., D. Shima, A. Squire, P. I. Bastiaens, S. Gschmeissner, M. J. Humphries, and P. J. Parker.1999. PKC␣regulates1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J.
18:3909–3923.
42. Ortlepp, S., P. E. Stephens, N. Hogg, C. G. Figdor, and M. K. Robinson.
1995. Antibodies that activate beta 2 integrins can generate different ligand binding states. Eur. J. Immunol.25:637–643.
43. Pesavento, J. B., A. M. Billingsley, E. J. Roberts, R. F. Ramig, and B. V. Prasad.2003. Structures of rotavirus reassortants demonstrate correlation of altered conformation of the VP4 spike and expression of unexpected VP4-associated phenotypes. J. Virol.77:3291–3296.
44. Ramachandran, M., J. R. Gentsch, U. D. Parashar, S. Jin, P. A. Woods, J. L. Holmes, C. D. Kirkwood, R. F. Bishop, H. B. Greenberg, S. Urasawa, G. Gerna, B. S. Coulson, K. Taniguchi, J. S. Bresee, and R. I. Glass.1998. Detection and characterization of novel rotavirus strains in the United States. J. Clin. Microbiol.36:3223–3229.
45. Rolsma, M. D., T. B. Kuhlenschmidt, H. B. Gelberg, and M. S. Kuhlen-schmidt.1998. Structure and function of a ganglioside receptor for porcine rotavirus. J. Virol.72:9079–9091.
46. Ruoslahti, E., and J. C. Reed.1994. Anchorage dependence, integrins, and apoptosis. Cell77:477–478.
47. Shaw, R. D., P. T. Vo, P. A. Offit, B. S. Coulson, and H. B. Greenberg.1986. Antigenic mapping of the surface proteins of rhesus rotavirus. Virology
155:434–451.
48. Staatz, W. D., K. F. Fok, M. M. Zutter, S. P. Adams, B. A. Rodriguez, and S. A. Santoro.1991. Identification of a tetrapeptide recognition sequence for the alpha 2 beta 1 integrin in collagen. J. Biol. Chem.266:7363–7367. 49. Steele, A. D., and B. Ivanoff.2003. Rotavirus strains circulating in Africa
during 1996–1999: emergence of G9 strains and P[6] strains. Vaccine21:
361–367.
50. Stupack, D. G., and D. A. Cheresh.2002. Get a ligand, get a life: integrins, signaling and cell survival. J. Cell Sci.115:3729–3738.
51. Tang, B., J. M. Gilbert, S. M. Matsui, and H. B. Greenberg.1997. Compar-ison of the rotavirus gene 6 from different species by sequence analysis and localization of subgroup-specific epitopes using site-directed mutagenesis. Virology237:89–96.
52. Thain, A., K. Gaston, O. Jenkins, and A. R. Clarke.1996. A method for the separation of GST fusion proteins from co-purifying GroEL. Trends Genet.
12:209–210.
53. Tyler, K. L., P. Clarke, R. L. DeBiasi, D. Kominsky, and G. J. Poggioli.2001. Reoviruses and the host cell. Trends Microbiol.9:560–564.
54. Wary, K. K., A. Mariotti, C. Zurzolo, and F. G. Giancotti.1998. A require-ment for caveolin-1 and associated kinase Fyn in integrin signaling and anchorage-dependent cell growth. Cell94:625–634.
55. Zarate, S., R. Espinosa, P. Romero, C. A. Guerrero, C. F. Arias, and S. Lopez.2000. Integrin␣21 mediates the cell attachment of the rotavirus neuraminidase-resistant variant nar3. Virology278:50–54.