Copyright © 1973 AmericanSociety for Microbiology Printed in U.S.A.
Characterization of
Nucleic Acid
of
Pichinde
Virus
MICHAEL F. CARTER, NILAMBAR BISWAL, AND WILLIAM E. RAWLS Department of Virologyand Epidemiology, BaylorCollege of Medicine, Houston, Texas 77025
Receivedforpublication22 September 1972
The nucleic acid ofPichinde virus was found to be single-stranded ribonucleic acid
(RNA)
asdetermined by
sensitivity toribonuclease, by alkaline
degrada-tion, by buoyant
density
in cesiumsulfate,
andby analysis
ofthe
base
compo-sition. TheRNA
ofthe
virioncould be
separated into five components which
had sedimentation coefficients
corresponding
to31S, 28S, 22S, 18S and
4to6S.
The
28S,
18S, and possibly the 4 to 6S RNAs appear to be derived from host cell components incorporated into the virion,whereas the
31S and 22S
componentsappear
torepresent
thegenome
ofthevirus.
The
arenaviruses aremorphologically unique
in
that they contain multiple electron-dense
granules instead
ofa
discernible core (6, 16,
17, 22). Thegranules
are similar in size and shape to cellribosomes,
and thepossibility
exists that host cell ribosomes may beincorporated
into
the
virion(16, 17, 19).
Pichinde virus,
amember
ofthe
arenavirusgroup,
isrelatively
stable,
and thevirus
replicates well
intissue
culture
(15).
The nucleic acids of Pichinde virus were, therefore,characterized
todeter-mine
whether
or not hostcell ribosomal
mate-rialwas incorporated
intothe
mature virions ofmembers
ofthe arenavirus
group.
MATERIALS AND METHODS Chemicals. Polyethylene glycol 6000 (PEG) was purchased from Union Carbide Corp., New York. Phenol, m-cresol and 8-hydroxyquinoline were ob-tained from Matheson, Coleman and Bell. Diethyl oxydiformate (DEP) and triisopropylnaphthalene sul-fonic acid sodium salt (TINS) were purchased from Eastman Organic Chemical Co., Rochester, N.Y. Electrophoretically purified bovine pancreatic deoxy-ribonuclease and deoxy-ribonuclease A were purchased from Worthington Biochemical Co., Freehold, N.J. Actino-mycin D in
200-lsg
quantities (lot 025007) was ob-tained from Calbiochem. Acrylamide, N,N'-methyl-enebisacrylamide, N,N,N',N' tetramethylene-diamine (TEMED), and ammonium persulfate were purchased from Canal Industrial Co., Rockville, Md. Agarose forelectrophoresis were obtained from Sigma Chemical Co.Uridine-5-3H(25Ci/mmole), uridine-2-I4C(50mCi/
mmole), and Aquasol liquid scintillation fluid were purchased from New England Nuclear Corp., Boston. L-Methionine-methyl-3H(3.3Ci/mmole) was purchased from Schwarz/Mann, Orangeburg, N.Y. Carrier-free orthophosphate 32P was obtained from Bionuclear, Inc., Friendswood, Texas.
61
Buffers and solutions. TNE buffer consisted of 0.01 M tris(hydroxymethyl)aminomethane (Tris)-hydrochloride, pH 7.4, 0.1 M NaCl, and 0.001 M ethylenediaminetetraacetic acid (EDTA). TNM buffer contained 0.01 M Tris, pH 7.4, 0.1 M NaCi,
and0.001 MMgCl2.PMQNwasutilizedasthe basic
deproteinizing agent and consisted ofthe following:
500 g offreshly distilled phenol, 70 ml ofdistilled m-cresol, 0.5 g of8-hydroxyquinoline and 55 ml of 0.15 M NaCl. E buffer used for electrophoresis,
describedpreviously (2), consistedof 0.4 MTris,pH
7.2, 0.02 M sodium acetate, and 0.001 M EDTA. The 3E buffer was three times the concentration of E buffer.Sucrose solutions utilized fordensity gradients weremadeinTNE buffer.
Media. Growth medium consisted ofEagle mini-mal medium supplemented with 10% fetal bovine serum (FBS), 0.75 g of sodium bicarbonate
(NaHCO,)/liter,
and antibiotics (100 unitsofpeni-cillin/ml and 100
Ag
ofstreptomycin/ml).Mainte-nance medium was composed of Eagle minimal
medium supplemented with 2% FBS, 1.50 g of
NaHCO,/liter,
and antibiotics.Virus. Pichinde virus strain AN3739 has been de-scribed (15, 28). The virus was passaged once in BHK-21 cells, and the extracellular fluid from in-fected cultures wasutilizedasthesourceof virus in all experiments. Thetiter ofthevirusstock, assayed
by the plaque counting method (15), was
approxi-mately2 x 108plaque-forming units(PFU)/ml.
Cell cultures.BHK-21cells(26)ingrowthmedium were seeded into 16-oz (ca. 0.47 liter) prescription bottles. Three daysafterseedingthe cells were
con-fluent monolayers with approximately 2 x 107 cells
perbottle. HeLacellswerepropagatedingrowth
me-diumaspreviously described(12).
Infection of cells andpreparationof radioactive
virus.Confluentmonolayerswereinfected withvirus
at amultiplicityof infection ofapproximately2PFU/
cell.Afteradsorption ofthevirusfor 90 minat37C,
the monolayers werewashedoncewith maintenance medium, and25 ml of fresh medium was added to
on November 10, 2019 by guest
http://jvi.asm.org/
each monolayer. Three different radioisotopes were
used to label the viral nucleicacid. 3H-uridine-labeled virions were grown in the presence of5 ,ACi of the radioisotope/ml and25
Ag
eachofdeoxycytidine and thymidine/ml. Methyl 3H-labeledvirions weregrown inmethionine-free maintenance medium containing methionine-methyl-3H (154Ci/ml).
32P-labeledvirions were grown in phosphate-free maintenance
medium supplemented with 25 ,uCi of 32P-ortho-phosphate/ml. Extracellular fluids were harvested 72hr after infection. Extracellular fluids from unin-fected cells servedascontrols.
In some experiments the radiolabeled virus was
grown in the medium containing actinomycin D. Maintenance medium supplemented with radioiso-tope and actinomycin D (0.05 ,ug/ml) was added to
the culturesafter virusadsorption. The extracellular
fluids containing thevirus wereharvested48hrafter
infection. Uninfected cultureswereusedascontrols.
In all experiments, however, label wasaddedatthe
time of infection and maintained throughout the
entire time ofinfection.
Concentration and purification of Pichinde virus. Extracellularfluids were harvested at 48 or 72 hr after
infection andclarifiedby centrifugationat 1,100 x g
for 20 min, and the virus was concentrated by
pre-cipitation with polyethylene glycol (13). PEG and NaClwereaddedto afinal concentration of 6.0%and 0.4 M, respectively, and the mixture was held over-nightat 4C. The precipitatewaspelleted by centrif-ugation at 11,000 x g for 45 min and resuspended in TNE(3mlofbuffer per100mloforiginal volume). After sonictreatment at 50kc for 30sec, the resus-pended material was layered on a discontinuous 20 and 50%(w/w) sucrose gradient and centrifuged for
90 min at30,000rev/min and4C ina Spinco SW50
rotor. A distinct band on the 50% sucrose was
ob-served, and it was collected by puncturing the side ofthe tube. The bandwas diluted to less than 20%
sucrose with TNE, and 10% FBS was addedto
sta-bilize thevirus.One milliliterofthe dilutedvirus was carefully layeredon a 20 to50%(w/w) linearsucrose density gradient and centrifuged at 35,000 rev/min
for 2hr at 4 C in the SW50rotor. After
centrifuga-tion, approximately 20 fractions (0.25 ml) were col-lected from the bottom by using a piercing unit (Buchler Instruments). The refractive index (n) of everyfifth fractionwasobtainedusingaBausch and Lomb refractometer, and the refractive index was
used to calculate the buoyant density. When the
infectious virus titer was to bedetermined, an 0.05-ml sample ofeach fraction was diluted 100-fold in maintenance medium and stored at -70 C until assayed.
Isolation of RNA from virus. The fractions of
the linear sucrosegradient whichcontainedthe virus
(density = 1.14 to 1.18 g/cm3) werepooled, and the nuclease inhibitors2-mercaptoethanol and DEP were added to 0.1% and 0.2% inthefinal volume, respec-tively. Sodium lauryl sarcosinate (SLS) was added
to 1% in the final volume, the mixture was diluted
fourfold inTNE,andthe nucleic acidwasextracted aspreviouslydescribed(4) usingthePMQNsolution fordeproteinization.
Isolation of RNA from cells. Confluent mono-layersofHeLacells in 16-ozbottles were labeled for 24 hr with 14C-uridine (2 ,ACi/ml). The cells were collected and suspended in 0.01 M acetate buffer, pH5.1,and the RNAwasextractedby the hot phenol procedure described by SchererandDarnell (24).
RNAwasextractedfromuninfected BHK-21 cells by using a method which was modified from that previouslydescribed by Biswal etal. (4). Cells from
four 16-ozbottlesweresuspended in 10ml ofTNM,
partiallylysed bytreatmentwith0.05 mlofSDS(10%) for5 minat roomtemp andtreated with deoxyribonu-clease(50
jg)
foranadditional 5min to remove most ofthe cell DNA. DEP (0.2 ml) and0.2 ml of 0.25 M EDTA were added, followed by the addition of 0.5 ml ofSLS (10%) and0.3mlofSDS. Aftershakingat 37 C for5 min,0.25g of TINS wasadded. An equal volume ofPMQN wasadded and gently shaken at0to4Cfor 10min.The aqueousphasewasseparated
by centrifugation (6,000 x g, 15 min) and adjusted
to 0.2 MNaCl, and the deproteinizationwithPMQN
was repeated until the white precipitate at the in-terface was no longer present. After the final ex-traction, the aqueous phase was collected, precipi-tated in twovolumes of 95%ethanolat -20C forat least 1 hr, and then centrifugedat 18,000 x gfor 20 min. The pelletwas resuspended in 2 ml ofTNM, deoxyribonuclease (50 Mg) wasagainadded, and the mixturewasincubatedat 37Cfor 15min. To remove the deoxyribonuclease, an equal volume of PMQN wasadded, and themixture wasgently shakenat 4C
for 10min. Thephaseswereseparatedby
centrifuga-tion, theribonucleic acid(RNA)inthe aqueousphase was precipitated, and the precipitate was dried in the cold andsuspendedin 1 mlof TNE.
Sensitivitytonucleases.ViralRNAsuspended in TNM buffer was tested for sensitivity to nucleases. Ribonuclease (20
Ag/ml)
ordeoxyribonuclease (50Ag/
ml) was added to the samples of the viral RNA prepa-rations, and, after 1 hr of incubation at 37 C, acid-precipitable counts were determined (4).Velocity sedimentation of viral and cell RNA. An0.2-ml sample of RNA was layered on top of 5.0
mlofa 5 to20%(w/w) linear sucrose density gradient.
A few drops of mineral oil was layered above the sample to insure uniform distribution of the sample onthe sucrose.
"4C-labeled
HeLa cell RNA was used as reference marker. After centrifugation, fractions (0.15 ml) were collected by bottom puncture of the tube, and the acid-precipitable radioactivity of each fraction was determined (4, 5).Buoyant density of viral RNA in cesium sulfate. Asolution of TNE(pH 4.5) containing 2% dimethyl-sulfoxidewas added tothe viralRNA preparation to a
totalvolumeof4.0 ml. Crystalline cesium sulfate (3.2
g)wasadded, the refractive index of the solution was adjusted to 1.3780, and 4.6 ml was covered with 0.6
ml ofmineral oilin polyallomertubes. After
centrifu-gation, therefractive indexofselected fractions and theacid-precipitable radioactivity of each fraction (4) weredetermined.
Determination of base composition. The base composition ofRNAspeciesfrom the virion or from
BHK-21 cellswasdetermined by a slightly modified
VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
NUCLEIC ACID OF PICHINDE VIRUS
method previously described by Biswal et al. (4). The 32P-labeled viral or cellular RNA species were mixed with 500
jug
ofyeast "carrier" RNA and di-gested in 0.05 ml of 0.5 M KOH for 16 hr at 37 C. The hydrolysates were spotted on Whatman 3M paper for electrophoresis, and the nucleotides were electrophoresed for 3 hr at 1,500 v in 0.05 M am-monium formate, pH 3.5. The ultraviolet light-ab-sorbing spots on the paper were cut out, and the radioactivity of each wasdetermined.Polyacrylamide gel electrophoresis. Composite gels (8 cm in length) of 2.4% polyacrylamide and 0.5% agarosewere prepared using methods previously de-scribed (7, 27). After polymerization for approxi-mately 4 hr, the gels were removed to E buffer solu-tioncontaining 0.2%SDS. Electrophoresis buffer was the E buffer containing 0.2% SDS. The gels were preelectrophoresed at 20 C for 30min at 5 mA per tube usinga Buchler DC power supply. For electro-phoresis, 0.05 ml of RNA sample was mixed with 0.005 ml ofbromophenol blue in 30% sucrose, and the mixture was carefully layered on the gel. Electropho-resis was usuallycarriedout at 20 C and 5 mA per gel for 2.5 hr. Afterelectrophoresis, the gels were sliced into 2-mm fractions by using a Gilson gel fractionator, model B100/GMA/GCB (Middleton, Wis.). A 10% solutionof BioSolv (Beckman Instruments, Palo Alto, Calif., wasused for elution of the isotopically labeled materials from minced gel fractions. Ten milliliters of scintillationfluid was added, and radioactivity of each gel fraction wasmeasured.
RESULTS
Purification
and
buoyant
density
ofPichinde
virus. When BHK-21 cells were in-fected at aninput
multiplicity of 1 to 2 PFU/cell,
maximum virus titers of107PFU/mlwereconsistently
observed at 48 and 72 hr afterin-fection.
Pichinde
virus was concentrated andpurified
from extracellular fluids as described above. To determine thebuoyant density,
purified Pichinde
viruswhich had been labeledwith 3H-uridine was
centrifuged
toequilibrium
in a sucrose
density
gradient (Fig. 1).
Fractions wereanalyzed
forbothinfectivity
andradioac-tivity.
Maximuminfectivity
andradioactivity
were recovered at a buoyant density of about 1.15 to 1.16
g/cm3.
Insome of the experiments maximum infectivity banded at a slightly greaterdensity
than maximum radioactivityalthough
the difference was never more than a single fraction whichrepresented a density dif-ference of 0.01g/cm3.
In all experiments no less than 15% of the original quantity of infec-tious virus wasrecovered in the fractions con-taining maximum virus. When culture fluids from uninfected cells were processed under identical conditions, anegligible amount of the radioactivity at the density of 1.16g/cm3
in the linear sucrose gradient was observed (Fig. 1).L-;5
l6bJ
I
w Z
10-a
I
1.20Zn
Z u
1.06
0-a:
I
CL.) Z2
L
5 a. 0 .
2 4 6 8 10 12 14 16 18
FRACTION NUMBER
FIG.1. Equilibrium density gradient centrifugation
of 3H-uridine-labeled Pichinde virus insucrose den-sity gradient. Partially purified virus (1.0 ml) was
layered onthe topofa4.0-ml 20to50%o(w/w) linear sucrose gradient. The virus was centrifuged in a Spinco SW50rotorat35,000 rev/minfor2 hrat4 C. Fractionswerecollectedfromthe bottomofthetube,
and theradioactivity(0)andinfectivity (0) ofeach fraction were determined. Radioactivity of purified
uninfected culturefluids (0)wasalso measured.
Sedimentation characteristics
of Pichinde
virus RNA.
The nucleic acid which wasiso-lated
from purified Pichinde virus wasana-lyzed by velocity
sedimentation in 5 to 20% sucrosedensity gradients (Fig.
2). The3H-uridine-labeled material was
separated
into four components, andthe
sedimentation co-efficient of each was calculatedusing
l4C-uridine-labeled
HeLa cell RNA as reference marker (14). The major componentcorrespond-ing to
28S
wasconsistently observed
as were the22S and 4 to6S
components. A minor18S
species was observed, but often this species
appeared
as ashoulder
on the22S
peak.
The sucrosesedimentation
pattern observed inFig.
2 represents RNA extracted from virusharvested
at 72 hr after infection.Identical
sedimentation patterns were observed
using
RNAfrom virus harvestedat 24, 48, and96hr afterinfection.
The fractions collected from the sucrose
density gradient
were tested for sensitivity toenzyme
digestion. Figure
2shows thatallfrac-tions were sensitive to ribonuclease but resis-tant to
deoxyribonuclease digestions.
These resultssuggested
that the Pichinde virusnu-cleicacid is
single-stranded
RNA.VOL. 11, 1973
5 o
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.495.257.449.64.285.2]AND
[image:4.495.54.243.328.560.2]Polyacrylamide
gel
electrophoresis
of
Pichinde virus RNA. When
3H-uridine-labeled Pichinde virus RNA was electropho-resed in 2.4% polyacrylamide gels for 2.5 hr, four distinct viral RNA components were ob-served (see Fig. 6). With HeLa cell ribosomal RNA as reference, the sizes of these RNA components
corresponded
to31S, 28S, 22S,
and 18S,
respectively
(2). The low-molecular-weight component (4 to6S) observed
in su-crosedensity gradients (Fig.
2)migrated
off thegel
under these conditions(unpublished
data).
Themajor peak corresponded to the size of28S, but
the31S, 22S,
and18S
segments werealso well resolved. These
data, plus those
obtained
by
sucrosevelocity
sedimentation,
indicate that the Pichinde virus RNA consists of at
least
five components.By
using themethod
ofSpirin (25),
the molecularweights
of the components were calculated to be 2.1 x106 (31S),
1.7 x106 (28S),
1.1 x 106(22S),
7 x 105(18S),
and 2.9 x 104(4S).
The28S,
18S,
and 4 to 6S components are similar in size to certainspecies of host cell RNA.
28S 18S
8
,05
Lu z
D
r3
13 17 20 25 29 33 37
FRACTION
NUMBER
[image:4.495.256.447.355.515.2]FIG. 2. Velocity sedimentation of RNA from
Pichinde virus labeled with 3H-uridine. Viral RNA
in TNE was layeredon the top ofa5to 20% (w/w)
linear sucrose gradient, and centrifugation was
carried out at43,000 rev/minfor3.25hrat 4C in a
Spinco SW50 rotor. Two 0.05-ml samples of each fraction were incubatedfor 1 hrat37 Cwith either ribonuclease (20 ,g/ml) or deoxyribonuclease (50
,gg/ml)
before determination of trichloroacetic acid-precipitable radioactivity. '4C-uridine-labeled HeLacell ribosomal RNA was centrifuged in a separate
Buoyant density of Pichinde virus RNA. The buoyant density of 3H-uridine-labeled Pichinde virus RNA in cesium sulfate was de-termined (Fig. 3). 14C-uridine-labeled HeLa cell ribosomal RNA was used as a density marker. When the buoyant density of the HeLa cell ribosomal RNA was 1.685 g/cm3, the den-sity of the viral RNA was found to be 1.677 g/cm 3, a value which corresponds to the density of single-stranded RNA. The density of the Pichinde virus RNA varied from 1.685 to 1.669 g/cm3 from experimentto experiment.
Identification of
host cell ribosomal RNA inPichinde
virus. Sincepart
ofthe RNA iso-lated from Pichinde virions consisted of28S
and 18S components, the possibility that these species were of host cell ribosomal RNA origin was examined by using actinomycin D to
in-hibit
the28S
and188
ribosomal RNAspro-duced
in the infected cell. Low concentrations of actinomycin D have been found to inhibitboth
the synthesis of the large precursor ribo-somal RNAs found in the cell nucleus and the appearance ofcytoplasmic 28S
and18S
ribo-somal RNAs (9, 21). Pichinde virus was repli-cated in medium containing actinomycin D,0
a-0
-1.76 E
-1.68 a -1.60 F
- c/)
z
-1.52 c
I-a
1.44 Z
co
m
FRACTION NUMBER
FIG. 3. Equilibrium density centrifugation of
Pichinde virus RNA in cesium sulfate. 3H-uridine-labeled Pichinde virus RNA (0) wascentrifuged for
72hrat30,000rev/minin aSpinco SW50rotor at20
C. Fractionswerecollected bybottompuncture,and the trichloroacetic acid-precipitable radioactivity of
each fraction was determined. 14C-uridine-labeled
HeLa cellribosomalRNA wascentrifuged ina
sepa-ratetubeand used as adensity marker(0).
tubeandusedas a marker. Symbols: 0,
3H-uridine-labeled PichindeRNA after no enzyme treatment or after deoxyribonuclease treatment; 0, 3H-uridine-labeled Pichinde RNA after treatment with ribonu-clease.
64 VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
and the RNA
fromresulting
virions wasana-lyzed.
It wasfound
that, when the culture me-dium was supplemented with 0.05gg
of ac-tinomycinD/ml,
the virus titersobtained
48 hr after
infection
wereconsistently
two- to fivefold greater than in cultures maintained in medium lacking the drug(unpublished
obser-vations).
The
effect of0.05 ugof actinomycin
D/ml
on RNAsynthesis
inuninfected cells
was mea-sured by examiningextracted RNA
by sucrose velocity centrifugation. Figure 4 shows thatthe
inhibitory
effect ofthe
drug
onRNA
syn-thesis
was quitemarked.
Inthe absence
of actinomycin Dthe
28S, 18S,
and 4S
species of hostcell RNA
weresynthesized.
Cells
that wereexposed
toactinomycinD,
however, failed tosynthesize the
28S
and18S ribosomal RNAs.
280-230
180-130 28S 18S
b 80
0
x30,
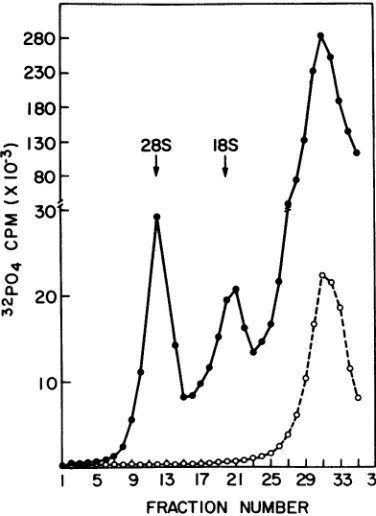
a-I5 9 13 17 21 25 29 33 37 FRACTION NUMBER
FIG. 4. Velocity sedimentation of 32P-labeled RNA from BHK-21 cells treated with actinomycin
D. Confluent monolayers were maintainedfor48hr ineitherEaglephosphate-freemaintenance medium supplementedwith 32pormaintenance medium sup-plemented with 32p andactinomycinD(0.05yg/ml).
The cellular RNA was extracted as described in Materials and Methods. A 0.2-ml
sample of
RNA from actinomycin D-treated (0) or untreated cells(0) was layered on top ofa5.0-ml,5 to 20% linear
sucrosegradientandcentrifugedas describedinFig.
2.Fractionswere collectedby bottom puncture, and the trichloroacetic acid-precipitable
radioactivity of
eachfractionwasmeasured.These
results confirmed the inhibitory effect of the actinomycin D for host cell ribosomal RNA (21).RNA was extracted from virions which were replicated in the presence or absence of 0.05
gg
ofactinomycin
D/ml.
The RNAswereana-lyzed by sucrose velocity centrifugation and
polyacrylamide
gel electrophoresis. The sedi-mentationpattern
of RNA from virions grown in the presence of the drug was considerablyaltered
(Fig. 5). The28S
RNA species was significantly reduced, and a smaller peak cor-responding to 30 to 31S was observed. The18S
species was also absent. However, the 22S and 4 to6S
peakswere essentially unchanged. The selective reduction in the 28S and18S
RNA species fromvirions grown in the presence of actinomycin D was also observed when the viral RNA was examined by gel electrophoresis
(Fig.
6).Replication of the virus in the presence ofactinomycin Dresulted in virions whose RNAresolved
into31S
and 22S components.Incorporation
ofmethyl-3H
group into Pichinde virus RNA. Cellular ribosomal and
I-N
b
x
a-u
a-Nv
I',
12
10
9
8
7
6
5
4
3
2
28S
I
I I I I
cI
II I
18S
13 17 21 25 29 33
FRACTION NUMBER
FIG. 5. Velocity sedimentation of 32P-labeled viral RNA
synthesized
in thepresenceof actinomycin
D. The32P-labeled RNAfromPichinde virions grown in the presence(0)
orabsence(0)
of actinomycin
D (seeMaterials andMethods) wascentrifuged
as de-scribed inFig.2.Fractions werecollectedby
bottom puncture of the tube, and the trichloroacetic acid-precipitable radioactivity ofeachfraction
was mea-sured.65
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.495.48.236.273.531.2] [image:5.495.248.438.321.577.2]transfer RNAs have been found to contain methylated
nucleotides
(10,29).
In contrast, however,there
isno evidence that thenucleo-tides
whichcomprise
the RNAs of animal vi-ruses aremethylated.
To obtain3H-methyl-ated RNA, Pichinde virus was grown in
me-dium containing
methionine-methyl-
3H
(see
above), and the
virionRNA
wasanalyzed
by
sucrose
velocity
centrifugation
(Fig.
7).
The
28S, 18S, and 4S
components ofthe viral RNA
contained
methylated
nucleotides. The 22S
component
observed when the RNA
wasla-beled with 3H-uridine
(see
Fig. 2)
was notde-tected under these
experimental
conditions.
The effect of
actinomycin D
onthe
incorpora-tion of
methyl-
3H
group
intoviral RNA is also
shown
inFig.
7.Labeled 28S and 18S RNA
components were
almost
completely
absent
from virions grown in
the
presence ofactino-mycin D.
The
4 to6S RNA
waslabeled,
how-ever,
and
incorporated
intothe virions.
Base
composition
of the RNA. BHK cell
RNA from
uninfected cells and viral RNA from
virions
replicated
in the presenceorabsence of
actinomycin
D werelabeled with
32P. Different
components of
BHK-21 cellular RNA
orviral
RNA were
separated
by
sucrosedensity
gradi-ent
centrifugation, and the base
composition
of
the various RNA
components
wasdetermined
(Table 1).
Thebase
composition
ofthe 28S
component from viruses
replicated
in the
absence
ofactinomycin
D wassimilar but
notidentical
tohost cell
28S
RNA. Both RNAshad
high guanosine and
cytosine
(GC)
content.The
base
composition
ofthe
virion22S
RNA
wasdistinct
fromeither host cell
28S
or18S RNAs.
When the
virus was grown inthe
presence ofactinomycin
D, the
virion31S and 22S RNAs
showed
adecreased GC
content.DISCUSSION
While these studies
were in progress, Peder-senreported that the RNA
oflymphocytic
cho-riomeningitis
viruscould be separated into fourcomponents corresponding
to31S, 28S,
22S, and 18S. Heconcluded,
on the basis of selec-tiveinhibition
of the 28S and18S
RNAs with actinomycin D, that the 31S and 22S RNAs were virusspecified and the 28S and18S
RNAs were derived from host cell ribosomes (19, 20).Our findings
withPichinde
virus, anothermem-ber
ofthe arenavirus group, in essence confirm theseobservations,
and weagree with the con-clusion that host cell ribosomal RNAisincor-porated
into the virion.The
nucleic acid of Pichinde virus clearly consists ofsingle-stranded
RNA. Thesensitiv-31S 28S 22S
+ 4 18S
0 10 20 30 40 50 60 70 - DISTANCE MIGRATED(mmI +
FIG. 6. Polyacrylamide gel electrophoresis of Pichinde virus RNA synthesized in the presence of actinomycin D. The 32P-labeled viral RNA synthe-sized in the presence (0) or absence (0) of actinomy-cin D wereelectrophoresed as described in Materials and Methods. "4C-uridine-labeled HeLa cell ribo-somalRNA was used as reference.
20
28S 18S
+
--1 C.10
1 5 1 0 1 5 20 25 30 35
FRACTION NUMBER
FIG. 7. Velocity sedimentation of
methyl-8H-labeled Pichinde virus RNA synthesized in the pres-enceof actinomycin D. Labeled RNA from Pichinde virus grown in the presence (0) or absence (0) of actinomycinD was centrifuged in 5 to
20%o
sucrose density gradients as described in Fig. 2. Fractions were collected by bottom puncture of the tube, and the trichloroacetic acid-precipitable radioactivity of eachfraction was measured.J. VIROL.
4
2
0
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.495.263.451.68.301.2] [image:6.495.262.450.373.616.2]~
ity
toribonuclease
andalkali,
buoyant density
ocq m co m in cesium
sulfate,
and
basecomposition
areall
H +-H
.
characteristics ofsingle-stranded
RNA. Wei: oo e > oo O CZ
,,:found
the RNA of Pichinde virus to becom-¢ Let-t o. posed of five discernible species when
analyzed
z- 5, 0 by sucrose gradient centrifugation and
poly--U
=acrylamide gel
electrophoresis. Three of the>. o o o o o
g0species,
28S,
18S
and 4 to6S,
appear, at least+
cif
++-H
-Hl -H inpart, to be ofhost
cell origin. Virions grown__e cnOvc
r0
inthe
presence of concentrations ofactinomy-cq c- m
o
cinD,
which inhibited ribosomal RNAsyn-thesis in host
cells,
yielded
RNA devoid of radiolabel in the28S
and 18S components. In addition,methylated
nucleotides were foundassociated with the
28S, 18S,
and 4 to6Sseg-ments of virions
replicated
in the presence ofa:~Co'
methionine-methyl-3H; methylation
ofnucleo-a,
tides
is
characteristic of cell ribosomal andZ= transfer RNAs
(21).
C|M:-0t Cl The data
obtained
upon
analysis
ofthe
base>zce + + + + + 0
composition
of the RNAcomponents
isolatedC _ 00 v s t C. 0 from virions
replicated
in thepresenceand
ab-Q:
X --ot
o -- sence ofactinomycin
Dalso
suggested
aribo-somal origin of the
28S
and 18S RNAs. The base composition of the viral28S
to 31Scom-ponent
observed
in virusgrowninthe presence>x +±+ + ++ , of the
drug
wascompletely
different than that.tt o > > e > = observed in virus grown in the absence of the
M co
0t
- CQQdrug;
the percent GC was markedly less. This4 _ is
compatible
with a significant portion ofthe
o e > oo s n =
28S
RNAcomponent
being
ribosomal RNA
o0 o
3which
civ
is rich inguanosine
andcytosine.
o ,,: +H +
,+
The22S
viral RNAcomponent
was alsoSo M Mm found to be altered when the RNA was
ex-tracted
from virionsreplicated
inthe
presenceof
actinomycin
D. Thedifferences
observedprobably
represent
technical difficulties inef-C;z
_o_ocq,
7; ficiently separating virion22S
and18S
RNAsZ ,,, + + + + + ¢^
by
sucrose centrifugation.The values
ofbase
Xcq
o0
0 CIOv~c
'It +,compositions
obtained from viruses grown inX> e6 M' t' M'Cn
the
presence ofactinomycin
D would appearm ,:= to more
accurately
portray
thecomposition
ofE_ Q) M.2
the viral
31S
and22S
RNAs. These RNAs(M 0 L -_ 0: = 0 appear to be viral-coded RNAs and correspond
x o o
_+o+c±
tEE
to molecular weights of 2.1 x106
and 1.1 x 106,OCeoo z n : .=>
trespectively.
The RNA responsible forviral-4 zc coded
products,
can,thus,
be estimated to becS
_n_coE >Xabout
3.2 x 106 daltons.The
origin
ofthe
4 to6S
virionRNA
isdiffi-cult to determine. Asimilarity of viral and host cell 4 to
6S
RNA wasfoundby
base composi-.0;;;;asCZ=tion
analysis,
and this RNA wasmethylated
Q SS S S + r <q5 o (observations
indicating
a host cellorigin).
L
Comparisons
of the basecomposition
of the0 > viral RNA
species
also suggest that the virion0
c: X ¢ ¢ 4 to 6S RNA is not the breakdown product of
.ne of the larger RNA components. A 4S RNA can also be extracted from the oncornaviruses
on November 10, 2019 by guest
http://jvi.asm.org/
(3, 8) and visna virus (11). This low-molecular-weight RNA in the oncornavirus is apparently a mixture ofhost cell andviral-specified RNA, whereas the
4S
RNA of visna virus is thoughtto be virus specified (11). Atpresent theorigin ofthe 4to6S RNA of Pichinde virus cannotbe stated withcertainty.
The nature ofthe arrangementofthe RNAs
within the virion is unknown. Ribosome-like particles havebeen observed in electron micro-graphs of sections of Pichinde virus (17). In addition, the replication of arenaviruses is inti-matelyassociated with ribosome-like inclusions in infected cells (1, 17), andaggregates of ribo-some-like particles have been observed at the site of virus maturation forseveral arenaviruses (6, 16, 17). These observations stronglysuggest
host cell ribosomes are incorporated into
ma-ture virions andserve as sourceof the 28S and
18S
RNA. The31S
and22S
components thencorrespond to the major viral-specific RNAs. It is not known if these two components are
present as distinct segments, as described for
orthomyxoviruses (20) and diplornaviruses (24), oriftheyare breakdownproductsfromalarger single-stranded molecule with specific weak points. These points will require further study.
ACKNOWLEDGMENTS
This work was supported by Public Health Service
re-search grant Al10,125 and training grant 5T1AI74fromthe
National Institute of Allergy and Infectious Diseases and researchgrantHE 05425 from the National Heart and Lung Institute.
LITERATURE CITED
1. Abelson, H. T., G. H. Smith, H. A. Hoffman, andW. P.
Rowe. 1969. Useofenzyme-labeled antibodyfor elec-tronmicroscope localizationofLCM virusantigens in
infectedcell cultures. J. Nat.CancerInst.42:497-502. 2. Bishop, D. H. L., J. R. Claybrook, and S. Spiegelman. 1967.Electrophoretic separation of viral nucleic acids
onpolyacrylamidegels. J.Mol. Biol.26:372-387. 3. Bishop, J. M., W.E.Levinson,N.Quintrell,D.Sullivan,
L. Fanshier, and J. Jackson. 1970. Thelowmolecular weight of RNAsofRoussarcoma virus. Virology 42: 182 -195.
4. Biswal, N., M. B. Grizzard, R. M. McCombs, and M.
Benyesh-Melnick. 1968. Characterization of
intracel-lularribonucleic acidspecificforthe murine
sarcoma-leukemia virus complex. J.Virol. 2:1346-1352. 5. Bollurn, F. J. 1968. Filterpaperdisctechniques for
as-saying macromolecules, p. 169-173. In L. Grossman and K. Moldave (ed.), Methods in enzymology, vol. 12(part B). Academic Press Inc., New York.
6. Dalton, A. J., W. P. Rowe, G.H.Smith, R.E.Wilsnack, and W. E. Pugh. 1968. Morphological and cytochem-ical studiesonlymphocytic choriomeningitis virus. J.
Virol. 2:1465-1478.
7. Dingman, C. W., and A. C. Peacock. 1968. Molecular
weight estimation and separation of ribonucleicacidby electrophoresis in agarose-acrylamidecomposite gels. Biochemistry. 7:668-674.
8. Erikson, R. L. 1969. Studies on the RNA from avian
myeloblastosisvirus.Virology 37:124-131.
9. Goldberg, I. H., and M. Rabinowitz. 1962.Actinomycin D inhibitionofdeoxyribonucleic acid-dependent syn-thesisofribonucleicacid. Science 136:315-316. 10. Greenberg. H., and S. Penman. 1966. Methylationand
processing ofribosomal RNA in HeLa cells. J. Mol. Biol. 21:527-535.
11. Lin, F. H., andH. Thormar. 1971. Characterization of ribonucleic acid from Visnavirus.J. Virol. 7:582-587. 12. McGregor, S., and H. D. Mayor. 1971. Biophysical and biochemical studies on rhinovirus and poliovirus. II. Chemical andhydrodynamic analysisofthe rhinovir-ion.J.Virol.7:41-46.
13. McSharry, J., and R. Benzinger. 1970. Concentration andpurificationofvesicularstomatitis virusby poly-ethylene glycol "precipitation." Virology 40:745-746. 14. Martin, R. G., andB. N. Ames. 1961. A method for de-termining the sedimentationbehaviorof enzymes: ap-plicationtoprotein mixtures. J. Biol. Chem. 236:1372-1379.
15. Mifune, K., M. Carter, and W. E. Rawls. 1971. Charac-terization studiesofthe Pichindevirus-a inember of
the arenavirus group.Proc. Soc. Exp. Biol. Med. 136: 637-644.
16. Murphy, F. A., P. A. Webb, K. M. Johnson, and S. G. Whitfield. 1969. Morphological comparison of Ma-chupo with lymphocytic choriomeningitisvirus: basis for a newtaxonomicgroup.J.Virol.4:535-541. 17. Murphy, F. A., P. A. Webb, K. M. Johnson, S. G.
Whitfield, and W. A. Chappell. 1970. Arenoviruses in Vero cells:ultrastructure studies.J.Virol.6:507-518. 18. Pedersen, I. 1970.Density gradient centrifugation
stud-ies onlymphocytic choriomeningitisvirusandonviral ribonucleic acid.J. Virol.6:414-420.
19. Pedersen, I. 1971. Lymphocytic choriomeningitis virus RNAs. Nature N. Biol.234:112-114.
20. Pons, M. W., and G.K. Hirst.1968.Polyacrylamide gel electrophoresis ofinfluenza virusRNA. Virology 34: 385-388.
21. Roberts, W. K., and J. F. E. Newman. 1966.Useof low concentrations ofactinomycinD inthestudyofRNA synthesisinEhrlich ascitescells. J. Mol. Biol. 20:63-73.
22. Rowe, W. P., F. A. Murphy,G. H. Bergold, J. Casals, J.Hotchin,K. M.Johnson, F. Lehmann-Grube. C. A. Mims, E.Traub, andP. A.Webb. 1970.Arenoviruses: proposed name for a newly defined virus group. J. Virol.5:651-652.
23. Scherrer, K., and J. E. Darnell. 1962. Sedimentation characteristics of rapidly labeled RNA from HeLa cells. Biochem. Biophys. Res. Commun. 7:486-490. 24. Shatkin,A.J.,J. D. Sipe, and P.Loh.1968.Separation
of 10 reovirus genome segmentsby polyacrylamide gel electrophoresis. J. Virol. 2:986-991.
25. Spirin,A.S.1963.Someproblems concerning the macro-molecular structure of ribonucleic acids, p. 301-345. InJ. N. Davidson and W. E. Cohn (ed.), Progress in nucleic acid research, vol. 1. Academic Press Inc., New York.
26. Stoker, M. G. P., andI. A. MacPherson. 1961. Studies ontransformation of hamster cellsbypolyomavirus in vitro.Virology14:359-370.
27. Summers,W. C. 1969. Theprocessofinfection with coli-phageT 7. I. Characterization ofT7RNA by
polya-crylamide gel electrophoresis analysis. Virology 39: 175-182.
28. Trapido, H., and C. Sanmartin. 1971. Pichinde virus: a new virus of the Tacaribe group from Colombia. Amer. J.Trop. Med. Hyg.20:631-641.
29. Vaughn, M. H., R. Soeiro, J. R. Warner, and J. E. Darnell. 1967. The effects of methionine deprivation on ribosome synthesis in HeLa cells. Proc. Nat.Acad. Sci.U.S.A. 58:1527-1534.
68