0022-538X/87/041136-11$02.00/0
CopyrightC) 1987, American Society forMicrobiology
Expression of Herpes Simplex Virus Type 1 Major
DNA-Binding
Protein, ICP8, in Transformed Cell Lines: Complementation of
Deletion
Mutants
and Inhibition of
Wild-Type Virus
PAULOK. ORBERG ANDPRISCILLA A. SCHAFFER*
Laboratory of Tumor Virus Genetics, Dana-Farber Cancer Institute, andDepartment of Microbiology and Molecular
Genetics, Harvard MedicalSchool, Boston, Massachusetts 02115
Received 16 September 1986/Accepted 11 December 1986
To minimizethecontributionof residualactivityassociated with thetemperature-sensitive (ts)form ofICP8
specified byavailable tsmutants, deletion mutations in this gene were constructed. Cellspermissivefor the
generation andpropagationof ICP8 deletion mutantswerefirstobtained. Vero cellswerecotransfectedwith
pKEF-P4, whichcontainsthegeneforICP8,andpSV2neoor ahybrid plasmid containingtheG418 resistance
gene linked to pKEF-P4. Of the 48 G418-resistant cell lines, 21 complemented ICP8 ts mutants in plaque
asssaysatthe nonpermissivetemperature. Four of thesewereexaminedbySouthernblotanalysis andshown
to contain 1 to 3 copies of the ICP8 gene per haploid genome equivalent. Cell line U-47 was used as the
permissive host for construction of ICP8 deletion mutants. In addition to cell lines which complementedts
mutants, twolines,U-27 and U-35, significantly inhibitedplaqueformation by wild-type virus,contained 30
and100 copiesof the ICP8gene perhaploidgenomeequivalent, respectively, andexpressed largeamountsof
ICP8 after infection withwild-type virus. At low but not high multiplicities ofinfection,this inhibition was
accompanied by underproductionof viralpolypeptidesof theearly, delayed-early,and late kinetic classes.For
construction of deletion mutants, a780-base-pair XhoIfragment was deleted from pSG18-SalIA, a plasmid
which contains thegenefor ICP8, toyield pDX. U-47 cellswerethen cotransfected with pDXand infectious
wild-type DNA. Mutant d61, isolated from the progeny ofcotransfection, was found to contain both the
engineereddeletion in the ICP8geneandanoriL-associated deletion ofapproximately55basepairs.Because
d61 containedtwomutations, asecond mutant,d21,whichcarried theengineeredICP8 deletion butanintact oriL, wasconstructedbycotransfection of U-47 cells with wild-typeDNA andanSalI-KpnI fragment purified
from pDX. Phenotypic analysisof d21 and d61 revealed thattheyweresimilar in allpropertiesexamined: both
exhibited efficientgrowthinU-47 cells butnotinVerocells;both inducedthesynthesisofanICP8polypeptide which wassmaller than thewild-typeform of theprotein andwhich, unlikethewild-type protein, wasfound in thecytoplasmand not the nucleus of infected Verocells;andnonpermissiveVero cells infected with either mutantfailed toexpresslate viralpolypeptides.
The major herpes simplex virus type 1 (HSV-1)
DNA-binding protein ICP8 is the product ofan early, or
3,
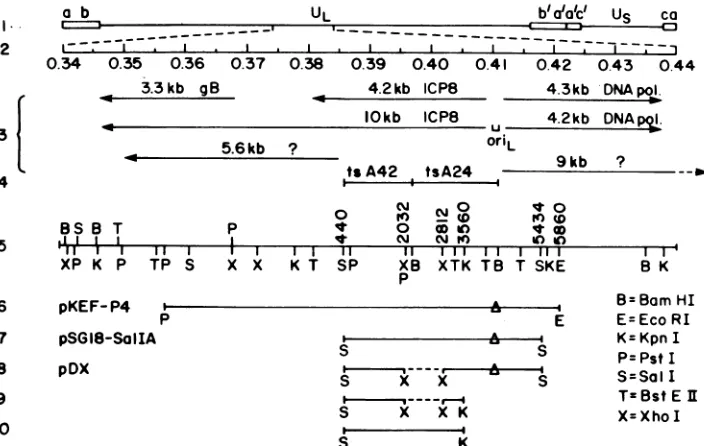
genelocated between map coordinates 0.380 and 0.410 on the
viral genome (Fig. 1) (16, 27, 39). This gene is transcribed
primarily asa4.2-kilobase (kb) mRNA species containinga
codingsequenceof3,591 bases (27). Amuchless abundant ICP8-specific mRNA, of approximately 10 kb, has also been
detected (16). The major DNA-binding proteinof HSV-1 is
composed of 1,196 amino acids of molecularweight128,341
(27).
During infection, ICP8 is transported to the nucleus (9, 18), where it associates with the nuclear matrix (26). Purified
ICP8 isable todestabilize apoly(dA:dt) helix (24)and hold
single-stranded DNAin an extended conformation (29). The
observation that temperature-sensitive (ts) mutants of ICP8
are phenotypically DNA negative at the nonpermissive
temperature demonstrates that ICP8 is required for viral
DNA synthesis (39). Indirect evidence that ICP8 interacts
with viral DNA polymerase to form a DNA synthetic com-plex (38) includes the following: (i) antibodies to ICP8 have
been reported to inhibit DNA polymerase activity in
chro-matin isolated from HSV-infected cells (24); (ii) viral DNA
polymerase andalkaline nuclease activities were found to be reduced in extracts prepared from cells infected with an
* Corresponding author.
ICP8 ts mutant at the nonpermissive temperature (21), and
addition ofpurifiedICP8 restored theability ofsuchextracts tosynthesize DNA; (iii) purified ICP8 hasbeenreported to
stimulate DNA polymerase activity in vitro (30); and (iv)
ICP8 ts mutants may exhibit altered sensitivity to DNA
polymeraseinhibitors (3).
In addition to its involvement in viral DNA synthesis,
ICP8plays an essential role in inhibitingthe expression of
viral genesof all kinetic classes, as shown by the fact that
ICP8 ts mutants overproduce certain a, ,
-Yl,
and Y2mRNAs and proteins at the nonpermissive temperature
(10-12).
Althoughts mutantshave provenextremely useful for the
functional analysis of HSV, they revert and may exhibit
residualwild-type activity(leak). Additionally, studies of ts
mutants of ICP4, an immediate-early (ao) transcriptional
regulatory protein, have shown that some ts forms of the
protein exhibit distinctactivities not characteristic of resid-ualwild-type activity(8). By contrast, ICP4 deletion mutants
exhibited a true null phenotype and were more efficiently
complementedin ICP4-expressingcells than was anICP4 ts mutant (7). This observation indicated that the ts
polypep-tide had aninhibitory effect on the activity of the wild-type
protein supplied by the cells. Because deletion mutants neither leak nor revert, they better reflect the null phenotype than ts mutants and therefore constitute useful tools for 1136
on November 10, 2019 by guest
http://jvi.asm.org/
DELETION MUTATIONS IN HSV-1 ICP8 1137
a b I
2 II
0.34 0.35
0.34
3.3 kb
3 4
BS B T
5
.n
IrI
1m
XP K P TP S
6 7
UL
6 0.37 gB
b'uaac' Us ca
~~
--
-I I I - - ;
0.38 0.39 0.40 0.41 0.42 0.43 0.44 4.2kb ICP8 4.3kb DNApoil
IOkb ICP8 4.2kb DNApQl.
OriL 5.6kb ?
ts A42 tsA24
i i
p
x x
0 N N g 4 0
t 0 O in t 0
q CN N ) IC) )
TII I II I III
K T SP XB XTK TB T SKE p
pKEF- P4
I-P pSGI8-SaIIA
8 pDX
9 10
S XX
S X X K
S K
9kb ?
B K
BBam HI E E=Eco RI
-4 K=Kpn I
s P= Pst I
-S
S=SalITzBstEl X=Xho I
FIG. 1. Genomic location of the ICP8 gene and plasmids and DNA fragments used in this study. The prototype arrangement of the HSV-1 genomeis shown in line 1. The map units of the expanded, central portion of the long unique region (UL) are shown in line 2. Transcripts known to map to this region of the genome (all of thep kinetic class [16]) and the location of a viral origin of DNA replication,oriL(14,40), areshown in line 3. The locations of the mutations in two ICP8 ts mutants used in this study, tsA24 and tsA42, appear in line 4 (39). Relevant restriction sites and the corresponding nucleotide numbers (27) are depicted in line 5. In lines 6 to 8 the symbol A represents deletions in OriL of-55bp which occur during cloning in bacteria. The fragment shown in line 9 was generated from pDX and used to constructd21bymarker transfer (see text). The fragment shown in line 10 was derived from
pSG18-SaIIA
and used as a probe forSouthernblotanalysis of cellular DNAs (Fig. 3).studies ofthefunctional roles ofspecific viral genes in the
life cycle ofthevirus.
Inthispaper we report thederivationand characterization
ofVerocell lines thatexpressICP8and the useofthese cells aspermissivehostsforthe construction and characterization
of HSV-1 mutantscarrying deletions in thegeneforICP8. In
additiontothecell lines used to generate deletion mutants, a
second class of ICP8-expressing cells was isolated and
shown to inhibit wild-type viral plaque formation and late
viral gene expression.
MATERIALS AND METHODS
Cells and viruses.Africangreenmonkey kidney(Vero) and
human embryonic lung (HEL) cells were propagated and
maintained as described previously (39). In some tests,
G418-resistant (Nero) cellswere used (7). Nero cells were
derived by G418
selection
ofVero cells transfected withpSV2neo.
The KOS strain ofHSV-1 was used as thewild-type virus from which ts (tsA24 and tsA42 [39]) and deletion mutants werederived. All viruses were propagated and assayed as
describedpreviously (33).
Plasmids and cloning. The map locations of viral DNA
inserts in plasmids used in this study are shown in Fig. 1.
pKEF-P4 (5) and a modified form of pSV2 neo (36) (see
below)
weregenerously provided
by N. DeLuca.pSG18-SalIA (19) was the
gift
of S. K. Weller(University
ofConnecticutHealth Center,
Farmington).
Restriction endonucleases and T4 DNA ligase were
ob-tained fromNewEngland Biolabs(Beverly, Mass.)and used as suggested bythe manufacturer.
Viral DNA isolation. Infected-cell DNA was
prepared
asdescribedpreviously (6). AfterproteinaseK
digestion,
viralDNAwasseparated from cellular DNA by centrifugationin
CsCl gradients (13). KOS DNAwas isolated frominfected
HEL cells, and deletion mutant DNA (d21 or d61) was
isolatedfrom infected U-47 cells (see below).
Isolation of DNA fragments. DNA fragments for use in
cotransfection experiments or as probes in Southern blot
hybridizations were isolated after restriction enzyme
diges-tion ofplasmid DNA and electrophoresis through a 0.4%
agarosegel. Afterbeing stained with ethidium bromide, the
bandsof interest were excised andsubmittedto three cycles
offreezing and thawing (34). Agarose was separated from
the DNAsolutionby centrifugation. The DNA in the
super-natant fluid was purified by passage through an Elutip-d
column (Schleicher & Schuell, Keene, N.H.) and ethanol
precipitation.
Blot hybridization. Specific DNA sequences in digests of
viral or cellular DNA were detected by the method of
Southern (22, 35). Probes werelabeled with [32P]dCTPand
[32P]dGTP (Amersham Corp. Arlington Heights, Ill.) by
nick-translation (22).
Transfection of Vero cells. Cells lines containingthe gene
for HSV-1ICP8 weregeneratedfrom Vero cells essentially
as described for ICP4-containing cell lines (7), except that
eitherpSV2neo (0.5 ,ug)wascoprecipitated and transfected
with 1.5 ,ug of pKEF-P4 or,alternatively, thehybrid plasmid
pKEF-P4-G418 (2.0 ,ug)wasused(Fig. 2).The concentration of G418 (Geneticin Sulfate; Gibco
Laboratories,
ChagrinFalls,Ohio)used for selection ofICP8-containingcells was
0.5 mg/ml. G418-resistant cell lines were
propagated
in thislevel ofdrugatalternatepassagesto maintain cellscarrying
the selectablemarker and ICP8 DNA sequences.
Marker transfer. Cotransfection of U-47 and Nero cells
with infectious KOS DNA and plasmidDNAfor
construc-VOL.61, 1987
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.122.474.75.298.2]tionofdeletion mutants wascarriedoutasdescribed
previ-ously for markerrescue (31).
Analysis of infected-cell polypeptides. [35S]methionine
la-beling of polypeptides produced in infected cells between 5
and 18 h postinfection was conducted as described
previ-ously (31).
Western blot analysis. Immunological detection of
ICP8-relatedpolypeptidesamonginfected cell polypeptides
sepa-ratedby sodium dodecylsulfate-polyacrylamide gel
electro-phoresis (SDS-PAGE) and transferred to a nitrocellulose
membrane (2)wasperformed asdescribed(7) with 0.2 ml of
polyclonal rabbit antiserumtoICP8(diluted 1:40 with Blotto
[17]), which was the generous gift of Richard Courtney
(Louisiana State University, Shreveport).
Immunofluorescence. Demonstration of the intracellular
location of wild-type and mutant ICP8 polyeptides was
performed essentially as described by Quinlan et al. (26).
Cellsgrown oncover slipswereinfectedatamultiplicityof
1 PFU/cell and incubated at 37°C for 12 h before fixation.
The ICP8 antiserum was the same as that used in the
Westernblot analysis described above, but itwasused ata
dilution of 1:5. Indirect staining was carried outwith
rho-damine-conjugated goat anti-rabbit immunoglobulins
(Coo-perBiomedical, Malvern, Pa.) ata 1:30 dilution.
RESULTS
Derivation of celllines containingthegenefor HSV-1 ICP8.
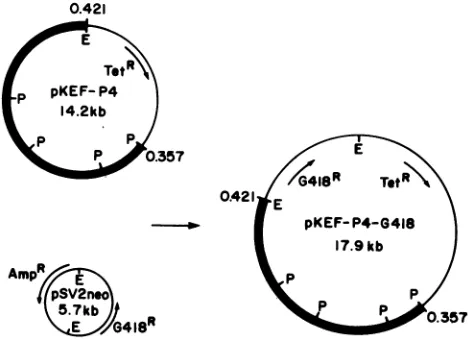
Plasmid pKEF-P4 (5) contains the viral DNA fragment with
mapcoordinates 0.357to0.421. Theonly intactgene
speci-fiedby this fragment is thegenefor the majorDNA-binding
protein, ICP8, under control of itsownpromoter(Fig. 1 and
2). Plasmid pSV2neo (Fig. 2) contains the gene which
specifiesresistancetoG418under control of the simian virus
40 (SV40) early promoter (36). pSV2neo was modified by
addition of an EcoRI linker at its single NdeI site (N.
DeLuca and A. McCarthy, personal communication) (Fig.
2). The G418 resistance moiety of EcoRI-cleaved pSV2neo
was cloned into the unique EcoRI site of pKEF-P4 to
generate pKEF-P4-G418(Fig. 2).
Vero cellswere transfected in suspension (7) with either
thehybrid plasmid pKEF-P4-G418 (linkedtransfection)ora
0.421
Tot
{ ~~Tet'\
P pKEF-P4 14.2kb
P_P>0.357
0.421 E
pKEF- P4-G418 17.9 kb
AmpR ,Pp
3pSV2neP .357
G418R
FIG. 2. Plasmids used to generate ICP8-expressing cell lines.
Cell lines resistant to G418 were obtained by cotransfection with
pSV2neo andpKEF-P4 (Fig. 1) or by transfection with
pKEF-P4-G418 alone. Heavy lines represent HSV-1 DNA sequences. E,
[image:3.612.326.567.92.155.2]EcoRI; P, PstI.
TABLE 1. Qualitative complementationtestsof ICP8tsmutants tsA24and tsA42inG418-resistantVero celllinesa
Plaqueformation(%) Derivation of cell line
Positive Negative
Linked transfection 7(41) 10(59)
Unlinked transfection 14(45) 17(55)
Total 21(44) 27(56)
aCell lines obtainedbylinked transfection with pKEF-P4-G418orunlinked cotransfection with pKEF-P4 and pSV2neo (Fig. 2) were tested for their abilitytosupportplaqueformationbyICP8tsmutants atthenonpermissive temperature(39.4°C).Testswerescoredaspositive (plaques)ornegative(no plaques).
mixture of pKEF-P4 and pSV2neo (unlinked
cotransfec-tion). TheresultingG418-resistant colonieswere designated
L for colonies derived from linked transfection or U for
coloniesderived from unlinked cotransfection.
After isolation and amplification of individual
G418-resistantcolonies, the48 resulting cell lineswere screened
for theabilitytocomplementthetsmutanttsA24at39.4°C.
tsA24 isanHSV-1mutantcarryingamutation in the gene for
ICP8 (Fig. 1) (39). Complementation was assessed
qualita-tively bythe ability oftsA24 to produce plaques on
G418-resistant cell linesat39.4°C relativetonontransformed Vero
cells.Wild-typeviruswasusedas acontrol. Cell lineswhich
supported plaque formation by tsA24 at 39.4°C were then
retested simultaneously with tsA24 and tsA42, a mutant
whose ts mutation maps immediately to the left of the
mutation in tsA24 in the ICP8 gene (Fig. 1) (39). In every
case, cell lines which complemented tsA24 also
comple-mentedtsA42. The results of complementation tests with the
48 cell linesare summarized in Table 1.Despite differences
in the transfectionprocedure usedtogenerateG418-resistant
cells,thefrequencyofisolation of lines abletocomplement
ICP8tsmutants at39.49Cwasquite similar: linked
transfec-tion, 41%;unlinked cotransfection, 44%.
Among the 21 cell lines which complemented the ts
mutants, 6exhibited efficient complementation as judgedby
the size and number of tsA24 plaques at 39.4°C. Ofthese,
line U-47 was chosen as the permissive host for the
con-struction andpropagation of deletion mutants. Prior to the
generation of deletion mutants, however, U-47 and other complementing cell lines were characterized further.
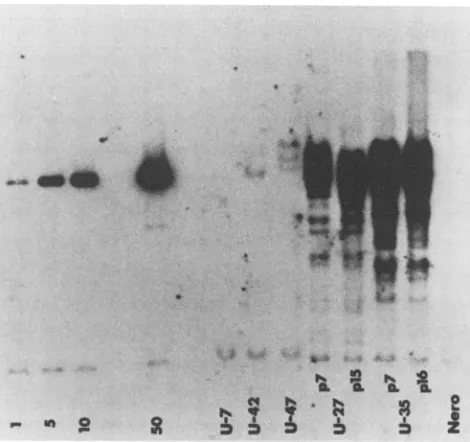
Presence of ICP8-related sequences in complementing cell
lines. Cell lines wereexaminedby Southern blot analysisto
establish the presence and copy number of the ICP8 gene.
For this purpose cellular DNAs were digested withEcoRI,
electrophoresed, and transferred to nitrocellulose filters.
The filterwasprobed witha32P-labeled SalI-KpnIfragment
(map coordinates 0.386 to 0.406) purified frompSG18-SalIA
(19) (Fig. 1). This probe comprises approximately 71% of the
ICP8 gene and is containedentirely within this gene (Fig. 1).
The results of thisanalysis are shown in Fig. 3.
The Southern analysis included pKEF-P4 standards
rep-resenting from 1 to 50copies of the ICP8 gene perhaploid
genome equivalent and U-7 and Nero cells as negative
controls. U-7 cells are G418-resistant cells selected after
cotransfection of Vero cells with pSV2neo and pKEF-P4
that failed to complement tsA24 or tsA42. Cells which
complemented the ICP8 tsmutants, such as U-42 andU-47,
contained from 1 (U-42cells) to 3 (U-47cells) copies of the
ICP8 gene per haploid genome equivalent. ICP8-associated
sequences in U-47 cells appeared to be associated with
cellular DNA,resulting in bands oflessermobility than that
of the viral DNA band in plasmid pKEF-P4. Additionally,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.69.305.504.674.2]DELETION MUTATIONS IN HSV-1 ICP8 1139
reduced
plating
efficiency
of KOS on U-27 and U-35 cells,plaque
sizes werequite heterogeneous.
Thedata inTable2 also indicate that a functional ICP8 polypeptide must beproduced
inbothU-27 andU-35cells, since theysupported
the
growth
of d61 several orders ofmagnitude
moreeffi-ciently
than did control Nerocells,
which donotcontain the ICP8gene.WhentheDNAs of U-27 and U-35 cellsweresubjectedto
Southernblot
analysis,
thetwocelltypeswereestimatedto 1 2 3 4 5 6 7 8 910 11 12 1314 1516_m 0
K- a
V%.
E
2
xX X
6
8
1
FIG. 3. Southern blot analysis ofthepresence and copynumber ofICP8genesequencesinselectedcelllines.AmountsofpKEF-P4 representing1, 5,10, or 50copies ofICP8 perhaploidgenome or 10
jigof total cellular DNAwasdigested withEcoRI,electrophoresed inanagarosegel,andtransferredto anitrocellulose filter.Thefilter was probed with theSalI-KpnI fragment shownin Fig. 1 (line 10) labeled with 32Pbynick translation. U-7 and U-47 were tested at passage 7 (p7) U-42 at passage 15, U-27 at passages 7 and 15, and U-35 atpassages7 and16.
thecopynumber and cellularDNAassociations of theICP8
gene were stable through eight passages in U-47 cells (not
shown).
ExpressionofICP8incomplementingcelllines.SDS-PAGE analysis showed that U-47 cells did not constitutively
ex-pressdetectable levels of ICP8 (Fig.4,lane 2).Infectionwith
HSV-1 KOS at a multiplicity of 0.2 PFU/cell revealed approximately equal amounts of ICP8 in U-47 (lane 6) and Nero cells (lane 5). At higher multiplicities, the amount of
ICP8 produced in KOS-infectedNerocellswasgreater than thatproduced in infectedU-47cells(lanes9 and10,lanes13
and 14).Thus, although theintensity of ICP8 and other viral polypeptide bands increased as a function oftheincreased multiplicity of KOS in Nero cells, no multiplicity depen-dence was seen in KOS-infected U-47 cells. In addition, KOS-infected U-47 cells exhibitedmore efficient shutoffof host protein synthesis at all multiplicities tested than did KOS-infected Nerocells (Fig. 4; seealso Fig. 8).
Characterization ofcellswhichfailedtocomplement ICP8ts
mutants andinhibitedplaque formation bywild-type HSV-1.
Among the cell lines that failed to complement tsA24 and
tsA42(Table 1),two,
designated
U-27andU-35,significantly
inhibited plaque formation by
wild-type
HSV-1 KOS. Thisphenomenon wasfurther investigated in a series of plaque assays ofwild-type virus and ofthe ICP8 deletion mutant
d61 (to be described
below)
on U-27 and U-35 cells atvarious passage levels (Table 2). This table shows a
repre-sentative
sampling
of titers ofasingle
KOS stock and asingle d61 stock obtainedby
plating
on U-27and U-35 cellsand on control Nero, U-7, and U-47 cells on different
occasions. Thereductionintiterof the KOS stock observed
onU-27 and U-35 cellsvaried fromtestto test, but inmost
cases was in the range of10- to 40-fold. In additionto the
U~~~~~~~~~~~~~~
- L'
->=1~~~~~~~~~~~~~~
._4 _
MOCK
1
0.2 1 1.05 6
8gEB
pgB
19
20
25
40
43
44
48
[image:4.612.67.302.74.295.2]1 10
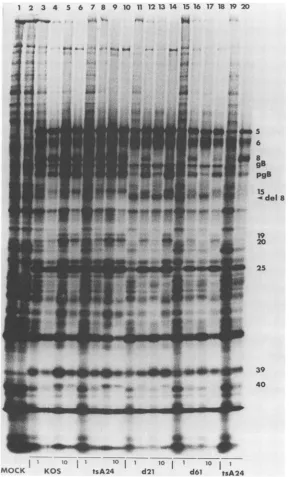
FIG. 4. SDS-PAGEanalysis of 35S-labeledpolypeptidesin cells infected withwild-type HSV-1 KOSatdifferentmultiplicities.Cells wereinfected with 0.2 (lanes 5to8), 1.0(lanes 9to12),or10 (lanes 13 to16)PFU/cellormock-infected(lanes 1to4).Polypeptideswere labeled with[35S]methionine from 5to18 hpostinfection.Cell lines: Nero(lanes 1, 5, 9, and 13), U-47 (lanes 2, 6, 10, and14), U-27(lanes 3,7, 11,and15), and U-35(lanes4,8, 12,and16).gB,Glycoprotein B;pgB,precursor toglycoprotein B.
VOL.61, 1987
''AMK.
,A..A;!.I::i
7
F
.41 41"s
40
to
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.325.560.180.649.2]TABLE 2. Plaqueassaysof virus stocks at 37°Condifferentcelllines' Titer(PFU/ml)b
Virus U-27 U-35
Nero U-7(p22) U-47(p9) p8 p21 p9 p21
KOS 5.4 x 107 5.4 x 107 6.3 x 107 4.0x 106 1.8 x 106 2.3 x 106 1.5 x 106
d61 1.0 x 102c NDd 2.3 x 107 3.9 x 105 ND 2.4 x 105 ND
aOne stock ofKOS and one stock of d61 were assayedatdifferent timesonthecelllines listed. b The passage numberof the celllines tested is indicated (p22 = passage22,etc.).
cTheappearance of d61 plaques on Nero cells was dueto thegeneration of occasionalrecombinantswhen themutant waspropagatedinICP8-containingU-47 cells.
dND, Not done.
contain from 50to 100copies ofthe ICP8gene perhaploid
genome,basedon the sumofhybridizing fragments of probe
size or greater (Fig. 3). This copy number did not vary
significantlyoverthe rangeof passagesexamined (passages
7 to 15and7 to 16forU-27and U-35cells,respectively). In
both cell lines, asignificant proportion of the ICP8-related
sequencesappearedasbands ofgreater and lessermobility
than that ofpKEF-P4 (the plasmid used to generate these
cell lines), indicatingthatrearrangements and deletions had occurred.
Polypeptide synthesisin U-27 and U-35cells infected with KOS and labeled from 5 to 18 h postinfection with
[35S]methionine
was examined by SDS-PAGE(Fig.
4). Itwasevident that while neither U-27 nor U-35 cellsexpressed
ICP8 constitutively (lanes 3 and 4; see also Fig. 7), large
amounts of ICP8 were detected in both cell types after
superinfection
withKOS. Thequantity of ICP8 detected inU-35 cells was noticeably greater than that seen in U-27
cells, which correlates with the greater copynumberofthe
ICP8geneinU-35cells indicated inFig.3. This observation
wasreproducibleinrepeatexperiments. Additionally, more
ICP8 was detected in these cells at a multiplicity of 1
PFU/cell thanat0.2 or 10PFU/cell.
Overproduction of ICP8 was
accompanied
by markedlyreducedsynthesisof viralpolypeptides ofthe ,B(gB, ICPs6
and40),
yl
(ICPs 5, 25, and 44),and _Y2(ICPs 19, 20, 43, and48)
kinetic classes. This effectwasmostpronouncedatlow multiplicity (0.2PFU/cell).
At high multiplicity (10PFU/cell), the
polypeptide
phenotype of KOS was mostsimilar in U-27, U-35, and Nero cells. The polypeptide profiles shown in Fig. 4 were obtained in U-47, U-27, and U-35cellsatpassage 5. Virtually identicalresults (including
the production of large amounts ofICP8) wereobtained in
the latter two cell lines at passage 20 (not shown). This is
consistentwith thestability ofthe ICP8gene copynumber observedinboth celllines (Fig. 3).
Construction ofdeletion mutants of ICP8. Plasmid
pSG18-TABLE 3. Burstsizesaofwild-typevirus, anICP8tsmutant, andtwoICP8deletionmutants oncomplementingand
noncomplementing cells Virusyield(PFU/cell)
Cells tsA24
KOS, 370C 340C
39.4°C
d21,37°C d61,370C Vero 250 23 7.8 x 10-3 5.2 x 10-3 7.2 x 10-3U-47 92 NDb 2.5 62 12
a6.0x105 cells were infected at a multiplicity of 1 PFU/cell, washed, incubatedattheindicatedtemperature for 18 h,harvested, and assayed for infectious virus.
bND, Not done.
SalIA
(19) contains theportion
of the HSV-1 genomebe-tween map units 0.386 and 0.418, which includes the 5'
two-thirds ofthe ICP8 gene (Fig. 1). A
780-base-pair
(bp) deletion in the ICP8coding
sequence inpSG18-SalIA
wasgenerated by
digestion
with XhoI andreligation
toyield
plasmidpDX(Fig.1).Analysis ofthenucleotidesequenceof
the ICP8 gene (27) indicates that the reading frame is maintained in pDX. This plasmid also carries a deletion of approximately 55 bp in oriL sequences. The deletion arose
spontaneouslyas aresultofinstability oftheoriL
palindrome
when cloned inbacteria (14, 40). The size of this deletion
was estimated by comparison with pKEF-P4, whose 55-bp deletion was determined by DNA
sequencing (40) (not
shown).
ICP8deletionmutantd61 wasconstructedby
cotransfec-tionofU-47cells withintact, infectious KOS DNA andpDX (linearized attheEcoRI site). Progeny of the
cotransfection
were plated on U-47 cells, and isolates were tested for growth on U-47 and Vero cells. Of240 plaque isolates, 1(d61)
grewefficiently in U-47 butnotin Verocells (Tables2and 3). Mutant
d61
wasplaquepurified
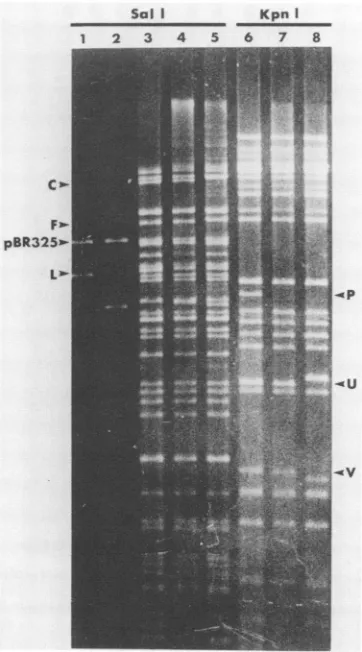
three times.Comparison
ofd61 and KOS DNAsby
restrictionenzymeanalysis andSouthern blot
hybridization
revealedthatthey
differedin several respects.
First,
asexpected,
theSail
Lfragment of
d61
DNA (map units 0.386to 0.418;equivalent
to
pSG18-SalIA
in LeeandKnipe
[19])comigrated
with theSalI
insert ofpDX,indicating
that the780-bp
deletion inICP8 in the plasmid had been incorporated into
d61
DNA(Fig. 5, lanes 2 and5;Fig. 6, lanes 2and5).
Second, and
unexpectedly,
theoriL-containing
KpnI Vfragment of
d61
DNA(0.406to0.419)wasapproximately
55bp shorter thanits KOS counterpart(Fig. 5, lanes 6and 8;
Fig. 6, lanes 6and 8). This indicated that the oriL deletion
present in pDX had alsobeen incorporated into
d61
DNA.The replication competence of
d61
in U-47 cells suggestedthat oriL may not be essential for replication of HSV-1 in
vitro. This interesting possibility has been explored in
greaterdetail and
will
beconsidered elsewhere(manuscriptinpreparation).
Third, and also unexpectedly,
d61
DNA was found tocontain an
insertion
orduplication of approximately 100bp in the "b" sequences ofthe inverted repeats surrounding UL.Thisinsertionorduplication canbeseeninFig. 5,lanes6 and 8, in which the dimolar KpnI U fragment (0.025 to
0.043 and 0.777 to 0.795 mapunits)waslarger in
d61
thaninKOS; the same was true forthe
SalI
F fragment (0.740to0.784)
(Fig.
5, lanes 3 and 5),although
this is not clearlyvisible in the gel. These diploid insertions or duplications
mapped
at or nearthe tandemreiterated sequence identifiedbyRixon et al. (28).
Fourth,
d61
DNAalso carriedan insertionorduplicationofabout 300bp, which mappedinUL betweencoordinates
on November 10, 2019 by guest
http://jvi.asm.org/
DELETION MUTATIONS IN HSV-1 ICP8 1141
Sol I KpnI
1 2 3 4 5 6 7 8
C
.-
Fp2-
pBR325*--.V
XhoI deletionin the ICP8
gene)
was of thesame size ind21
and d61.
Importantly,
d21 DNA exhibited no detectablechange
in themobility
of theoriL-containing KpnI
Vfrag-mentoranyother
fragment
relativetoKOS DNA(Fig.5and6).
Phenotypic characterization ofICP8 deletion mutants. (i)
Growth properties. As anticipated, the burst sizes of both
d21 and d61 mutants in nonpermissive Vero cells were
negligible
andsimilartothat of theICP8mutanttsA24 in thesame cells at the nonpermissive temperature (Table
3).
Clearly,
expressionof the resident ICP8 gene in U-47 cellswas required for growth of d21 and d61, although virus replicationwasless efficientthan that of KOS in either Vero
orU-47 cells. The
nearly
threefold-lowerburst size ofKOSin U-47 cells than in Vero cells may reflect an
inhibitory
effect ofthe ICP8 inducedfollowing infection, such asthat
seenin U-27 and U-35 cells. Consistentwiththese
observa-tions, the plaques produced bythe wild-type virus in U-47
cellswere largerthan thoseproduced by eitherd21or
d61,
withd61 producingthe smallestplaques.
(ii) ICP8 polypeptide synthesis. The ICP8 polypeptides
producedin U-47 and Verocells, mock-infectedorinfected
with either KOS or d61, were examined by Western blot
analysis
(Fig. 7).
As mentioned above, U-47 cells do notappear to express ICP8 constitutively; however, an ICP8
polypeptideofthe expected size was detected in U-47 cells
infectedwithd61 (lane7), as was the truncated form of ICP8
specified by d61. The larger form of ICP8 presumably
constitutes the
product
of theresidentwild-type
ICP8 gene.Sal I KpnI
1 2 3 4 5 6 7 8
FIG. 5. Agarose gel electrophoresis of plasmid andviral DNA digests. Lanes: 1 to 5, Sall digests; 6 to 8, KpnI digests; 1,
pSG18-SalIA;2, pDX; 3 and 6, KOS;4and 7, d21;5and8, d61.
RelevantSalI (left)orKpnI(right) fragmentsareindicated.
0.060 and 0.063. As a result ofthis -300-bp insertion or
duplication and the -100-bp insertion or duplication
men-tioned above, the Sall C fragment (coordinates 0.036 to
0.095) ofd61 migrated noticeablymore slowly than its KOS
counterpart (Fig. 5,lanes 3 and 5). Digestions with several
restriction enzymes were used to fine-map the -100- and
-300-bp alterations in d61 DNA(datanotshown).
The presence ofmultiple alterations in the DNA ofd61 made it unacceptable forgenetic and phenotypic analysis,
necessitating the construction of a second ICP8 deletion
mutant. Forthis purpose pDX was digested with
SalI
andKpnI, and the 2,340-bp SalI-KpnI fragment was purified
(Fig. 1). This fragment, with map coordinates 0.386-0.406, includestheXhoI deletion withintheICP8gene but excludes
oriL-related
sequences. U-47 cells were cotransfected withthis
SalI-KpnI
fragment and infectious KOS DNA, and adeletion mutant,
d21,
wasisolatedbythe samestrategy used toidentify d61.Itis notablethathomologyconsisting of748bp to the right ofthe 780-bp deletion in the cotransfected
fragment was sufficient to ensure incorporation of the
de-letedfragmentinto the viral genome at afrequencysimilar to thatobservedwhen moreextensivehomologywasavailable
(i.e., pDXand theresultingmutant
d61).
Restrictionenzymedigestion,
agarose gelelectrophoresis,
and Southern blotanalyses revealed that d21 had indeed incorporated the
780-bpdeletion within ICP8codingsequences. As shown in
Fig. 6, the
KpnI
Pfragment(whichcontains theengineered
LoQ *
_
~~~~~~~
~P
[image:6.612.91.272.74.400.2]._ V
FIG. 6. Southern blot analysis of thegel shown inFig. 5. The probe used was 32P-labeled pSG18-SalIA (Fig. 1). The Sall L fragment of d21 (lane 4) migrated slightly behind the equivalent fragments of pDX and d61 (lanes 2 and 5), sinceonlythe lattertwo contain a deletion inoriL.In theKpnIdigests (lanes6to8)theOriL and ICP8 deletions occur in separate fragments (V and P, respectively).
VOL.61, 1987
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.345.528.374.658.2]In Vero cells infectedwithd61, onlyan ICP8
polypeptide
ofreduced size was detected(lane 4). Theobserved size of the
truncatedpolypeptideproduced by d61
(lanes
7and4)
agreeswell with thatpredicted fromthe nucleotidesequenceof the
ICP8 gene and the sizeofthedeletion in this gene ind61
(27).
Similar resultswere obtained withd21
(data
notshown).
Asmentioned previously, the XhoI deletion introduced into pDX (the plasmid used in the construction of
d61)
should maintainthe ICP8reading
frame.Moreover,
the loss of780bp is equivalent to the deletion of 260 amino acids at the
protein level, yielding a
polypeptide
smaller thanwild-type
ICP8 by approximately 31,000 daltons. This is in fact the
case, as shown in Fig. 7.
Additionally,
as shown inFig.
8,
the mutantform of ICP8
specified by
d61,
which should haveapredictedmolecularweight of 97,000,hada
slightly
greaterelectrophoretic mobility than
ICP15,
103,000
molecular weight(23).Mutantd21
specified
anICP8polypeptide
ofthe samesizeas that specified by d61
(Fig. 8).
The presence of thewild-type form ofICP8 in U-47 cells but not in Vero cells
infected withd21ord61 was also evident
(Fig. 8,
lanes 11to18). At amultiplicity of1
PFU/cell,
less ICP8waspresentinU-47cells infectedwith d21ord61thanin U-47 cellsinfected
with KOS (lanes 4, 12, and 16). This observation was
consistent with the differences in
plaque
and burst sizesdescribed above. The contribution ofthe resident
wild-type
ICP8gene was
required
forlate geneexpression
by
d21andd61, as shownbythefactthat
polypeptides
ofthe Y2 kineticclass such as ICPs 15, 19, and 20were induced
by
d21andd61onlyin U-47andnotin Verocells
(Fig. 8,
lanes 11to18).
It is alsonotable that less
wild-type
ICP8 was detectable in U-47 cells infectedwith 10 PFU than with 1 PFU ofd21ord61 per cell, whereas in KOS infection of either Vero or U-47 cells there was no decrease in the
quantity
of ICP8detected as the
multiplicity
was increased from 1 to 10PFU/cell.
(iii) Intracellular localization of ICP8. To determine the
effect ofthe780-bp deletiononthe
ability
of ICP8tolocalizeto the nucleus, infected cells were
analyzed
by
immunoflu-orescence with
polyclonal
antibody
to ICP8. Unlike thewild-type form of ICP8, the mutant ICP8
polypeptides
specified by d21and d61 were present in the
cytoplasm
andnotinthe nucleus.Bycontrast, ICP8
expressed
inU-47cellsafterinfectionwith either d21ord61localizedtothe
nucleus,
andlittle ifanyICP8 couldbedetectedin the
cytoplasm
(Fig.
9).
DISCUSSION
Derivation of ICP8-expressing cell lines. Sandri-Goldin et
al. (32)werethe firsttoreporttransformationofmammalian
cells with ICP8 and
expression
of this and other ,B and -yHSV-1genes in these cells. These authors transfected
Ltk-cells withplasmids
containing
the HSV-1EcoRI Ffragment
linked to the HSV-1thymidinekinase(tk)gene and selected
transformed cells exhibiting the TK+ phenotype. In the
presentstudyboth linked transfections
(pKEF-P4-G418)
andunlinked cotransfections (pKEF-P4 plus pSV2neo) were
used in the generation of Vero cells
containing
afunctionalHSV-1 ICP8 gene. The rationale forconstructing
pKEF-P4-G418 was that covalent
linkage
of the two genesmight
increase theprobability of isolatingcell lines
containing
theunselected marker(ICP8 gene). On the other
hand,
because expression of the ICP8 gene in thehybrid plasmid
couldconceivably be affected by the
proximity
ofthe G418 geneand its strongSV40earlypromoter, unlinked cotransfections
2 3 4 5 6 7 8 910
.-
-a
,a,
.4w
41
=
M_Iq)
FIG. 7. Westernblotanalysisof ICP8polypeptides.Infected-cell
polypeptideswerenotlabeled(lanes2to7)orlabeled(lanes 1and 8to10)from 5to18 hpostinfectionwith
[35S]methionine,
separatedby SDS-PAGE, andelectroblottedonto anitrocellulose filter. The
filter was then probed with polyclonal rabbit antiserum to ICP8. Boundantibody was subsequently taggedwith
'251-protein
A, and thefilterwasautoradiographed.Cell lines: Nero(lanes 1to4and8),U-47 (lanes5to7), U-27 (lane9),orU-35 (lane 10). viruses: KOS
(lanes 1, 3, 6,and8), d61(lanes4and7),ormockinfection(lanes 2,
5, 9, and 10). Themultiplicity of infection used was 10 PFU/cell,
exceptfor 20PFU/cell in lane 1.
were
performed simultaneously.
Thesimilarity
in thefre-quency ofisolation of cell lines able to
complement
ICP8ts mutantsby
thetwotransfectionprocedures
demonstratednoclear
advantage
tothe linked system.Complementation
of the ICP8 mutant tsl8by
cellscon-taining stably integrated
copies
of theHSV-1ICP8genehasbeen
reported by
Sandri-Goldin et al.(32).
These authorsassessed
complementation by determining
virusyields
after48 h of
growth
atthenonpermissive
temperaturerather thanby
plaque
assay,as inthisstudy.
Noconstitutivesynthesis
of ICP8 (or other
polypeptides
of theP
andy1
classes)
wasdetected in transformed cells
by
theseinvestigators,
and thestability
of the copy number ofintegrated
genes wassug-gested by widely spaced
assayswhich showednoalterationin the
complementing
capacity
of the transformed cells(32).We report similar
findings
here.Cell lines
inhibitory
towild-type
virusplaque
formation andlategene
expression.
The isolation of the U-27 and U-35 celllines was of
special
interest. Theamplification
of theICP8
sequences transfected into these cells was spontaneous in the sense that an unselected marker which was not
physi-cally
linked to the selected marker wasamplified.
Theamplification
of the transfected ICP8 sequences was not arareoccurrencein that itwasdetected in4% of the cell lines
tested (2 of 48).
Puilm
andKnippers
(25) transfectedLtk-cells with
plasmids carrying
theHSV-1 tkgene and studiedon November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.363.515.76.347.2]DELETION MUTATIONS IN HSV-1 ICP8 1143
1 2 3 4 5 6
:44
4
7 8 9 10 11 12 13 14 15 16 17 18 19 20
*O.4 %, ',-.
*-* 5 I . a.4."
^tr.t- H _
F4
t-7
-~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
I0 1 1 10 I i I0 1 1 10 I I
[image:8.612.165.456.73.550.2]MOCK KOS tsA24 d21 d651 tsA24
FIG. 8. SDS-PAGE of infected-cell polypeptides: comparison of KOS, tsA24, d21, and d61 in Vero and U-47 cells. Infected-cell
polypeptides werelabeled with [35S]methionine from 5 to18 h postinfection. Odd-numbered lanes contain polypeptides from Vero cells; even-numbered lanes containpolypeptides from U-47(ICP8-expressing) cells.Themultiplicity of infection usedwas1 PFU/cell (lanes 3, 4, 7,8, 11, 12, 15, 16, 19, and 20)or10PFU/cell (lanes 5, 6, 9, 10, 13, 14, 17, and 18). Thetemperatureof infectionwas34°C(39.4°Cin lanes
19and20). Lanes 1 and 2, mockinfection; lanes 3to6,KOS; lanes 7to10,tsA24;lanes 11 to14,d21; lanes 15to18, d61;lanes 19 and 20, tsA24. TheICPsarenumberedonthe right. The smaller ICP8polypeptide specified by d21 andd61 isindicatedbyan arrow(del 8).
the number and structure ofintegrated gene copies in 65
resulting cell lines. One of these lines exhibited wide
varia-tioninthe size ofintegrated tksequencesandwasestimated
tocontain85copiesoftkpercell(25). These findings closely
resemble our findings for U-27 and U-35 cell lines, except
that inthepresentcase anunselected markerwasamplified.
Toour knowledge, inhibition of HSV replication in cells
carrying multiple copies of an HSV gene has not been
reported previously. U-27 and U-35 cells offer a unique
opportunity to examine the regulatory role of ICP8 in the
HSV-1 growth cycle. The data presented here constitute
independent evidence for the participation of ICP8 in the
negative regulation of
P,
-yl,and Y2viralgenespostulatedbyothers (10-12), since viral polypeptides of these kinetic
classes were markedly underproduced concomitantly with
overproduction of ICP8.
It isnotable that theinhibitoryeffect of the residentICP8
genein U-27 and U-35 cellswasreducedatahigh
multiplic-ityof infection and that less ICP8wasactuallymade inthese
cellsat thismultiplicitythan atamultiplicityof1 PFU/cell.
i-5 _6
h8
r gB isy
*i pgB
5del8
19 20
014 0 25
I
1* 39 40
VOL.61, 1987
on November 10, 2019 by guest
http://jvi.asm.org/
U-7
MOCK
KOS
d2l
d61
U-47
I~-I
I
i
IL
FIG. 9. Immunofluorescenceanalysis of cellularlocalizationof ICP8polypeptides.Thecell lineusedas acontrol,U-7,wasG418resistant andobtainedaftercotransfection withpSV2neoandpKEF-P4, butit didnotcomplementeitherICP8tsmutant(tsA24ortsA42),nordidit containanyICP8gene sequences(Fig. 3). Infectionand staining conditionsaredescribed in thetext.
Thesameeffectwasobservedin U-47cells. Inasimilarvein, Leiden et al. observed that more tk was expressed in
transformedLtk+ cells infected withatk- mutant ofHSV-1
atamultiplicity of2PFU/cell thanat5 or 10PFU/cell(20).
Sandri-Goldin et al. (32) also reported that the induction of
glycoprotein gB and ICP8 from resident genes in cells
transfected with theEcoRI F fragment of HSV-1 decreased
as the multiplicity of infection increased. Similarly, the
optimum multiplicity ofinfection of induction of
glycopro-tein gC from a gene integrated into mouse cells was 2
PFU/cell,and no gC couldbe detected at a multiplicity of 10
PFU/cell (1). Sandri-Goldin etal. (32) suggested that these
effectscould be due to the virus-induced early hostprotein
shutoff function (15), which might decrease expression of
integratedviral genesasit wouldfor bona fide cellular genes.
On the otherhand, it is notable that at low multiplicity,
unexpectedly large amounts of ICP8 polypeptide were
ex-pressed in U-27 and U-35 cells (even considering the high
gene copynumber), since ICP8 is known torepress itsown
synthesis in the contextof the viral genome(12). This may
reflect the need for an additional viral protein(s) for the
shutoff of ICP8. This protein(s) would not be present in
sufficientamounts tomediatesuppressionduringinfection at
lowmultiplicity.
In additiontotheir utility for studies of the regulation of
HSV-1geneexpression, the U-27 and U-35cell linesshould
prove tobe excellent sourcesofwild-typeICP8protein for
usein antiserum production orother studies.
ICP8 deletion mutants. The primary reason for initiating
the present study was the desire to examine HSV-1 gene
expression in the absence of functional ICP8 without the
complicating factors of leak and reversion. DeLuca et al.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.612.127.492.70.506.2]DELETION MUTATIONS IN HSV-1 ICP8 1145
have shown that a ts form of ICP4 can lower theefficiency of
complementation by ICP4expressed from a wild-type ICP4
gene (7). This interference was not observed in two ICP4
deletion mutants isolated and studied by those authors. Because a similar situation could conceivably occur with respect to ICP8, we constructed the deletion mutants
de-scribed here and compared them with the ICP8 ts mutant
tsA24. Under nonpermissive conditions, the phenotypes
were equallyrestricted for both types of mutant.
Addition-ally, complementation of tsA24, d21, and d61 by resident
ICP8 as seen in thepolypeptidephenotype was much more
efficient at 1 PFU/cell than at 10 PFU/cell. However, unlike
thesituation observedwith ICP4 (7), there was nosignificant
difference in the polypeptide phenotypes of tsA24, d21, or
d61 at either multiplicity of infection. This suggests that the ICP8polypeptide encodedby tsA24 has no significant inhib-itory effectoncomplementationby the wild-type ICP8made
in U-47 cells. An alternative explanation is that if such an
effectexists, itcannotbedetectedbecauseathigh multiplic-ity-when an inhibitory effect would be most readily ob-served-expression of resident wild-type ICP8 is turned
down and therefore complementation is already too
inef-ficentfor further impairment to be detected. This hypothesis
is supported by the fact for all three ICP8 mutants, the
polypeptide phenotypesat amultiplicity of 10 PFU/cell were
similar in complementing (U-47) and innoncomplementing
(Vero) cells (Fig. 8).
Thedefective ICP8 polypeptides specifiedby d21 andd61
were excluded from the cellular nucleus, as were the ts
forms ofICP8 expressed by tsA24 and most other ICP8 ts
mutants(39). For this reason it is unlikely that d21 andd61,
like tsA24 at 39°C, induce the synthesis ofviral DNA. By
contrast to these mutants, tsHAl (4) produces an ICP8
molecule that can associate with the cell nucleus (19),
although abnormally (26). This is consistent with the fact
that there is only a small overlap between the engineered
deletion in d21 and d61 (which eliminates amino acids 328 through587on theICP8molecule[27]) and the mutationsite
in tsHAl (between amino acids 563 and 685; D. Knipe,
personal communication). tsHAl is also DNA negative at
the nonpermissive temperature, indicatingthat this
pheno-type is aconsequenceof alterations inICP8notassociated
withnuclearlocalization.
Whether
the780-bp deletion ind21and d61 includes the nuclearlocalization signalor whether
failuretoreachthe nucleus reflectsafolding problem in the
truncated form of the protein retnains to be determined. Interestingly, virtually no cytoplasmic ICP8 could be
de-tected in U-47 cells infected with either deletion mutant,
suggesting that the wild-type form ofthe polypeptide may
have facilitated thenuclear localization ofthedefective form
ofthe
protein.
Thedeletionmutantsgenerated in this study shouldprove
useful for studies of the in vivo biology of HSV-1, the
influence of the deleted domain on ICP8 DNA-binding
capacity
(19, 30), and interactions of ICP8 with other viralproteins (3, 38).
ACKNOWLEDGMENTS
We thank N. DeLuca for valuable discussions, R. Byrn, N. DeLuca,M.Polvino-Bodnar, andW. Sacks for assistancein many
oftheexperiments,D. Coenfor criticalreadingof themanuscript,
andM. Datzformanuscript preparation.
Thisinvestigation wassupportedby PublicHealthServicegrant CA-21082 from the National Cancer Institute. P.K.O. was
sup-ported by Postdoctoral Fellowship DRG-840 from the Damon
Runyon-Walter Winchell CancerFund.
LITERATURECITED
1. Arsenakis, M., L. F. Tomasi, V. Speziali, B. Roizman, ahdG.
Campadelli-Fiume.1986.Expressionandregulationof
glycopro-tein C gene ofherpessimplex virus1 resident inaclonal L-cell line.J. Virol. 58:367-376.
2. Burnette,W.N.1981.Westernblotting:electrophoretictransfer
ofproteinsfromsodiumdodecylsulfate-polyacrylamide gelsto
unmodified nitrocellulose and radiographic detection with an
antibody and radioiodinated protein A. Anal. Biochem. 112:
195-203.
3. Chiou,H.C.,S. K.Weller,and D. M.Coen. 1985.Mutations in
the herpes simplex virus major DNA-binding protein gene leading to altered sensitivity to DNA polymerase inhibitors. Virology145:213-226.
4. Conley, A.J.,D. M.Knipe,P.C.Jones,and B.Roizman.1981. Moleculargeneticsofherpes simplexvirus. VII.
Characteriza-tion of a temperature-sensitive mutant produced by in vitro
mutagenesisand defective inDNAsynthesisandaccumulation
of-y polypeptides. J. Virol. 37:191-206.
5. DeLuca,N., D. Bzik, V. C. Bond, S. Person, and W. Snipes. 1982. Nucleotide sequences of herpes simplex virus type 1
(HSV-1) affecting virus entry, cell fusion, and production of
glycoprotein gB (VP7). Virology 122:411-423.
6. DeLuca, N. A., M. A. Courtney, and P. A. Schaffer. 1984.
Temperature-sensitive mutants in herpes simplexvirustype 1
ICP4permissiveforearlygeneexpression.J.Virol. 52:767-776.
7. DeLuca,N.A.,A.McCarthy,and P. A.Schaffer.1985. Isolation andcharacterization of deletionmutantsof HSV-1 in the gene
encodingtheimmediate-early regulatory proteinICP4. J. Virol.
56:558-570.
8. DeLuca,N.A.,and P. A. Schaffer. 1985.Activation of
immedi-ate-early, early, and late promoters by temperature-sensitive
and wild-type forms of herpes simplex virus type 1 protein
ICP4. Mol. Cell. Biol. 5:1997-2008.
9. Fenwick,M.L.,M.J. Walker,andJ.M.Petkevich. 1978. On the association of virus proteins with the nucleus of cellsinfected with herpes simplex virus.J. Gen. Virol.39:519-529.
10. Godowski,P.J.,and D. M.Knipe.1983.Mutations in themajor
DNA-bindingproteingene ofherpessimplexvirustype1result
in increased levels of viral gene expression. J. Virol. 47:478-486.
11. Godowski, P. J., and D. M. Knipe. 1985. Identification ofa
herpes simplexvirusfunction thatrepresseslategene
expres-sionpriortoDNAreplication.J. Virol. 55:357-365.
12. Godowski,P.J.,and D.M.Knipe.1986.Transcriptionalcontrol
of herpesvirus gene expression: gene functions required for
positive and negative regulation. Proc. Natl. Acad. Sci. USA
83:256-260.
13. Goldin, A. L., R. M. Sandri-Goldin, M. Levine, and J. C. Glorioso. 1981. Cloning of herpes simplex virus type 1 se-quencesrepresentingthewhole genome. J.Virol.38:50-58.
14. Gray,C.P.,andH.C.Kaerner. 1984.Sequenceof theputative
origin ofreplication in the ULregion ofherpes simplex virus
type 1ANG DNA. J.Gen. Virol. 65:2109-2119.
15. Hill, T. M., R. R. Sinden, and J. R. Sadler. 1983. Herpes
simplex virus types 1 and 2 induce shutoff of host protein
synthesis by different mechanisms in Friend
erythroleukemia
cells. J.Virol. 45:241-250.16. Holland, L. E., R. M. Sandri-Goldin, A. L. Goldin, J. C.
Glorioso, and M. Levine. 1984. Transcriptional and
genetic
analysisof theherpessimplexvirus type1genome:coordinates 0.29to0.45. J.Virol. 49:957-959.
17. Johnson, D. A., J. W. Gautsch, J. R. Sportsman, and J. H. Elder. 1984. Improvedtechnique utilizingnonfactdry milk for
analysisofproteinsand nucleic acids transferredto
nitrocellu-lose.Gene Anal.Tech. 1:3-8.
18. Knipe, D. M.,and A. E. Spang. 1982. Definitionofaseties of
stages in the association oftwo herpes viral
proteins
with the cell nucleus. J. Virol. 43:314-324.19. Lee, C. K., and D. M. Knipe. 1983. Thermotabile in vivo
DNA-bindingproteingene ofherpessimplexvirustype1results
in increased levels of viral gene
expression.
J. Virol. 46: VOL.61,1987on November 10, 2019 by guest
http://jvi.asm.org/
909-919.
20. Lelden, J. M., R. Buttyan, and P. G. Spear. 1976. Herpes simplex virusgeneexpression in transformed cells. I. Regula-tion oftheviralthymidine kinasegeneintransformedLcells by productsof superinfectingvirus. J. Virol.20:413-424. 21. Ljttler, E., D. Purifoy, A. Minson, and K. L. Powell. 1983.
Herpes simplex virus nonstructural proteins. III. Function of themajor DNA-bindingprotein.J. Gen. Virol. 64:983-995. 22. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular
cloning: alaboratorymanual, p. 382-389. Cold Spring Harbor Laboratory, Cold Spring Harbor, N. Y.
23. Morse, L. S., L. Pereira, B. Roizman, and P. A.Schaffer. 1978. Anatomyofherpessimplexvirus(HSV)DNA. XI.Mapping of viralgenesby analysis of polypeptides andfunctions specified by HSV-1 x HSV-2 recombinants.J.Virol.26:389-410. 24. Powell, K. L., E. Littler, and D. J. M. Purifoy. 1981.
Nonstructural proteins of herpes simplex virus.II.Major virus-specific DNA-binding protein.J. Virol.39:894-902.
25. Pfiln, W., and R. Knippers. 1985. Transfection of mouse fibroblast cells withapromoterless herpes simplex virus thymi-dine kinasegene: number of integrated genecopiesand struc-ture of single and amplified gene sequences. Mol. Cell. Biol. 5:295-304.
26. Quillan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location ofaherpesviral DNA-binding protein is determined by the status of viral DNA replication. Cell 36: 857-868.
27. Quinn, J. P., and D. J. McGeoch. 1985. DNAsequenceofthe region inthegenomeofherpessimplex virustype 1containing the genes for DNA polymerase and DNA-binding protein. Nucleic Acids Res. 13:8143-8163.
28. Rixon, F. J., M, E. Campbell, and J. B. Clements. 1984. A tandemly reiteratedDNA sequencein thelongrepeatregion of herpes simplexvirustype 1foundinclose proximityto imme-diate-early mRNA 1.J. Virol. 52:715-718.
29. Ruyechan, W. T. 1983. Themajorherpes simplex virus DNA-binding protein holds single-stranded DNA in an extended configuration. J.Virol.46:661-666.
30. Ruyechan, W. T., and A. C. Weir.1984. Interaction withnucleic acids and stimulation of the viral DNA polymerase by the herpes simplex virus type 1 major DNA-binding protein. J.
Virol. 52:727-733.
31. Sacks, W. R., C. C. Greene, D. P.Aschman, and P. A.Schaffer. 1985.Herpes simplex virustype 1ICP27 isanessential regula-toryprotein. J.Virol.55:796-805.
32. Sandri-Goldin, R. M., A. L. Goldin, L. E. Holland, J. C. Glorioso, and M. Levine. 1983. Expression of herpes simplex virus P and -y genes integrated in mammalian cells and their induction byana geneproduct, Mol. Cell. Biol. 3:2028-2044. 33. Schaffer, P. A., V. C. Carter, and M. C. Timbury. 1978.
Collaborativecomplementation study oftemperature-sensitive mutants ofherpes simplex virus types 1 and 2. J. Virol. 27: 490-504.
34. Smith, H. 0. 1980. Recovery of DNA from gels. Methods Enzymol. 65:371-380.
35. Southern, K. M. 1975. Detection of specific sequences among DNAfragments separated by gel electrophoresis.J.Mol. Biol. 98:503-517.
36. Southern, P. J., and P. Berg. 1982. Transformationof mamma-lian cells to antibiotic resistance with a bacterial gene under control of theSV40 earlyregionpromoter.J. Mol.Appl. Genet. 1:327-341.
37. Spear,P.G.,and B.Roizman.1980.Herpessimplexviruses,p. 615-745. In J. Tooze(ed.), DNA tumor viruses. ColdSpring HarborLaboratory,Cold Spring Harbor. N.Y.
38. Vaughan, P.J.,L. M.Banks, D. J. M.Purifoy, and K. L. Powell. 1984.Interactions between herpes simplex virusDNA-binding proteins.J.Gen. Virol. 65:2033-2041.
39. Weller, S. K., K. J. Lee, D. J.Sabourin, and P. A. Schaffer. 1983. Genetic analysis of temperature-sensitive mutantswhich define the gene for the major herpes simplex virus type 1 DNA-binding protein. J. Virol. 45:354-366.
40. Weller, S.K.,A.Spadaro,J. E.Schaffer, A. W. Murray, A. M.
Maxamn, and P. A. Schaffer. 1985. Cloning, sequencing and
functional analysis of oriL,anHSV-1 origin ofDNAsynthesis. Mol. Cell. Biol.5:930-942.