0022-538X/92/042195-13$02.00/0
Copyright ©)1992, AmericanSocietyforMicrobiology
A
Deletion
in
the Simian Virus 40
Large
T
Antigen Impairs
Lytic
Replication
in
Monkey
Cells In Vivo but Enhances
DNA
Replication
In
Vitro:
New
Complementation
Function of
T
Antigen
CATHARINAMAULBECKER,"12t IANMOHR,3* YAKOV GLUZMAN,3§ JAMES
BARTHOLOMEW,1
ANDMICHAELBOTCHAN2*Laboratory of Chemical Biodynamics, LawrenceBerkeleyLaboratory,1andDepartment ofMolecular and CellularBiology,2 University of California, Berkeley, Califonia 94720, andColdSpringHarborLaboratory,
ColdSpring Harbor, New York J7243 Received 21 August 1991/Accepted6January1992
Wedescribe a new complementationfunction within the simian virus 40 (SV40) Agene. This functionis required for viral DNAreplication and virusproduction in vivobut, surprisingly, does notaffectanyof the intrinsic enzymatic functions of T antigen directly required for in vitro DNA replication. Other well-characterizedSV40 T-antigenmutants, whetherexpressedstablyfromintegratedgenomesorincotransfection experiments, complementthesemutantsfor in vivo DNAreplication and plaqueformation. ThesenewSV40 mutantswereisolatedandclonedfrom human cells whichstablycarrythe viral DNA. The alteration in the large-T-antigengenewasshownbymarkerrescueand nucleotidesequenceanalysistobeadeletion of 322bp spanningthesplice-donorsite of the firstexon,creatinga 14-amino-aciddeletion in thelargeTantigen.The mutantgene wasexpressed in H293 human cells fromanadenovirus vector,and theproteinwaspurified by immunoaffinitychromatography. The mutantprotein directs greater levels of DNA replicationin vitro than does thewild-type protein. Moreover,themutantprotein reduces thelagtime for in vitro DNAsynthesisand
canbe diluted tolower levels thanwild-type Tantigen and stillpromote good replication,which is in clear contrast tothe in vivo situation. These biochemical features of theproteinareindependentof thesourceof the cellularreplicationfactors(i.e., HeLa, H293,COS7,orCV1cells)and the cells from which theTantigenswere purified.ThemutantTantigendoesnottransform Rat-2 cells.Several different models whichmightreconcile thedifferences observed in vivo and in vitroareoutlined. Weproposethat the function of Tantigen affected
preparescells forSV40 replication by activationofalimitingcellularreplicationfactor.Furthermore, alink
between the induction ofacellular replicationfactor and transformation by SV40is discussed.
Simian virus 40(SV40)wasfirst isolated from cultures of rhesus monkey kidney cells, where it was described as a
"nondetectable" passenger. Indeed, it was originally be-lieved thatSV40was analmostubiquitous contaminant ofall such primary or established lines and that no detectable cytopathiceffects could beobserved(61). The titers of virus producedwereatbest 2to3 orders ofmagnitude lowerinthe homologousrhesussystemrelativetovirusproductioninthe Africangreenmonkeyheterologoussystem.Furthermore,in another isolation of the virus, it was reported that SV40
could persist formany months after infection of virus-free homologous or heterologous primate cells such as human
cells without having any visible effect (27). These early studies indicated that theSV40 lytic replicationresponse can
behighlyattenuated andby implication maybearegulated
process.Little is understood ofthese attenuationprocesses,
which in principle could be achieved by the production of large numbers of defective "interfering" viral templates or bymodulationoflimiting viralor hosttrans-acting
replica-tion factors. While someevidence in support of theformer
* Correspondingauthor.
tPresent address: Department ofMolecularCellBiology,
Max-Planck Institute for Biophysical Chemistry, 3400 Gottingen, Ger-many.
t Presentaddress:DepartmentofMolecular andCellularBiology,
UniversityofCalifornia,Berkeley, CA 94720.
§Present address: Lederle Laboratories, American Cyanamide
Co.,PearlRiver,NY10965.
possibility is available(46, 57), most speculation about this invivo attenuation of the natural (often called semipermis-sive) SV40 replication process is based on the enormous
amount of biochemical information concerning the lytic growth of SV40 in the permissive African green monkey kidney cells. These many points for regulation can be incorporated into putative scenarios for the regulation of virus production but give little evidence of the host cells' involvement in the regulation of the viruses' replicative functions thatwere evident from theearly studies.
Thekey viral replication protein is the large T antigen. It autoregulates itsownsynthesis andis itselftranslated from different mRNAs whichhave intrinsically different transla-tion efficiencies (3, 31). The rateor amount of synthesis of thissite-specific DNA-binding protein, which is involvedin the unwinding of the DNA at the viral origin of DNA replication (30), could bepart of the modulation processes
referred to above. Phosphorylation and dephosphorylation oflargeTantigen bycellularenzymes maybepivotal control points for regulation (43b). T antigen produced in Esche-richia coli cells is weak in certain assays in its abilities to
bind to the SV40 origin ofDNA replication and to aid in initiating DNA replication in vitro (43a). After specific phosphorylation at threonine 124 by cellular maturation-promoting factor kinase activity, the T-antigen protein is efficient at DNAbinding and showsrobustactivity in initi-ating DNA replication (42). Moreover, it has been shown that certain phosphorylation sites must negatively regulate theactivityof Tantigen (43b), as the cellularprotein called
2195
on November 9, 2019 by guest
http://jvi.asm.org/
replication factor C required in the early or lag period steps of in vitroSV40 DNA synthesis is a phosphatase (63). These findings suggest that in vivo, persistent SV40 infection relies on a virus-cellinteraction more in tune within a normal cell cycle control than would necessarily be an a priori extrapo-lation from the rather immediate response well characterized in the lytic cycle.
Modulation of host cell factors by SV40 infection, the involvement of these factors in the control of viral DNA synthesis, and the link between these functions and cellular transformation form an area of intense interest. For exam-ple, the well-known and well-studied role of the virus in oncogenic transformation may be a direct consequence of the ability of the large T and small t antigens to prepare a resting cell for S phase by inducing cellular factors necessary for cellular DNA replication. These same cellular factors probably participate in viral DNA replication, as it is only in S phase that the viral DNA replicates efficiently (6, 51). Here, it may be interesting to point out that the large T antigen can bind to at least five cellularproteins. Among the cellular proteins that interact with the T antigen are the retinoblastoma (Rb) gene product(7),p118/120 (10), and the p53 protein (34). There isevidence that these three cellular proteins play some role in repressing cellular progression intothe S phase (8), which in the case of the Rb protein was suggested to be mediated by differential phosphorylation throughout the cell cycle (2, 4, 8, 64). Thus, an interesting antagonism between the Tantigen's innateability to stimu-latesynthesis of DNA and these cellular protein's abilities to indirectly, or directly, in the case ofp53 (14, 60, 65), inhibit DNA synthesis may play afurther role in attenuating viral DNA synthesis. However, to date, mutations in T antigen which reduce or abolish Rb or p53 interactions have no measured effects oninvivo virus production(28,34, 49, 69). It is, however, entirely possible that these functions are redundant for high-level viral replication in cell culture and thatdouble mutants in the "Rb pocket" region of T and the p53-binding site would have a phenotype for virus
produc-tion. Alternately, such mutants may become lytic in infec-tions of rhesus cells.
In addition to the ability of T antigen to modulate the activity of cellular factors by direct binding to cellular proteins, both T antigens are known to activate the tran-scription of cellular genesby transactivation(29,37, 44,69).
This trans activation may enhance or be necessary for transformation and DNAreplication. Akeyfocus, then, for the presentstudiesofSV40DNAreplication is what cellular
factors involved in viral replication are indeed modulated during the cell cycle and which of these activitiesare inturn affected by the well known trans-activation functions of large Tantigen.
From the above perspective, we have been interested in the SV40 genomes carried by the human cell lines GM637 and XP12ROSV. Unlike most postcrisis SV40-transformed human cell lines, which contain replication-defective inte-grated SV40 genomes, both of theselines have beenreported
to carry persistent multicopy viral plasmids (33, 41, 52)
capable of replication in human cells. These established SV40 cell lines originate from a viral infection of the host cells. Because the free viral DNA replicates extrachromo-somally(41), the viralrepliconper se mustindeed beactive. Wehadpresupposed that if these cellsremainpermissivefor viral DNA replication, either interesting new mutations in the viral replication systemhad occurred orthe cells them-selves had established some potent way ofattenuatingviral runaway DNA replication. Studies with both stably
trans-formed simian cells (19, 39) and human cells(16) show that mutations whichinactivate thereplicationfunctions arisein response to this pressure and lead to defective integrated
viral genomesincapable of spontaneous excision. Selection for such mutants is clearly provided by these systems, as viralreplication leadsto celldeath. Somehuman cell lines maintainviral DNAextrachromosomally inaprecrisisstate (15, 70), and if such disabling mutations do notarise,the cell lines do not survive (16,45).
Thus, stably transformed lines such as GM637 and XP12ROSV carrying replication-competent SV40 genomes are interesting material for investigating attenuation of a replication-competent SV40 genome. We have cloned the free SV40 genomes from these human cell lines and found
that they retain a limited replication efficiency. They are indeedmutantforms of the viruswithdramaticallyreduced DNA replication effectiveness when introduced into the normally permissive monkey cells. Moreover, persistent plasmid DNA can be detected in transfected CV1 cells, though lysates ofthese culturesdonotproduce virusstock
witha measurable titer. By avariety of geneticrescue and
inactivation studies,weshow thatthe defect forreplication
in monkey cells lies within the first exon of the A gene.
Unexpectedly, themutant T-antigen protein showsactivity
better than that of wild type forreplicating SV40 DNA in cell-free in vitro assays.Therefore,this function of Tantigen
does not seem to involve roles of the protein normally
addressed by present in vitro DNAreplication assays, and we propose that the alleles of the gene define aninductive function of Tantigenwhichcanelevate ormobilizeabasal
cellular activityneeded forDNAreplication.Asthemutant genomes are defective for transformation, it seems likely that this function linksanimportant(andunknown) function
necessaryfor both activities.
MATERIALS ANDMETHODS
Cell lines. The GM637-b cell line wasobtained from the National Institute of General Medical Sciences
(Human
Genetic Mutant cell
repository).
Thesecells will be referred tohereasGM637cells. GM637 cellswereproduced
by
SV40transformation of human skin fibroblasts and isolated as a clonal cell line
(National
Institute of General MedicalSci-ences catalog of human cell
lines).
XP12ROSV cells were obtained from J. Cleaver;theyare asubclone of the cell line called XP12RO (52). The cell lines were derivedby
SV40 transformation of skin fibroblasts(XP12RO
cells)
from a patientcarryingtheautosomal recessive disease xeroderma pigmentosum(47, 52). CV1-P,COS7,H293,and HeLa cells were obtained from the Cold Spring Harbor Cell Culture Facility.The GM638cellline has beendescribedpreviously
(16). CV1-H cells were agiftfromMelvin
DePamphilis.
All cells were passaged in Dulbecco modifiedEagle
mediumsupplementedwith 10%fetal calfserum.
GMSV40 and XPSV40 DNA refer to the extrachromo-somal DNAisolated byHirtextraction
(26)
ofGM637 and XP12RO cells,respectively.
COS 7 cells are SV40-trans-formed CV1 cells that containreplication-defective
SV40genomes but
constitutively produce
large
amounts of Tantigen (17). Human 293
(H293)
cellsare humanembryonic
kidney cells transformed by transfection with adenovirus 5
fragments (21). H293 cells express the
early
adenovirus transforming genes ElA and E1B. HeLa cells arecervical carcinoma cells that express humanpapillomavirus
type 18transforming proteinsE6 and E7
(55).
Plasmids and constructs. The recombinant
plasmids
on November 9, 2019 by guest
http://jvi.asm.org/
pGMSV40 and pXPSV40 were constructed by cloning the
extrachromosomal SV40 genomes isolated and gel purified
from the Hirt supernatant ofGM637 and XP12ROSVcells, respectively, intopUC19 (obtainedfrom NewEngland
Bio-Labs) at the BamHI site. pJYM is
wild-type
SV40 clonedinto pML1 attheBamHI sites (38). To create adenovirus-SV40 hybridvirus, amodified adenovirus vector wasused (16a). pAC4C+pTGX1 is an adenovirus 5 derivative that lacksElA andE1Bcodingregions
(18, 59)
butcontainsthe T antigen under the control of the adenovirus major late promoter.Toreplace thewild-type T-antigensequencewith the GMSV40 T-antigen sequence, GMSV40 DNA was cleaved with StuI, BamHI, andNciI. The DNA sequenceextending from SV40 positions 5190 to 2534 was inserted into the vector. The vector was first hydrolyzed with HindIII, and then the ends were filled in with Klenow
polymerase.Then thevector wascleaved withBamHI,and themutantfragmentwas ligatedinto thevector. Forthe in vitro replication studies, a wild-type SV40 plasmid called
pwtSV40 (which contains the entire SV40 genome) and a mutant derivative (p8-4) of pwtSV40 (which contains a defect in the origin of replication) were used as templates (59).
Transfections. For DNAtransfections, we used the
cal-ciumphosphatecoprecipitation method (22)with modifica-tions suggested by Weber and Schaffner (66). Cells were transferred1daypriortotransfectiontogiveamonolayerof cells 80% confluent on the day they were transfected. Plasmid DNAwasdigestedtorelease andlinearize theSV40
insertpriortotransfection of theunligatedDNA. In cotrans-fectionexperiments, 2 ,ug each ofthe DNAsof interestwas used,while inexperimentstoproduce infectiousvirus(e.g.,
Table 1), 1 ,ug wasused. TheDNAprecipitatewasexposed tothe cells for 10 h. The cellswerethen shocked with 15% glycerol-Dulbecco modified Eagle medium for 1 to 3 min
depending on the cell line used, rinsed with phosphate-buffered saline solution, and incubated with fresh medium. At12 hpostshock,the cellsweresplitif necessary. Thefirst
time point is calculated from the time elapsed after the
glycerolshock.
Extrachromosomal DNA was isolated from the Hirt
su-pernatantandpurified byRNase andproteinase digestsand repeated phenol extractions following standard procedures. One quarter of the DNAwasdigested overnightwith MboI
(New England BioLabs)andanalyzed bySouthern analysis of 1% agarose gels using nick-translated 32P-labeled SV40 DNAas aprobe.
Intransformation assays, Rat-2 cells(5 x 105 cells) were transfected with 1 p,g of SV40 DNA and stained with methylene bluefor focus formation at 3 weeks posttransfec-tion.
Purification of GMSV40 and wild-type SV40 T antigens. TheT-antigen expression plasmidwascotransfectedwith a
fragment of adenovirus DNA containing sequences from map units 4 to 100 into H293 cells by calcium phosphate
coprecipitation. Virus plaques were picked from the H293 cell infectedmedium, and thelysate was assayed for
T-an-tigen protein by radioimmunoassay with PAb 405 anti-T
antibody (24, 25)and for SV40 DNA by dot blot hybridiza-tion with labeled SV40 DNA. The H293 lysate, which contained eitherAd5SVRII2 (wild-type T-antigen
recombi-nant) or Ad5SVRII2/GM (mutant T-antigen recombinant), was used to infect H293 cells for protein expression. The mutant virus stock was plaque purified on H293 cells. To extract Tantigen from HeLa Spinner cells, the cells were coinfected with the wild-type adenovirus helper and the
defective virus stock. Inacontrolexperiment,Tantigenwas expressed by transfection ofpGMSV40 and pwtSV40 into CV1 cells. T-antigen localization and production of the mutant andwild-type proteinswere thus measured sideby side. The protein waspurified byimmunoaffinity chromatog-raphy as described previously (56, 59). Protein concentra-tions were measuredbythe Bradford assay(la)with bovine serumalbumin as astandard.
Preparation of in vitro replication extracts. Cytoplasmic replication extracts were prepared from HeLa and H293 cells bysuspension inhypotonic buffer and Dounce homog-enization (59). CV1-H cell replication extracts were
pre-pared as described byDecker et al. (9) with the following modifications. Cellswere preincubated for 5 min at 4°C in hypotonic buffer and thenscrapedfree witharubber police-manandlysedinaDouncehomogenizerwith 15 strokes of pestle B. The hypotonicbuffer containedacetate instead of chloride salts(20mMHEPES
[N-2-hydroxyethylpiperazine-N'-2-ethanesulfonicacid;pH7.5],5 mMpotassiumacetate, 1.5 mMmagnesiumacetate,0.5 mMdithiothreitol).The cells werelefton icetoincubate for 45 min and thencentrifuged in aSorvallHB4 rotor at10,000 rpm for 10 min. The protein
concentration (5 to 8 mg/ml) was determined as described above. The supernatant, which contains the cytoplasmic
extract, was frozenon dry ice-ethanol and stored in liquid
nitrogen. Prior to use in the in vitroreplicationreactions,the extracts were spun for 10 min at 12,000 rpm, and the supernatantwas used for thereplication assay.
In vitro replication reactions. Reactions were performed
essentially as described by Stillman and Gluzman (59).
Replication reaction mixture volumes of 50
Ru
contained40mMHEPES (pH 7.5); 8 mMMgCl2;0.5 mMdithiothreitol;
100,uMeachdCTP, dGTP,and dTP;2.5 ,uMdATP; 25 ,uM
[a-32P]dATP
(1,000 cpm/pmol); 200 FM CTP, GTP, and UTP; 40 mM creatine phosphate; and 1jig
of creatinephosphokinase(Sigma) asan ATP-generating system. Typ-ical reaction mixtures contained 300 ng of template DNA
(pSV origin plus andpSV origin minus), 1
jig
of Tantigen,and150 jigofcytoplasmicextracts. pSV origin-minus
plas-midwasusedto measurethe amountofrepair synthesisand to correct the number of picomoles obtained from pSV
origin-plus replicationto obtain absolutereplicationvalues. The reaction mixtures were incubated at 37°C for various amounts of time, and reactions were terminated by the addition of EDTA to 10 mM and carrier DNA(50 jig).The DNA was precipitated by the addition of ice-cold 8%
tri-chloroaceticacid-1%sodiumpyrophosphateontoWhatman
GF-C glass fiber filters. The specific activity of the newly
synthesized DNA was determined by scintillation counting and corrected for the amount ofrepair synthesis obtained
withpSVorigin-minus plasmid. RESULTS
Replication properties of plasmid DNA. The free plasmid DNAs persisting in the human cell lines GM637 and XP12ROSV arehomogeneous but different from each other. This wasfirst indicated by the electrophoretic behavior of the DNAs inSouthern blotexperiments, in which only single species of supercoiled DNAs could be detected. We noticed thatrestriction fragments spanning the A gene were smaller thanthose of wild type. The HindIII-B fragment in particular was diagnosticfor a deletion of approximately 320 bp. The HindIII-B fragments of both the GM637 and XP12ROSV genomes were distinct from each other in that the GM637 HindIIIfragmenthadamobilitycorrespondingtothat ofan
on November 9, 2019 by guest
http://jvi.asm.org/
TABLE 1. Plaque assays onCV1 cellsa
SV40 No.of
construct plaques/dishb
pA... 0
pB... 11,400
pC...0
pD... 11,000
pGMSV40... 0
pXPSV40... 0
pwtSV40... 12,000
pJYM... 21,000
Medium fromuntransfected cells ... ... 0 aIndicated plasmid DNAs (1 jigeach)were hydrolyzedwithBamHI to
releasetheSV40DNAfromtheplasmidvector andtransfectedinto CV1 cells asindicatedinMaterials and Methods. At 120 hposttransfection, the plates wereharvestedand afreeze-thawlysateof virusstockwas obtained. Plaque
assays wereperformedonCV1 cells.Inthecase ofpwtSV40,pJYM, pB, and
pDconstructs, a100-folddilution of the medium fromthe120-h time pointof
the transient replication assay (Fig. 1)wasusedto infect CV1 cells. The
number of plaques obtained was corrected for the 100-fold dilution.
pGMSV40, pXPSV40, pA, and pC mediawere usedundiluted.
bFromtransfectedcells.
A
GM wt sv8012 24 3641182018 2243648120 2 2436 120
Inptu
L;nears-.
B GM4 SV80
12 24 36 48 120 189
wt+ SV80
12 243648 120
CVGM
1 0weeks
846-bp fragment, while the XP12ROSV fragment
electro-phoresed as an 864-bp fragment. Thewild-type fragment is
1,169 bp. The homogeneity of the Hirt-extracted plasmid DNAspersisted overcontinued cell passages and was con-firmed by sequence analysis across the end points of the
HindIII-B fragment. To analyze SV40 plasmid DNAs in the human cell lines GM637 and XP12ROSV, we cloned the DNAs extracted from Hirt supernatants after hydrolysis withBamHI. We cloned the plasmidDNAs from
indepen-dent Hirt extracts, and these results substantiatedthe
homo-geneity of the extrachromosomal DNA. We refer to these DNAs as GMSV40 from GM637 cells and XPSV40 from XP12ROSV cells.
To characterize the biological potential of these cloned genomes,weasked if the cloned DNAs could supportplaque production in CV1 cells. The cloned viral DNAs were released from the plasmids and transfected into CV1 cells in anattempttoproduceinfectious stocks. Lysates from these cultures were then assayed for infectious virus by plaque
assay(Table 1).Whileneitherplasmidwould form infectious virusasmeasured by plaqueassayon CV1cells, wefound that the plasmids could establish themselves as a latent
episome without apparent cell
lysis
for at least 10 serial passagesof the transfected cultures(Fig. 1B[CVGM]).
For this laterexperiment,weharvested the transfectedCV1 cells andsplitthem1:3 each week for 10 weeks.Again,infectious viruswere not detected inthe medium from these cultures afterthis mediumwasused inplaqueassayson CV1 cells.We assayed the
replication potential
of the cloned GMSV40 and XPSV40DNA in CV1 cellsby using a tran-sient replication assay. The input unreplicated DNA was distinguished from replicated DNA by digestion with the methylation-sensitive restriction enzymeMboI.TheplasmidDNAreplicatedin theE. coli hostcannotbehydrolyzed by
MboI
because ofspecific methylation. Afterreplication
ineucaryotic cells, the methylation pattern is altered and the DNA becomes susceptible to MboI digestion. Replication wasmonitored by the appearance of MboI restriction frag-ments on Southern blots thatwere probedwith nick-trans-lated SV40 DNA.
The autoradiograph in Fig. 1A shows transfections into CV1 cells of cloned wild-type SV40, SV80, and GMSV40 DNAs.pSV80isaclonedDNAcodingin part foraTantigen
defective in origin binding. The clone was
originally
con-FIG. 1. Transient replication assays in CV1 cells ofGMSV40 andwild-type SV40genomesand acotransfectionwith the replica-tion-incompetent SV80 genome. (A) Kinetics ofDNAreplication when4pug each of GMSV40, wild-typeSV40,andSV80formIII was transfected into CV1 cells. TheDNAisolated from the Hirt super-natants was digested withMboI.Two of the MboIcleavage sites present inwild-type SV40aredeletedinGMSV40suchthat the two types of DNA can easily be distinguished from each other. The DNA wasanalyzed by gelelectrophoresison1%agarosegels and Southern blothybridizationto32P-labeled SV40DNA.Input MboI-insensitive form III DNA is visible in the lanes asthe5,243-bpband in thewild-typetransfection and the4,920-bpband in theGMSV40 transfection. Replicated SV40DNA iscleavedby MboI. (B)Data for complementation by SV80 cotransfection. Cotransfections of GMSV40orwild-type SV40 with SV80 contained2,ugof each ofthe SV40 genomes per dish. Lane CVGM shows the Hirt-extracted DNA 10 weeksposttransfection byGMSV40 DNAalone.
structed from the integratedSV40 sequences rescued from thehuman cell lineSV80(16).At 12hposttransfection,there was no SV40 replication in either wild-type SV80 or GMSV40-transfected CV1 cells. At 24 and 36 h, prior to virion formation, both wild type and GMSV40 began to replicate,butGMSV40replicationwassignificantlyreduced
comparedwith that of wild type, despite equalamounts of input DNA. The SV80DNAfailstoreplicate aspreviously reported (16). Inmultiple experimentsof this sort,wefound that wild-type SV40 was at least 20-fold better than the mutant in its ability to replicate in CV1 cells. Similar deficiencieswerefound for theXPSV40mutantgenome(see Fig. 8).Inall theseexperiments,itwasimportantto measure
w &M
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.64.308.79.197.2]5243J0
BamH Hi
4550Q1007
524310
Bam H m Fll
4558
O1007
P
A D GMSV4C wiSV40 pJYt XPSV40
2472120 169 210 24 72 120 169 210 24 72120169 210 24 72 120169 210 24 72120169210247212D0169210 24 72120 16921024 72 120169 210
-_
_
.
_
Ff
HindIII
a..A
400
I,
5172 5172
4558 (1X
FIG. 2. Construction ofhybrid clonesfrompGMSV40andpJY. Identicalconstructs weremadewithXPSV40genomes.The resultswere
identicalfor both pXPSV40 andpGMSV40, and forsimplicity,weshowonly the resultsforpGMSV40. Bothplasmidsweredigestedwith PflmI,andthetwobackbonevectorfragmentswereisolatedtogetherwith the4,558-to1,007-bpPflmISV40fragmentfromlow-melting-point agarose.ThePflmIfragment ofpGMSV40wasinserted into thepJYvectorbackbone(pC subclone),and thewild-type pJY-PflmIfragment wasinserted intothepGMSV40backbone(pDsubclone) by cuttingtheappropriatebands from thegel,mixing them,and ligatingthem in low-melting-point agarose. pC and pD were distinguishable from their parent plasmid backbone byPvuII digests, since the vector,in pGMSV40ispUCwhereasthepJYvectorispBR322, while the PflmI inserts differ because of the deletion in the Ageneand the creation ofa newPvuII siteinthepromoter-enhancerregion of GMSV40(68a).ThepC and pJYplasmidsweredigestedwithPflmI,and the isolatedPflmI fragmentsweredigestedwithHindIII. pAwasconstructedby ligatingthepJYbackbonefragmentwith the4,558-to5,172-bpPflmI-HindIII fragment of pCand the5,172-to1,007-bpHindIII-PfimIfragmentofpJY. pBwasconstructed inasimilarmanner, exceptthat thesourceof the PflmI-HindIII fragmentwaspJY,while theHindIII-PflmI fragment camefrompC.PvuIIdigestionwasusedtoconfirmthe identityof the fragments. The GMSV40sequencesin thefigure arerepresented as thicklines, and wild-type sequencesarerepresented bythinner lines. Numberingis withrespecttotheSV40genome.TheBamHIsitesatwhichwild-typeandGMSV40werecloned intopUC(pGMSV40)and pBR322(pwtSV40)areindicated. Therestrictionenzymesindicatedin thefigure simplydenotethe borders offragmentsused in the swaps. The autoradiogram shows the transientreplication assaysof subclones ofpGMSV40 and pwtSV40 in CV1 cells. pA, pB, pC, pD, pGMSV40, pXPSV40, pJY,andpwtSV40weredigested with BamHItoreleasetheSV40 insert andtransfectedby calcium phosphatecoprecipitationinCV1 cells. SV40genomesweretransfected(2 p.g/60-mm-diameterdish), andthereplicatedDNAwasanalyzedby MboIdigestion.
replication rates prior to virion assembly and reinfection. Therefore,weemphasizethecomparisonsbetweenmutants and wildtype atthe 24- and36-hpoints. By120h,reinfection andasecond round ofreplicationcould be detected for the
wild type(Fig. 1;seeFig. 8). It is clear fromFig. 1(seealso Fig. 8) that the GM mutant isdefective for replication, but the data do not preclude a secondary defect in virion
formation. Capsid protein can be detected in CVGMlines,
and virions are produced (40). However, thesevirions are notinfectiousatlowmultiplicity, presumablybecause of the T-antigendefect characterized below. Wenote that lack of titratable infectious viruscan be the resultofa
replication-defective Tantigenor arole for Tantigeninvirionassembly. The datapresentedhere indicate that the DNA is defective forreplication.
Mapping the lesion for DNAreplication in CV1 cells. To geneticallymapthe lesions inthe clonedDNAs,avariety of recombinant SV40molecules weremade. The experiments
whose resultsaregiveninFig.2 and Table 1mapthe lesions
for DNAreplicationandproductionof infectiousvirustothe sameregion,whichspansnucleotides4558to5172ofSV40. Wenotethatthereplicationassayhere issensitivetovirion formation, as the time points are extended compared with those in Fig. 1. This result substantiates the plaque assay
data of Table 1. Interestingly, both the GMSV40 and XPSV40DNAs haddisablingmutations in this region (data notshown). Thus, whenaPflmI-HindIII restriction fragment which spans nucleotides 4558 to 5172was placed into the wild-type SV40 genome (construct pA), 20-fold-reduced
replication levels and no plaque formation were detected. Moreover, when the wild-type fragments spanning this re-gion were inserted into the mutant backbone, wild-type
replication was detected (construct pD), as was plaque
formation (Table 1).
Inordertoidentifytheprecisenatureof themutations,the deleted forms of the mutant HindIII-B fragmentwere
sub-clonedintoM13mpl9vectors,and theSV40 sequenceswere determined. This studyshowed that nucleotides, including
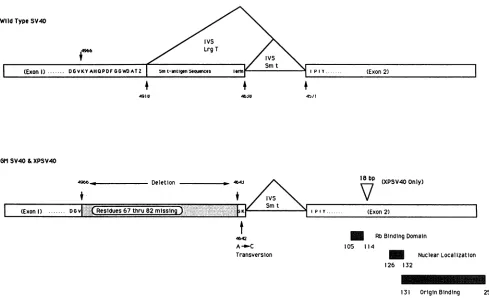
nucleotides 4965 to 4644, are deleted in bothGMSV40 and XPSV40. Thus, both DNAscontainadeletion of 322
nucle-otides that deletes part ofthe codingsequencesforlarge T
antigen and all the sequences that are unique for small t antigen
(Fig.
3). The deletion destroys the large-T-antigensplicedonor but stops short of thesmall-t-antigendonor site. The deletion alsoplaces thesmall-t-antigen stopcodon out of frame with respect to the coding sequence, and thus, a modified large T antigen can be produced. New mRNA
species predicted by the deletion have indeed been verified by
Si
analysis (data notshown). The sequence shows that smalltantigen cannotbeproducedand that largeTantigencontainsadeletion of 16 amino acids (residues 67to82) from the region encoded by the end of the first exon and an additionof 2newamino acids(Tyr-Lys)from sequences that would normally be in the large-T-antigen intron. The net effect isareduction of 14 amino acids in thelargeTantigen. Interestingly, the nucleotides of the large-T-antigen gene thataredeletedatthe5' border of the deletion would code for a Lys-Tyr sequence, and they arereplaced by
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.54.552.70.272.2]WildType SV40
GM SV40&XPSV40
18bp (XPSV40Only)
(Exon I) ... D 6v ..Resldues67 thru 82missin t.MF KS Sm
t
4642
A-n-C
Transversion
I p Iy... (Exon 2)
_ Rb BindingDomain
105 114
m
Nuclear Localization126 132
131 OriginBinding 259
FIG. 3. Genotypesof GMSV40andXPSV40 in comparison with wild-type SV40 small- and large-T-antigen-coding sequences. The map shows theearly region ofSV40with the nucleotidenumbersreferring to positions in the wild-type genome. Letters inside boxes refer to amino acids and indicate both the amino acidslost fromthe wildtypeby the deletion inGMSV40andXPSV40 and theaminoacid changes at the C-terminal end of the firstexon. The nucleotides between nucleotides4966and4643 aredeleted,and two codons from theC-terminalendof the small-t-antigen regionjust before its donor siteareplaced in frame with the large-T-antigen-coding sequencesin both genomes. The deletion of 322bp, thepoint mutationat4,642 bpthatchangesan A to aC,andthe 18-bp duplication at positions 4461 to 4479 in XPSV40
areindicated. Theexonsandintrons of thewild-typesmall andlargeTantigensaregiven for comparison, and exon 2 domains discussed in the text are indicated. IVS,intervening sequence.
tides which encode for the amino acids Tyr-Lys in the mutant protein. This tyrosine codon is constructed by a cytosine-to-adenosine transversion, as shown in Fig. 3. AlthoughtheXPSV40genomecarries the samedeletionas
the GMSV40genome,there isaduplicationof 6 amino acid codons within the secondexon, resultinginanetreduction of 8 amino acids for theXPSV40Tantigenrelativetothat of wildtype.Thisduplicationdoesnotappeartoplayarole in
themutantphenotype, asthe markerrescuefragmentwhich defined the phenotype was the PflmI-to-HindIII fragment (nucleotides4558to5172) stopping just5' of theduplication. To corroborate the predictionsof the sequence data, we
transfected the cloned GMSV40 DNA into CV1 cells in side-by-side experimentswithwild-typeSV40andanalyzed the T antigen produced byboth alleles. Both thewild-type and mutantTantigens accumulated in the nucleus, and no
differences in T-antigen levels could be detectedby immu-nofluorescence. However, asshown in Fig. 4,themutantT antigenwhichwaspurified by immunoaffinity chromatogra-phyfrom CV1 cells(seeMaterials andMethods)wassmaller than thewild-type protein byabout2,000Da.Furthermore, immunoprecipitation using PAb 108 antibody, which is di-rectedat the amino-terminal endof Tantigen (23), showed that neitherGMSV40norXPSV40 producessmall tantigen (datanotshown).
Small t antigen is known to be a transactivator ofgene expression (37).Giventhat thedeletionsin theGMSV40and
XPSV40DNAs eliminate theproductionofsmallt antigen,
we wereinterestedtoknow ifexpressionofsmalltantigenin
transcould rescue wild-type replicationof the two DNAs. Cotransfection with small-t-antigen expression plasmid pW2-t/cDNA (37)didnotrescue or increase thereplication activityof themutants.In theseexperiments, small-t-antigen expression was positive when monitored with PAb 108 in parallel transfections in whichonlypW2-t/cDNAwas used (data not shown). We conclude that the primarydefect of theseSV40genomes lies in thelarge Tantigen andnotin a
lack ofsmall-t-antigen production.
In vitro DNAreplication directedbytheGMSV40T
anti-gen. Several studies have shown that mutations in the first exon oflargeTantigen result inmarkedlyreduced replica-tion rates for the mutant genomes (49, 53, 58). The only known function encoded in the first exon related to DNA replication is abinding domain for ot-polymerase, and it is notknown whether this interaction is critical forreplication (30a).As all of the known biochemical activities oflargeT antigen that are critical for the enzymatic functions of the protein in SV40 DNA replication (e.g., ATP hydrolysis, helicaseactivity, andsite-specificDNAbinding)maptothe second exon, we were interested to explore the in vitro properties of themutantTantigen. To thisend,wecreated adefective adenovirus-SV40 hybridviruswhichcould
pro-duce themutantGMSV40allele oflargeTantigenin human cells (see Materials and Methods). In vivo labeling studies
4b4i
ivs
4910 46JO 45 /
4966 Deletion so
I
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.68.557.70.368.2]MW
GM
wt
MW
...
... ..0
0
0
0
0
E a. 116
97
66
60
...g....
::s 42
FIG. 4. GMSV40Tantigen(lane GM) andwild-typeTantigen (lane wt) isolatedfrommonkey cells. Each laneonthe 5% sodium
dodecyl sulfate-polyacrylamide gel, which was stained with Coomassie blue, contains 1 p,g of T antigen that was produced
throughtransfection of CV1-H cells. Thewild-type-T-antigen pro-teinmigrates slightlyfasterthan the97-kDamolecular sizemarker,
in agreement withapredictedsize of 96 kDa.GMSV40runsatabout 94kDa,presumablybecause of the deletion of amino acids 67to82
(2newaminoacidsareaddedalso,which results inanetlossof 14 amino acids in GMSV40 T antigen). The molecular sizes of the
commercial Bio-Radmarkers(lanes MW)aregiveninkilodaltonsat
theright.
with H293 cells infected with the mutant and wild-type hybrid viruses showed no difference in apparent stability between themutantandwild-typeTantigens. As the appar-entmutationmappedforboth theGMSV40and theXPSV40
genomes wereidentical,wechosetofocuson theGMSV40
Tantigen.
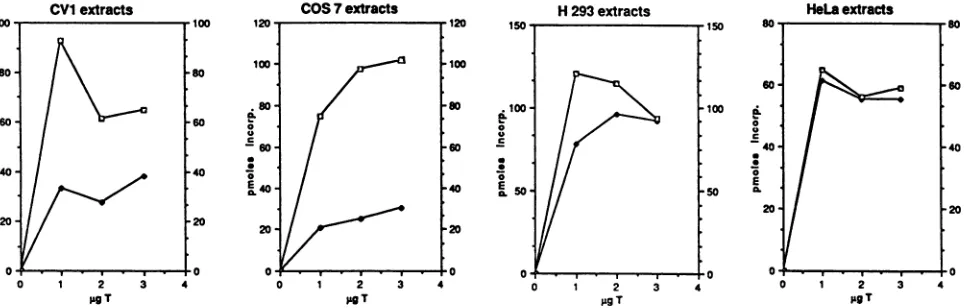
TheabilityofGMSV40and wild- typeSV40Tantigensto direct DNA synthesis in H293 cytoplasmic extracts was
examined (Fig. 5). In this experiment, we determined the
kinetics ofSV40DNAreplicationatdifferentconcentrations of Tantigen. As seen from this data, 0.25 ,ugof T antigen represents a suboptimal protein concentration for stimulat-ingin vitro replication by wild-type Tantigen. The rateof GMSV40 T-antigen-directed invitro replicationat this
con-centration is approximately 25-fold greaterthan wild-type-T-antigen-directed replication. Increasing the amount of T antigento0.5 ,ugperreaction enabledwild-typeTantigento supportreplication. However,GMSV40Tantigenstill sup-ported replication fivefold better than wild-type T antigen after 60 min of incubation. At optimal wild-type-T-antigen concentrations of 1 ,ug, the difference in replication rates between GMSV40 T antigen- and wild-type-T-antigen-di-rected DNAsynthesiswasonly2.5-foldat60min.GMSV40 Tantigen purifiedfromHeLa cells thus isupto25-foldmore
'-0
.5 E
c
a
0
0
E
C.
0
0
E a.
60-
40-
20-04
0
time In min
tinm Inmin
1
gg
20 40
timeInmin
60
40
20
60
FIG. 5. T-antigen concentration-dependentinvitro DNA replica-tion. The numbers ofpicomolesused for thisgraphwerecorrected for theamountofrepair synthesisas describedinMaterialsand Meth-ods. Values below1pmolarelower thanthe value forrepair synthesis and represent background. Symbols:
l,
GMSV40-directed replica-tion; *,wild-type SV40-directed replication.efficient in directing SV40DNA replication in vitro at low T-antigen concentrations.
Alag of 15 to25 minpriortotheonsetofDNAsynthesis wasobserved with 1 ,ugofwild-type T antigen per reaction
__z /
Ir
on November 9, 2019 by guest
http://jvi.asm.org/
CVI exctracts
100- 100
80:
80l60- 60
c
E40- 40
E
20 -20
0 ,, , ,, 0
0 1 2 3 4
pgT
COS7extracts
1 2
pagT
H 293 extracts
2
;igT
HeLa extracts
2 jigT
FIG. 6. Invitro replication inCV1,COS7,H293,andHeLa cell cytoplasmic extracts supported by eitherGMSV40orwild-type SV40 T antigens.Reaction mixes contained 200 ,ug of cytoplasmic extracts, 300 ng of DNA template, and 1, 2, or 3igof T antigen and were incubated for 2 h at 37°C. The picomol values for replication were corrected for repair synthesis. The open squares represent GMSV40 T-antigen-directed in vitro replication, and the solid squares represent wild-type-T-antigen-directed replication.
(Fig. 5), a result in agreement with those of published reports
(59, 67, 68). Only after 30 min at any concentration does wild-type T antigen promote measurable amounts of DNA
synthesis. In contrast, the lag period in DNA replication
observed forsynthesis initiated by GMSV40 T antigen was considerably shortened (Fig. 5). Thus, invitro, GMSV40 T
antigenseemstobemoreefficient thanwild-type T antigen for DNAreplication when comparedonthebasis of concen-trationdependence and effect on presynthesis lag time.
Theseresultsweresurprising, given thestrikingdefects of GMSV40 DNA in vivo in monkey cells. To test whether the replication defects might be specific to simian cells, we prepared replication extracts from a variety of cells: CV1 andCOS 7(simiancells)andH293and HeLa(humancells). In vitro replication was measured at different T-antigen
concentrations, and the results obtained with the different cellular replication extracts are shown in Fig. 6. Standard protocols suggest that optimal replication rates in monkey cellsrequire2to4 ,ugofTantigenperreaction(35).Human cells reach levels ofoptimalDNAsynthesisat0.5to 1 ,ugof T antigen per reaction (59, 67). The monkey cell extracts
CV1and COS 7 contained0.5, 1, 2, and 3 ,ug of Tantigen.
The human cellextractscontained0.25,0.5, 1,and 2 ,ug ofT antigen per reaction. The difference in replication
efficien-cies between GMSV40 and wild-type SV40 T antigens is greatest atsuboptimal proteinconcentrations. We conclude thatat allT-antigen concentrations employed in both mon-key and human cell extracts, the GMSV40 T antigen was moreactive inpromoting in vitro DNAreplication than the
wild-typeTantigen, although thedifferences weregreatest withthesimian extracts(Fig. 7).
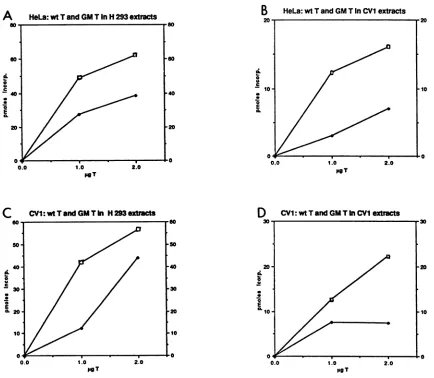
Although the cell extracts used in the above experiment werepreparedfromdifferent cell types, theTantigenswere prepared only from human cells. Apossiblewayto explain
thebehavior of themutantGMSV40 Tantigen in vivo isto invoke specific modifications of the T antigen when it is produced in a specific host cell. Thus, modifications found
specificallyinCV1 cellsmight crippletheGMSV40 Tantigen. Alternatively,specificmodificationsoccurringinthe infected HeLacellsmight activate the Tantigen, which would other-wise bemutantif translated andproduced in simian cells. To
explore thispoint, the adenovirusvector was usedto infect both human and simiancells, and the Tantigens from these sources werepurifiedandassayedasbefore.Asshown inFig.
7, the T antigen from GMSV40 was still more active than
wild-type Tantigen, regardless of the source of the protein. To testthe influence of adenovirus-induced T-antigen
modi-fications, we prepared mutant and wild-type Tantigens by transfection of therespectivepUCcloned genomesinto CV1 cells. Again, the mutant Tantigen was more effective than the wild typeatinitiatinginvitroreplication.
First exondeletion defines a genetically distinct function in the largeTantigen.Althoughthe large-T-antigenprotein isa multifunctionalpolypeptidewith discrete domain activities, intracistronic complementation by different mutants for functions required for DNA replication has not been re-ported. Theseresults may indicate that althoughthe active T-antigen complex at the replication origin functions as an
oligomer, most activities (e.g., ATP hydrolysis, ATP bind-ing,helicaseactivity,andDNAbinding) must be presenton a single polypeptide chain. In fact, some mutations in the secondexonof the Tantigengiveatrans-dominant negative effect,downmodulatingreplication promoted by wild-typeT
antigen(11).Therewas ahint that thismightnotoccurwith thenew mutants characterized here. We reasoned that the
replication effect defined by the GMSV40 and XPSV40 genomes might affect an indirect function of T antigen requiredin vivo.Thiswassuggestedby theunique position
ofthedeletion in the firstexonof the large-T-antigengene. For example, it is conceivable that a unique or specific
function of the first exon of the large-T-antigen gene is carried by those amino acids which are shared by the
small-t-antigen gene, which is not directly involved in the mechanics ofDNAreplication. This notion would then be
compatible with the existence ofa mutant
large-T-antigen
proteinthat is indeedmoreactive thanthewild-typeprotein
with respect to all directfunctions required for DNA
repli-cation. Wewerethus interested in
exploring
thequestion
of separate activities within theT-antigen gene, asother indi-rectreplicationfunctions of Tantigen couldbesupplied by
separate polypeptides incapable of direct replication func-tions.Aprecedencefor
multiple
functionalcomplementation
groupsin the A genewasestablishedbyTornowetal.
(62).
However, the function they mapped was at the
carboxyl
terminus of the protein, is important for
capsid
protein
synthesis, and thus affects a
postreplication
function. To determine whether the mutants described here define a different activity,twodifferent assaysweredone.First,wetransfected theGMSV40 and XPSV40 genomes into SV40-transformed permissive cells harboring
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.76.558.78.232.2]A HeLa: wt T and GM TInH 293extracts
a
0
a.
0 0 -9
10-E a.
poT
C CVI:wtTandGMTIn H293extracts
a.
0
0
E
0.0 1.0 2.0
[image:9.612.90.524.77.455.2]pgT pgT
FIG. 7. Comparison ofDNAreplicationin H293and CV1 cellextractspromotedby either GMSV40(GM T)orwild-typeT(wt T) antigens produced inHeLaorCV1 cells.(AandC)DNAreplicationin H293 cellextracts.GraphA comparesreplicationwithGMSV40andwild-type SV40Tantigens produced in HeLa cellsusingH293extractsin vitro.Graph C showsreplication inH293extractspromoted bybothT-antigen proteins produced in CV1 cells. (B and D) replication rates of the HeLa cell-produced T antigens in CV1 extracts (B) and the CV1 cell-produced T antigens in CV1 cell extracts (D). Open squares represent GMSV40 T antigen-directed replication, and solid squares representwild-type-T-antigen-directed replication rates.
fined T-antigen mutants. C2 cells are simian cells trans-formed byareplication-incompetent SV40 genome (39), as are the human cell lines GM638 and SV80(16). The SV80
line carriesaTantigenwithasingle amino acidsubstitution in theDNA-binding domain oftheprotein,while theC2 and GM638 proteins carry single but different changes in the
ATP-binding-helicase domains of the protein. These cell lines will support wild-type SV40 replication but will not provide a T-antigen source which cancomplement exon 2
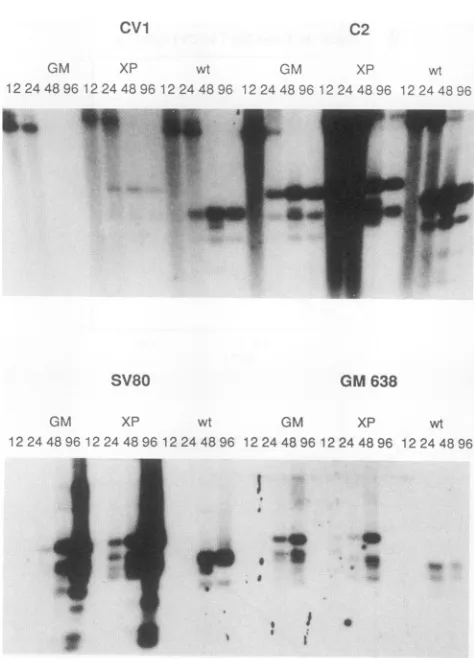
lesions inDNAreplication functions (for example,seeFig.2 in reference 16). As can be seen in Fig. 8, these cell lines
supported the replication of the mutant XPSV40 and
GMSV40genomestolevelsatleastasgreatasifnotgreater thanwhat is found forwild-typeSV40. While recombination between circular DNA and the integrated genomes can occur, this recombination is too rare to account for the immediate and wild-type levels of replication detected in these assays. Furthermore, we note that the pattern of
amplificationdetected issolelythat of the mutant genomes. Forthetransfected C2cells,visiblelysisand cell death with
classical SV40-induced cytopathic effect occurred 1 week after transfection. The harvested virus would not form plaquesonCV1 cells.
Next, we asked whether cotransfection of the GMSV40 DNAwould be complemented bythe replication defect in the cloned mutant SV80 allele of T antigen in a direct transfection of CV1 cells. As can be seen in the bottom section ofFig. 1,the SV80plasmidandtheGMSV40plasmid effectively complement eachother. Multiplerounds of rein-fectionbyvirions andreplicationoccurredduringthecourse of these cotransfection experiments, and the entire mono-layerof CV1 cellslysedat200 hposttransfection.Wecannot effectivelycomparethe relativereplicationefficiencies of the twogenomes, given the resolution of the autoradiogram in
Fig. 1B. However, DNA sequencingof the viral origin of both viral DNAs showednodeviations in thecoreonregion
and immediately flanking sequences for either genome,
although some differences in the 72-bp enhancer were de-tected(data notshown). Theseparticular resultswere sub-stantiated in separate experiments wherein the GMSV40
on November 9, 2019 by guest
http://jvi.asm.org/
cvi C2
GM XP wt GM XP wt
12 24 489612 244896 12 24 4896 12 24 489612 244896 12 244896
MOLALwaImX
SV80
GM XP
12 24 48 96 12 24 4896 12 2
I
I_
GM 638
wt GM XP wt
44896 12 244896 12 24 4896 12 2448 96
..
[image:10.612.66.303.67.397.2]I :
FIG. 8. TransfectionofGMSV40, XP,and wild-typeSV40 into recipient C2, CV1, GM638, and SV80 cells. GMSV40, XP, or
wild-type SV40 DNA (2 ,ug) was transfected into C2 or CV1 (monkey)orSV80orGM638(human) cells.DNAwascollectedby
Hirt extraction at 12, 24, and 120 h after the glycerol shock. One-fourth of the DNAateach timepointwasdigestedwithMboI.
C2 transfections of both GMSV40 and wild-type SV40 DNAs resultedinlysisafter 120 h ofincubation.SV80 cells showed slower celldeath,whichafter 1week resulted incomplete destruction of the monolayer, a response that is morecharacteristic ofcrisis in humancells.CV1 cells showed celllysisandcytopathiceffectsonly withwild-typeSV40DNA.
genomeswerecotransfected with thesmall-t-antigenvector
described above. This vector contains an effective SV40 originof DNAreplication and is intactacross theregionof deletion in theGMSV40genome.Complementationofthese replication defects ofGMSV40 genome was not detected, nor did cell lysis occur. In a parallel transfection with the
SV80genomeplustheGMSV40genome,celllysisoccurred
asdescribed above. This comparison impliesthat recombi-nationwas notplayingarole intheseassays.
GMSV40and XPSV40genomes aredefective for
transfor-mation of rodent cells. The results described above were
consistent with the hypothesis that the SV40 T antigens encodedby themutantgenomesweredefective in functions
needed forlyticSV40DNAreplicationinanindirectmanner or in replication functions not measured by the in vitro replicationsystem.We thus looked for known functions of T antigen which might have an indirect impact on in vivo replication. From immunoprecipitation data, it was clear
thatthemutantTantigensbindtothecellularretinoblastoma (p107) proteinandtop53,asdoes thewild-type protein (9a).
Wealso asked if theGMSV40Tantigencouldtransactivate
TABLE 2. Transformationassay
Source of No. of foci inexpa:
DNA 1 2 3 4
pSV40 118 72 63 56
pGMSV40 0 0 0 0
pXPSV40 0 4 0 3
pBR322 0 0 ND ND
IFoci were counted on a Rat-2 cell monolayer at 3 weeks posttransfection
of 5 x 105 cells with 1 ,ug of DNA. ND, no datum. Experiments were performed at different times. Recipient cells in experiments 3 and 4 were rapidlyproliferating.
the SV40 latepromoter. Todo this, we asked if pGMSV40 could activatetheplasmid pL2N-CAT (29) in a cotransfec-tion of CV1 cells. pL2NCAT contains the chloramphenicol
transacetylase gene reporter linked to the SV40 late pro-moter(with a small deletion at the origin of SV40
replica-tion).Inside-by-sideassayswith theplasmid6-1(20) (which
provides a wild-type Tantigen), no differences in transacti-vation were detected (data not shown). In these experi-ments, aninternal
3-galactosidase
reporterplasmidwasused tomonitor transfection efficiencies.These experiments thusdo notprovide evidence that these in vivo functions of the
mutantTantigen areaffected. However, as T-antigen
acti-vation of the late SV40 promoter is complex, involving
multiple pathways, someof whichrequire SV40 DNA
bind-ing andothers ofwhichdo not, this experimentisdifficultto interpret, as one pathway could be affected and the other not. The recent workofZhu et al.(69) iscertainlyconsistent
with the concept thatmultipletrans-activatingdomains of T
antigen arerequiredforfullfunction. Thoseresultsarethus
consistent with the notions that T antigen can stimulate
transcription through multiple pathways and that critical
cellular promoters may respond differently from the SV40 late promotertoTantigen.
The mutant SV40 genomes rescued from these human cells areclearly defectivefortransformation ofrodentcells. These data are shown inTable 2. In multiple independent parallel assays, the mutant genomes were at least 20- to 100-fold reduced in their abilities to morphologically trans-form the established Rat-2 fibroblast indicator line. This reduction was observed when the DNAs were transfected
into confluent or growing Rat-2 cells. In any case, these
particularmutantsof Tantigen maybemoredependenton small-t-antigen functionsfortransformationthanawild-type large T antigen would be. We would like to know if the mutant T antigens retain their abilities to immortalize or transform other indicatorcelllines, particularlybecause the genomeswererescued from transformed human cells.
Nev-ertheless,theresultsclearlyshow that themutantT
antigens
have lost some unknown activity that manifests in the
pleiotropic SV40 transformation phenotype. These results
reinforce a point recently made by Montano et al.
(44).
Those workers showed thatthe large T
antigen
and smallt antigenhaveacommontransformationdomain, atleastone of which is required for SV40 function in transformation.Themutantgenomesdescribedhere maylack this function. DISCUSSION
The transforming and replication functions of the SV40
earlyproteinshave beenwidelystudied,yetnoclearinsights
into therelationshipsbetweentheseactivitieshave
emerged.
Specifically, ithaslongbeenassumed that the
transforming
on November 9, 2019 by guest
http://jvi.asm.org/
[image:10.612.320.560.79.163.2]functions serve the virus either positively or negatively to
regulate
itsvegetative propagation
in permissive environ-ments. Transformation can thus be seen as an abortive infection in a nonpermissive cell. The large T antigen is acomplex protein,
and it has beenclearlyshown that at least two distincttransforming
functions areencoded in thepro-tein
(see
reference 58 and the references therein). One of these activities is believed to be manifested as a result oflarge
Tantigen's ability
to interact with the cellular Rb orp107 proteins.
Yet mutations in SV40 largeTantigen whichcripple
the virusfor transformationand incapacitate large Tantigen's
abilities tobind these cellular proteins areappar-ently
wild type for DNAreplication andvirion production.Large
Tantigen
can also associate with the cellular p53protein
through
theauspices
of a second large-T-antigenprotein
domain. As cellularp53canactas anantioncogenein some assays, an attractive hypothesis would be that this association betweenlarge
Tantigenand p53 suppresses viralreplication
(60, 65).
The deletions present in GMSV40 andXPSV40 may delete a third distinct domain of T antigen
required
for transformation. However, as pointed out by Montanoetal.(44),
the firstexonof Tantigenmayinfluenceits
wild-type
interactionwith Rborp53.The deletionmutantsdescribed in thisstudyareinteresting in that
they provide
support for the notion that an indirect function oflarge
Tantigen
isrequiredboth forlyticreplication and for transformation. The deletion of amino acids 67through
82of the firstexonof thelarge-T-antigengeneresults inaprotein
thatis defective forviral replication invivo,yet thepurified
protein
itselfseemstobeaveryactive Tantigen forreplication
invitro. These results argue thatthe affected function is distinct from the known replication activities oflarge
Tantigen.
In addition, the deletion mutant can becomplemented
forreplication by
other known SV40 A-gene mutants. This isconsistentwiththeputativerequirement for theprotein
toact atsomesite other than thereplication fork. Other SV40 mutants whichdirectly
affect replication func-tions showtrans-negative
effects on coinfection with wild-typeDNA. We suggest that theGMSV40Tantigenis actuallya
highly
active Tantigen
forthereplication processitself invivobut is defective in some earlystepwhich prepares the cells for
replication.
Thecomplementing
viralgenomes(SV80or
C2)
aredefective forknownreplicationfunctions (43), and wesuggestthatthey
canprovidetheearlypre-DNA syntheticfunction.
Possibly,
Tantigen
functionsasamonomerfor thisearly
function and is thus notpoisoned bymutantproteins. That this deletion inactivates SV40 transformation arguesdirectly
thatthisparticular
T-antigenallele is defective in itsinteractionswithin the cell. Moreover,todate, mutations that disable direct
replication
functions such as DNA binding or helicaseorATPaseactivitiesarewild type for transformation. This laststatementistruewiththeprovisothat the mutationdoes not also affect nuclear localization or stability; the mutantT
antigens
fromGMSV40andXPSV40arewild typefor bothoftheseaspects.
If the deletion mutants described here encode for a T
antigen
more active than that of wild type in in vitro DNAreplication,
what couldaccountforits defect for replication in vivo? We suggest that the function affected concernsSV40's
knownability
to trans activate gene expression, which could elevate eitherdirectlyorindirectly the activity ofalimiting
cellularreplication
factor. This factor may besequestered
in the cytoplasm and mobilized to a higher nuclear concentrationas aresult of viral gene action or may be modified to increase its specific activity. For example, Kraussetal.(32)
have shown thatphosphorylation of DNApolymerase a increases its catalytic properties. In either
case, the in vitro DNA replication systems may not be sensitive to these effects, since the cellular extracts are adjusted foroptimal activity. Furthermore, itseems reason-able that atrans-activation functionwould beimportant for viral transformation as well as for DNA replication. An implication of this hypothesis would be that neither Rb
binding nor p53 binding is sufficient for transformation without this trans-activation function (69) because the GMSV40mutantisdefective fortransformation even
though
it binds Rb andp53 proteins aswild type (datanot
shown).
Itis known that a trans-activation function maps to thefirst exonoflargeTantigen (69).The smallt antigenalsoencodes for a trans-activationfunction(37),andthesequencesdeleted inthe defective genomesdescribedhereoverlapthecommon regionshared by large T antigen and small t antigen. Forthis
reason, we asked whether ectopic expression of small t antigen could complement the GMSV40genome for in vivo
replication. That it could not suggests thatthis domaincarried by the large Tantigen may be influenced by the presence of theexon 2-encodedprotein and may be morereadilyassayed
in our cell system. It will be interesting to know whetherthe small-t-antigen gene can effectively help the deletion mutant tooncogenically transform established rodent cells. We make a simple suggestion to link the trans-activation functions of large and small t antigens to the transformation helpereffects
and a replication requirement for a first exon domain. It remains tobe shown wherein the first exon these functions reside genetically (69). Linking the known trans-activation and transformation functions of thefirst exon tothe
replica-tion defect shown by the GM (XP) mutants is of course speculation. One could alternatively explain our data by suggesting that the separate complementation function pro-vided by the first exon entails a process not assayed by in vitro replication yet directly involved in replication in vivo. For example, T antigen may need to interact with chromatin-associated DNA in a way not mimicked by the in vitro system.Or, even though the mutant proteins showapparently normal nuclear localization and in vivo stabilities (data not shown), a special nuclear replication compartment which cannot be found by the mutants may be used. We favor our hypothesis mainly because it fits nicely with known activities ofT antigen (5, 12, 13). Perhaps our unpublished observations concerning the phosphorylation state of the GM T antigenwill provide a key for understanding the mutated functions of T antigen(see below).
Another interesting and perhaps paradoxical finding re-ported here is that these T-antigen mutants, thoughdeficient invivo, are better than wild type in all of our in vitro assays. In fact, the mutant protein shows a phenotype in vitro similar to that of adephosphorylated T antigen. The mutant T antigen shows a shortened lag in its kinetics of initiation and can function at a reduced T-antigen concentration. Theseproperties of an underphosphorylated T antigen (43b, 63) are substantiated by our findings that the adenovirus vector produced GM T-antigen labels less effectively per mass with phosphate than does wild-type T antigen (1). Consistent with these observations, Jim Roberts (Seattle, Wash.) has reported (50) that the purified GMSV40 large T antigen cannot be further activated by replication factor C, which isthe catalytic subunit of phosphatase hIa (63). These observations could explain the increased activity in vitro but do not readily explain decreased replication in vivo. It is tempting to link these observations with the recentdiscovery thatSV40 small t antigen binds to phosphatase
Ila
and inhibits its activity (54). It is conceivable that with the wild-type Ton November 9, 2019 by guest
http://jvi.asm.org/
antigen, the serine-phosphorylatedstateis necessary fortrans activation and that the phosphatase IIaconvertsthe protein from one that trans activates to one that is active in DNA replication. Adeletionofthis domain(and of smalltantigen
entirely) might lead to a protein incapable of transactivation but acting as an apparent "super" Tantigen in replication.
Reciprocally, specificphosphorylation may be required fora trans-activationfunction for which the GMSV40 allele maybe defective. Aswitch between a trans-activator Tantigen which functions early in the lytic cycle and a replication Tantigen
which functions later may thusbegovernedby such cycles of phosphorylation.This hypothesis, whichcanexplainall of the
data, can of course bedirectlytested.
Finally, we come backto a point addressed in the Intro-duction. The GMSV40-XPSV40 A-gene mutants described here do not completely inactivate the in vivo replication potentialof the virus. Inasense,then,thedeletion described creates an attenuated form of the viral replicon in that the DNA can persist without obvious lyticeffects in the trans-formed cells. Thispoint is consistent withprevious findings
which show thatpermissive orsemipermissive cells cannot tolerate stable transformation by wild-type SV40. Further-more, though the deletion cripples the SV40 for transforma-tion in focus assays, it is possible that other oncogenic
domains (Rb binding and p53 binding) may contribute to immortalization activities which may haveplayed a role in the initialisolation of the human cells from whichtheywere cloned. We assume that the viralvariants preexisted in the wild-type stocks of SV40 used to establish the human cell linesGM637 andXP12ROSV, because bothmutants(which
result fromanapparentlyillegitimaterecombination) harbor precisely thesamedeletion. Inany case, thesemutantsmay provide a fortuitous tool for studying the cellular compo-nents involved in the control of transformation and viral
replication invivo.
ACKNOWLEDGMENTS
WethankGreg Armstrong forreadingthemanuscriptandS. Kim forhelpwith thetransformation experiments.
C.M. was supported in part bya Fulbright, by aUniversity of California Regents Scholarship, and by the Office of Energy Re-search, U.S. Department of Energy, under contract DE-AC03-76SF00098 toJ.B. This researchwas supported by PublicHealth Service grant CA 30490 from the NIH to M.B. and by NIEHS Center grantES01896.
REFERENCES
1. Bartholomew,J., and M. Botchan.Unpublisheddata.
la.Bradford, M. 1976. Arapidandsensitive method for quantita-tion ofmicrogramquantitiesofproteinutilizingtheprincipleof protein-dyebinding. Anal. Biochem. 72:248-254.
2. Buchkovich, K.,L. A.Duffy,and E. Harlow.1989. The retino-blastomaproteinisphosphorylated duringthespecific phasesof the cellcycle. Cell58:1097-1105.
3. Buchman, A. R., M. Fromm, and P. Berg. 1984. Complex regulation of simian virus 40 early-region transcription from differentoverlappingpromoters. Mol. Cell. Biol. 4:1900-1914. 4. Chen, P.-L.,P. Scully, J.-Y. Shew, J.Y.J. Wang, and W.-H.
Lee.1989.Phosphorylationof the retinoblastoma geneproduct is modulated duringthe cellcycle and cellular differentiation. Cell 58:1193-1198.
5. Cherington, V.,M.Brown,E.Paucha,J.St. Louis,B. Spiegel-man,and T. Roberts.1988.Separationof simian virus 40largeT antigen transforming and origin-binding functions from the abilitytoblockdifferentiation. Mol. Cell. Biol. 8:1380-1384. 6. Chou,J., and R.Martin.1975. DNAinfectivityand the
induc-tion of host DNAsynthesiswithtemperature-sensitivemutants
of simianvirus 40. J. Virol. 15:145-150.
7. DeCaprio, J. A., J. W. Ludlow, J. Figge, J.-Y. Shew, C.-M. Huang,W.-H.Lee,E. Marsilio,E. Paucha,and D. M.
Living-ston. 1988. SV40largetumorantigenformsaspecificcomplex
with theproductofthe retinoblastomasusceptibilitygene.Cell 54:275-283.
8. DeCaprio, J. A., J. W. Ludlow, D. Lynch, Y. Furukawa,J. Griffin,H.Piwnica-Worms,C.-M.Huang,and D. M.Livingston. 1989. Theproductof theretinoblastomasusceptibilitygenehas propertiesofacellcycle regulatoryelement.Cell 58:1085-1095. 9. Decker, S., M. Yamaguchi, R.Possenti, and M. DePamphilis. 1986. Initiation of simian virus 40 DNA replication in vitro: aphidocolincausesaccumulationofearly-replicating
intermedi-atesand allows determination of the initial direction ofDNA synthesis.Mol. Cell. Biol. 6:3815-3825.
9a.Dyson,N.,andC. Maulbecker.Unpublisheddata.
10. Ewen,M.E., J.W.Ludlow,E.Marsilio, J.A.DeCaprio,R.C. Millikan,S. H.Cheng,E.Paucha,and D. M. Livingston.1989. An N-terminal transformation-governing sequence of SV40 largeTantigen contributestothebindingofbothpllOrb anda secondcellularprotein,p120.Cell 58:257-267.
11. Farber, J., K. Peden, and D. Nathans. 1987. trans-Dominant defectivemutantsof simianvirus 40 Tantigen.J.Virol. 61:436-445.
12. Fried, M.,andC. Prives. 1986. Thebiologyofsimianvirus 40 andpolyomavirus,p. 1-16. In M.Botchan,T.Grodzicker,and P.Sharp(ed.),DNAtumorviruses,vol.4.ColdSpringHarbor Laboratory,Cold Spring Harbor,N.Y.
13. Gallo,G.J.,G.Gilinger,andJ.C. Alwine. 1988.Simianvirus 40 Tantigen alters the bindingcharacteristics of specific DNA-bindingfactors. Mol. Cell. Biol. 8:1648-1656.
14. Gannon, J. V.,and D. P. Lane. 1987.p53and DNApolymerase acomplete for bindingto SV40 T antigen. Nature (London)
329:456-458.
15. Gimbrone, M.,andG. Fareed.1976.Transformation of cultured humanvascular endotheliumbySV40 DNA.Cell 9:685-691. 16. Gish, W.,and M. Botchan. 1987. Simianvirus 40-transformed
human cells that express large T antigens defective for viral DNAreplication.J.Virol.61:2864-2876.
16a.Gluzman,Y.Unpublisheddata.
17. Gluzman,Y. 1981. SV40 transformed simian cellssupport the replicationofearlySV40 mutants. Cell 23:175-182.
18. Gluzman,Y. (ed.). 1982. Eukaryoticviralvectors, p. 187-189. ColdSpringHarborLaboratory,ColdSpring Harbor,N.Y. 19. Gluzman, Y.,and B. Ahrens.1982. SV40earlymutantsthatare
defective forviral DNAsynthesisbut competent for transfor-mationof culturedrat andsimian cells.Virology22:78-92. 20. Gluzman,Y.,J.Frisque,andJ.Sambrook. 1979. Origin
defec-tivemutantsofSV40. ColdSpringHarborSymp. Quant.Biol.
44:293-300.
21. Graham,F., J. Smiley,W.Russel,and R. Nairu. 1977. Charac-teristicsofahuman cell line transformedbyDNAfromhuman adenovirustype5. J. Gen. Virol.36:59-77.
22. Graham,F.,andA. Van der Eb.1973.Anewtechniqueforthe assay of infectivity of human adenovirus 5 DNA.
Virology
52:456-467.
23. Gurney,E.G., S.Tamowski,and W.Deppert.1986.
Antigenic
bindingsites ofmonoclonal antibodiesspecificforsimian virus 40largeT antigen.J. Virol. 57:1168-1171.
24. Harlow, E., L. Crawford, D. Pim, and N. William. 1981. Monoclonalantibodies specificforsimianvirus 40tumor anti-gens. J. Virol. 39:861-869.
25. Harlow, E.,andD.Lane.1988.Antibodies. Alaboratorymanual. ColdSpringHarborLaboratory, ColdSpring Harbor,N.Y. 26. Hirt, B. 1967. Selective extraction of polyoma DNA from
infectedmousecultures. J. Mol. Biol. 26:365-369.
27. Hsiung,G. D., and W.D.Gaylord, Jr. 1961. Thevacuolating virus of monkeys. I. Isolation, growth characteristics, and inclusionbodyformation. J. Exp.Med. 114:975-986.
28. Kalderon, D., and A. Smith. 1984. In vitro
mutagenesis
ofaputativeDNAbindingdomain ofSV40largeTantigen.
Virology
139:109-137.29. Keller, J.M.,andJ.C. Alwine. 1985.Analysisofanactivatable promoter: sequences in the simian virus 40 late promoter