Copyright ©1971 American Society for Microbiology
Unilateral
Synthesis of
Reovirus Double-Stranded
Ribonucleic Acid
by
a
Cell-Free
Replicase System
S. SAKUMA AND Y. WATANABE
The WistarInstituteof AnatomyandBiology,Philadelphia, Pennsylvania19104
Received for publication19 March 1971
Alarge-particle fraction obtained from reovirus-infected Lcells contained both
replicase and transcriptaseactivity. The in vitro replicase reaction slowed downsoon
after initiation, whereas the transcriptase reaction proceeded at anunabatedrate.
The replicase and transcriptasewereboth template-bound and could beseparated
fromoneanotherby controlled chymotryptic digestion followed by centrifugation
inaCsClgradient. Thetranscriptasewasrecoveredasasharpband (p = 1.43) and
resembled viruscorederived frommaturevirions. Incontrast,replicase activitywas
distributed throughout the gradient, indicating that replicase is associated with structures of various density in CsCl. In subsequent experiments, the replicase
productwasfoundtobeindistinguishable from the double-stranded ribonucleic acid
(RNA) reovirusgenomewithrespect toitsbuoyant densityincesium-salt gradients
and denaturation-annealing characteristics. A "hybridization-competition"
experi-mentin which thereplicase productwasdenatured and annealed in thepresenceof
anexcessofplus-RNA indicated that the in vitro replicase reaction proceeded by meansofaunilateralsynthesis of minus-RNAupona preexisting plus-RNA
tem-plate,presumably of single-stranded form.
We have previously shown that in
reovirus-infected L cells, a ribonucleic acid (RNA)
polymeraseis induced which synthesizes
double-stranded RNA (dsRNA) segmentsresemblingthe
genomic segments ofreovirus(12).Inthe present
paper, this polymerase will be tentatively
desig-nated "replicase," to distinguish it from the
reovirus-bound "transcriptase." Transcriptase is
found inthe subviralparticles of reovirus (virus
core) and synthesizes single-stranded RNA
(ssRNA) by transcribing only one strand of
reovirus dsRNA (1, 4, 5, 10, 11). Forour
pur-poses, the ssRNA synthesized by transcriptase
will be referred to as plus-RNA, and the RNA
complementary to it, asminus-RNA.
Recently, Schonberg etal. (9) found that the
reovirus dsRNA obtained from pulse-labeled,
infected cells contained label only in the minus
strand. They concluded from this invivo study
that thecomplementarystrands of reovirus dsRNA
are formed in an asymmetric manner; i.e., the
minus strand is synthesized upon a preformed
plus-strand template. We decided to further
investigate the mechanism by which reovirus
dsRNA issynthesized under themorecontrolled
conditions provided by a cell-free replicase
system. We therefore wished to determine (i)
whether replicase and transcriptasepresentin cell
extract are two distinct entities and (ii) whether
the cell-free replicase reaction involved the
simultaneous synthesis of plus- and minus-RNA
strands or, as in replication in vivo, the
asym-metric("unilateral") synthesis of eitheraplus-or
minus-RNA strand. To investigate the latter
point,we conducted ahybridization-competition
experiment employing the in vitro replicase
product.
MATERIALS AND METHODS
Buffers and chemicals. The following buffers were
used: 0.1 M STE buffer, containing 0.1 M NaCI,
0.05 M tris(hydroxymethyl)amninomethane (Tris)-hydrochloride (pH7.4) and0.001M
ethylenediamine-tetraacetate (EDTA);0.3 and 0.01 M STEbuffers, as above, but containing 0.3 and 0.01 M NaCl, respec-tively; 0.1 M STES buffer, containing 0.5% sodium dodecyl sulfate (SDS) in 0.1 M STE buffer; TMS buffer, containing 0.25 M sucrose, 0.001 M MgC12,
and 0.01 M Tris-hydrochloride, pH 8.2; TM buffer, as above, but without sucrose. Tritiated uridine
tri-phosphate (3H-UTP; specific activity, 17 Ci/mmole)
and 32P-orthophosphoric acid were purchased from New England Nuclear Corp., Boston, Mass.;
a-chymotrypsin and ribonuclease (pancreatic), from Worthington Biochemical Corp., Freehold, N.J.;
190
Printed in U.S.A.
on November 11, 2019 by guest
http://jvi.asm.org/
and unlabeled nucleoside triphosphates, from
Cal-biochem Co., LosAngeles,Calif.
Cells,virus, and viral RNA. Conditions for the sus-pension culture of L cells and their infection with
reovirus type 3 [a strain producing only complete
virions (8)] have been describedpreviously (13, 15). Reovirus highly labeled with 32p was prepared by
incubating the infected cells in a phosphate- and
serum-free medium containing 32P-orthophosphate (10 MCi/ml) throughoutthe infectionprocess (about
20 hr). The 32P-labeled virus was purified by a
pro-ceduredescribed previously (15),andthe virion RNA
wasextracted with phenol. Adenine-rich ssRNA (2)
waseliminated from the viral RNAextract by filtra-tionthroughacolumn ofSephadexG-100gel (1.5 by
30 cm) equilibrated with 0.1 M STES buffer. The purifiedviriondsRNAelutedas adistinctpeakafter
avoid volume of buffer.
Preparation of RNApolymerase-template complexes from infected cells. A modification ofa procedure
describedpreviously (12)wasused. At 7to8 hr
post-infection, the cells were centrifuged, washed once
with cold phosphate-buffered saline, suspended in fivevolumes ofTMS buffer containingthe nonionic
detergent, Nonidet P-40 [1% (v/v)], and disrupted
inaDouncehomogenizer.Theextentof cell
destruc-tionwasmonitored bymicroscopy,andthe
homogeni-zation continued until 80% of the cells were
dis-rupted. This and all subsequent procedures were
carried out at 4 C. Nuclei and largecellular debris
were removed after each of three centrifugations at 860Xgfor10mininaSorvall refrigerated centrifuge.
The supernatant fluidwasnextcentrifugedat20,000X gfor 30 min, and the resulting sedimentwas resus-pended inTM buffer containing 1% sodium
deoxy-cholate (volume ofbuffer was equal to 10-3 of the volume of the original infected cell culture). This suspensionwasthoroughly homogenizedinaDounce homogenizerandcentrifugedat3,000X gfor 10min toremovelarge particulate material, afterwhich the
supernatant was centrifuged in a Beckman
ultra-centrifuge (SW50Lrotor) at200,000X gfor60min.
After centrifugation, the supernatant was discarded
and the gelatinous pellet was resuspended in TM buffer. This material shall be referredtoasthe large-particle (LP) fraction. At this point, chymotrypsin was addedto the fraction at aconcentration of200
jug/mi, and the mixturewasincubatedat37 C for 30 min with occasional shaking. The resulting turbid
solution was centrifuged in a clinical centrifuge at
1,000 X gfor 3 min, and the supernatant fluidwas
either used immediately for polymerase assay or
stored at-70C. Theproteincontentofthis
prepara-tionwas2.5to3mg/ml,asdeterminedbythemethod ofLowryetal. (6).
During digestion with chymotrypsin, replicase activitywaslittleaffected for the first 60min,andthen
it declined gradually. Transcriptase activity, on the
otherhand,increasedduringthe first 30minof
diges-tion and thenremainedunchangedforatleast90min.
This increase varied, ranging from 1.3- to 3.0-fold
fromoneLP fractiontoanother.
RNA polymerase assay. RNA polymerase activity was determinedby measuring the conversion of 3H-UTP into acid-insoluble form. The standard assay
system contained, in a total volume of 0.25 ml, the
LP fraction (200 jug of protein), 5 MACi of3H-UTP
(0.3 nmole), 0.2 Mmoleeach of the other three
ribo-nucleoside triphosphates, 3
ptmoles
of MgCl2, 20moles ofTris-hydrochloride (pH 8.2), and 2 Lug of actinomycin D. The reactionwascarriedout at37C.
In the "chased" polymerase assay, 0.2 Mmole of
unlabeledUTP(0.01ml)wasaddedatspecified times,
and the mixture was incubated for anadditional 20 min. Thereactionwasthenterminated bychillingthe reaction tube in an ice bath and adding 1.75 ml of cold 0.3 M STE buffersupplemented withEDTAto a
final concentration of 10 mm. The mixture was
di-vided into two 1-ml portions, and one portion
re-ceived 10
jg
ofribonuclease (0.01 ml) priortoincu-bationat 37Cfor 30 min. Synthesized 3H-RNAwas
precipitated by adding carrier yeast RNA, trichloro-acetic acid (10%). and sodium pyrophosphate (0.02
M).Theprecipitatewasthencollectedon amembrane
filter, washedwith cold 5%trichloroacetic acid
con-taining 0.02 M sodium pyrophosphate, dried, and
counted in a liquid scintillation counter, by using toluene-Liquifluor scintillator.
Preparation of single-stranded plus-RNA. Single-strandedplus-RNA of threesize-classeswasobtained by the methoddescribed byBanerjeeandShatkin(1).
Thesubviral particles derivedfrom 10 mgofapurified
virion preparation by digestion with chymotrypsin
were incubated ina 25-ml reaction mixturefor 5 hr
at37C.After thesynthesis,thesubviralparticleswere
removed bycentrifugationat165,000X gfor 90 min.
The single-stranded plus-RNA synthesized was
ob-tained byextractionfrom the supernatantwith phenol and filtration through Sephadex G-25.
Approxi-mately 300 ,ug of plus-RNA was obtained [40jug of
RNA per 1 optical density (OD) unit].
Denaturation and annealing of dsRNA. The pro-cedure fordenaturatingdsRNA in dimethylsulfoxide
(DMSO) and subsequent annealing was essentially
thesame asthatdescribedpreviously(13, 15).Briefly,
the labeled dsRNA dissolved in 0.01 M STE buffer
wasmixed with9 volumesof DMSO. [Inthe
compe-tition experiments, single-stranded plus-RNA was
addedatthistime.] Themixturewas heatedat 70C for 20 min, and the denatured RNA wasprecipitated bytheaddition of carrier yeast RNA (200
M&g),
adrop of saturatedNaClsolution,and5volumes ofalcohol.The precipitate was collected by centrifugation and
washed with 95% alcohol and then acetone. After
drying,the precipitatewas dissolved in 0.3 ml ofcold
0.3 M STE bufferand heated at 70 C for annealing. Theextentofannealingwasdetermined by digestion
with ribonucleaseasdescribed previously(13).
Thermal denaturation of the labeled dsRNA in 0.01 M STE buffer was carried out by heating the
samplesatvarious temperatures for 10 min andthen
quickly chilling in an ice bath. The extent of
de-naturation was determined by the loss of acid-in soluble label after digestion with 10 Ag of ribonu-clease in1 ml of 0.3 M STE bufferat37Cfor20min.
RESULTS
Kinetics of replicase reaction. Replicase was
assayed by determining the incorporation of
on November 11, 2019 by guest
http://jvi.asm.org/
3H-UTP into a ribonuclease-resistant product.
When the determination was made without
"chase" (see above), the incorporation of label was greater into ribonuclease-resistant product thaninto ssRNA for thefirst 3 to 10 minof the
reaction. After about 10
min,
it declined to alevel below that for ssRNA, whereas ssRNA synthesis maintained an unabated rate of in-corporation (Fig. 1). Wefoundthat aportionof the label in the ribonuclease-resistant product was unstable and could be "chased out" upon addition of unlabeled UTP. In the experiment shown in Fig. la, unlabeled UTP (a 660-fold excess) was added to the reaction mixture 10 min after the polymerase reaction began, and incubation continued for the periods of time shown in the diagram. (This particular concen-tration ofunlabeled UTPblocked >90% of the incorporation of
label,
but nevertheless stimu-lated the rate of the transcriptase reaction.) As shown, the amount oflabel in the ribonuclease-resistant fraction decreased considerably 5 to 10 minafter theaddition of unlabeledUTP, whereas the label in the ssRNA fraction increased. Thepresence of an "unstable" portion in the final
ribonuclease-resistant product, therefore, was
effectively avoided
by"chasing,"
and, aswill be shownlater,
theremaining
product demonstrated thecharacteristics
only
ofdsRNA.In our
investigation
of the kinetics of the-
20-0
16- a-w
<
12-
I-0
a.
0
0 8
Z
z 4' a
CHASE
I A, ^'.:' ,.° .C.,°
/~~~~~1
/~~~~~01
o=~~~~~~
l""~~~~~~~~~~~~
replicasereaction, we accordingly added an excess
of unlabeled UTP as chaser prior to measuring the incorporation of label into
ribonuclease-resistant product and incubated the mixture for
20-min periods before each point of
determina-tionduring the course of the polymerase reaction
(Fig. Ib). We believe thebottom line in Fig. lb
validly represents the actual kinetics ofreplicate reaction. The kinetics of replicase reaction differ
from transcriptase in the rapid slow-down of the
replicase reaction. These data are compatible
with the view that the replication and
transcrip-tion of reovirusgenome are two distinctprocesses. (This matter will be dealt with more directly in thefollowingsection.)
The nature of the "unstable" portion of
ribonuclease-resistant product which became ribonuclease-sensitiveafter chase is unknown. We have observed thatasimilaramountof unchased ribonuclease-resistant product became ribo-nuclease-sensitive when extracted with phenolor heated at 80 C for 5 min in 0.3 M STE buffer.
Thissuggeststhataportion ofthe nascent ssRNA
was
protected
from ribonucleasedigestion by its association with protein, by the existence of a short region of base-pairing with the templateRNA, orby both.
Partial dissociation of replicase from
trans-criptase.To
verify
thatreplicase
andtranscriptase
are two distinct entities, the LP fraction was
20
16
12
4-10 20 30 40
b
..
,**, A.
C
0 20 30 40l0 20 30 40O
TIME IN MIN
FIG. 1. Kineticsofreplicaseandtranscriptasereactions.RNApolymeraseassaywascarriedoutinourstandard
assaysystem. (a) UnlabeledUTP(0.2,umoleper0.25mlofreactionmixture) wasaddedat10 minafterthe
polym-erase reactions began, and at the timesindicatedsampleswereanalyzedfor 3Hincorporated into
ribonuclease-sensitiveand resistant RNA. Acontrol experimentwascarriedoutinthesame mannerwithout theadldition of
un-labeled UTP. (b) Unlabeled UTP (0.21imolepertube) wasaddedateachpointindicated and incubatedforan
additional 20 min.3H incorporationwasdetermined.Symbols:(0)3HindsRNA withoutchase;(A) 3Hin dsRNA afterchase. (0) 3HinssRNA withoutchase; (A)3HinssRNAafterchase.
on November 11, 2019 by guest
http://jvi.asm.org/
[image:3.484.119.410.383.578.2]centrifugedthroughapreformedgradientofCsCI (Fig. 2). A sharp,
readily
visibleband,
wasob-served at a buoyant density of 1.43 g/cm3. This
band coincided with a virus core marker (not shown) which was derived from purified virions by chymotryptic digestion. Electron microscopy couldnotdistinguish betweenthe
particles
presentinthis band and virus core derived from mature
virions (8).
A majority of the
transcriptase
activity
wasrecovered in the subviralparticleband (p = 1.43
141 9- P.143 I
S. ~~~~~~~16.5s
7' IL ~~~~~248
IL
o I
W
4-
5-0
otto4 1 10 1.203 30 40
F.3 D rP.ad
frcto (dgete wih20 f ~yotrp# o ,0
4O
.Co.0o
%.doo
/'0oA
0
\/P
BOTTOM 5 10 I 20 TOP
FRACTIONS
FIG. 2. Distribution ofreplicase and transcriptase
activity ina CsCI gradient. A 1-miamowit ofthe LP
fraction (digested with 200 pg ofchymotrypsin for 90minat37C)wascentrifugedinapreformed gradient
of CsCl(p = 1.29to1.46g/cm3) inTMbufferfor2hr
at 165,000 X g (SW50L Beckman ultracentrifuge rotor). The gradient was then fractionated into 24 tubes. Eachfraction was immediately diluted to S ml with coldTMbuffer andcentrifuged at 165,000 X g
for I hr to pellet the polymerases. The pellet was
resuspendedin0.45 mlof TMbuffer, and 0.15 ml was removedfor polymerase assay in the standard assay system. Incubation was for 30 milt and chase with
unlabeledUTPwasfor20 min.Symbols:(0)replicase
activity; (0) transcriptase activity. Recoveries of the activities were 80 to 100% for transcriptase and 60
to 80% for replicate in four repeated experiments. The inset isa sedimentation profile of ssRNA
synthe-sized with the subviral particle fraction of density
1.43g/cm3. This fraction was further dialyzed against
TMbufferovernightandincubated in a reaction mixture
reported by Borsa (3) for 30 min at 37 C. The product
RNA was extracted by the SDS-phenol method and
centrifugedin a sucrose gradient (5to 20% in 0.1 m
STESbuffer)withasedimentation markerat200,000 X
gfor150minat15 Cin aSW50Lrotor.
g/cm3) andminoractivitywasdetectedina more diffuse band distributed around the density of
1.32 g/cm3. This lighter peak was reduced when
the LP fraction was digested with
chymotrypsin
fora
longer period
oftime beforecentrifugation,
suggesting that the presence of transcriptase in
thisbandwasdueto
incomplete
digestionof theLP material. When the heavier subviral particle
fraction was incubated in a reaction mixture
suitable fortranscriptase reaction (3),three
size-classesofssRNAresemblingthe messenger RNA (mRNA)formedininfected cellsweresynthesized (Fig. 2, inset). This result is consistent with the conclusion of others that transcriptase is asso-ciated with the subviral particles ofreovirus
(1,
4, 5, 10, 11). In contrast, replicase activity was
notassociatedwith the subviral particles butwas depositedvariously over the gradient. Exogenous virus-specific RNA (dsRNA, denatureddsRNA, or plus-ssRNA) was not required as a template
for the replicase present in these fractions. In
addition,thedsRNAsynthesizedbythedifferent fractionsof the gradient was similarto that syn-thesizedbythe unfractionated replicase
prepara-tion,
i.e., the synthesis of three major classes of dsRNA,asshown inaprevious paper(12).Theseresultsindicatethat thereplicasewasbound to a
template and that the complex was associated
with structures of various density in CsCI. We
conclude from these data that replicase and
transcriptase exist as two distinct forms of an
enzyme-templatecomplex.
Physical properties of the replicase product.
The replicase product was extracted with SDS andphenol fromthe reaction mixture which had
been incubated for 30 min and chased for 30min.
Ethanol-precipitated RNAwasthendissolvedin
1 ml of 0.3 M STE buffer containing 10 jug of
ribonuclease, incubated at 37 C for 20
min,
and finally deproteinized by phenol and filtered through acolumnofSephadex G-100 (1.5 by 30cm; reference 12). The ribonuclease-resistant
replicase product elutedas adistinctpeak after a
void volume ofbuffer.
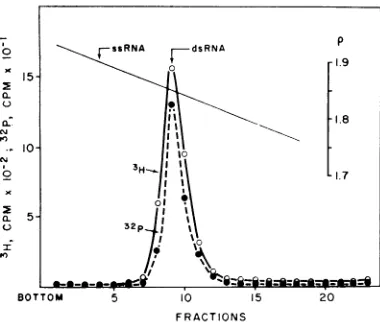
Buoyant density. Thereplicase product obtained
in this manner was found to have a buoyant
densityof 1.60g/cm3 inCs2SO4 (not shown) and
1.85 g/cm3 in an 8:1 CsCl-Cs2SO4 mixture (7),
as determined by equilibrium density gradient
centrifugation (Fig. 3). The 3H-labeled replicate product banded coincidently with the 32P-labeled virion dsRNA.
Thermal denaturation and annealing. The replicase productandvirion dsRNA both under-went sharp thermal denaturation (Tm of 92 C)
and were rendered completely
ribonuclease-sensitive at 95 C (Fig. 4). Denaturation also
resulted from heating at 70 C in 90% DMSO
on November 11, 2019 by guest
http://jvi.asm.org/
[image:4.484.39.229.187.407.2]P
ossRNA dsRNA
19
-1.9
0~~~~~~~0
BOTTOM 5 l0 15 20
[image:5.484.60.250.63.224.2]FRACTIONS
FIG. 3. Equilibrium density gra.dietcentrifugation of the 3H-labeled replicase product and 32P-labeled
virion dsRNA ina CSCI-Cs2SO4 (8:1) mixture.
Cen-trifugation wascarriedout at84,000 X gfor96 hrat
22
Cy
by using an SW50.1 rotor. The position ofssRNA (arrow) tvasdetermined in a separate
experi-ment by using theplus-RNA synthesized by reoviru~s
transcriptase.
100-80
w
z 60
w
cn
0 in z 40
z
20-60O 70 80 90 100
TEMPERATURE
FIG. 4. Thermal denaturation of the 3H-labeled replicase product and 32P-labeled virion dsRNA. The analysis wasconducted in0.7mlof 0.01M STEbuffer
containing 3H-dsRNA (3,000 count/min, 0.3 pig) and 32P-dsRNA (1,800 count/min, 0.01 jig).
(Fig. 5). When the 3H-labeled replicase product
andthe 32P-labeled virion dsRNAwerecombined
ina27:1massratioanddenaturedandannealed,
the annealing of both 3H and 3Ip radioactivities
proceeded gradually and was near completion
after 22 hr. The anealing kinetics of both the
replicase product and virion dsRNAweresimilar
(Fig. 5). In contrast, the self-annealing of only
the 32P-labeled virion RNA (i.e., in the absence
ofreplicase product) proceeded ata
significantly
slower rate (Fig. 5), because of the decreased
concentration (13). The fact that the mixed
annealing of the 3H- and 32P-labeled RNA
fol-lowed similar kinetics suggested that the
com-plementary strands of each species behaved
identically with respect to annealing and that
some strand interchangeability between the two
occurred.
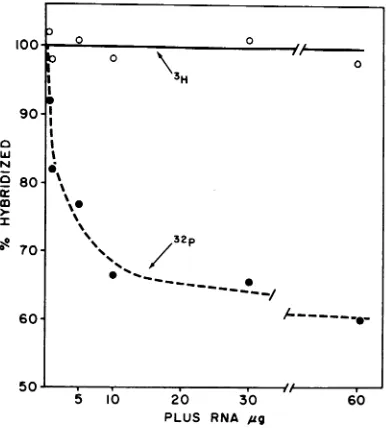
Mixed annealing inthepresenceofanexcessof
plus-RNA. The denaturation and subsequent
annealingofamixture ofthe 2H-labeledreplicase
product and 32P-labeled virion dsRNA were
carried out in the presence of an increasing
amount ofunlabeled plus-RNA which had been
synthesized in vitro by the reovirus-core
tran-scriptase. If the 3H labelwasuniformlydistributed
between the plus and minus strands of the
replicase product, the labeled plus-RNA would
be diluted upon denaturation by the unlabeled
plus-RNA that was added. Thus, most of the
original labeled plus-RNA might be expected to
belost from the annealed product. On the other
hand, the labeled minus-RNA originally present
inthe replicase product would be retained
com-pletely in the annealed product. The theoretical
value for the retention of label in the annealed
product would therefore be 50%. This was the
100-
80-
20-2 3 4
HOURS
5 6 22
FIG. 5. Annealing kinetics of the denatured IH-labeledreplicase productand32P-labeledviriondsRNA. The denaturation of RNA in DMSO and subsequent annealing were carried out as described in the text. The mixedannealingwasperformed in0.3 mlof0.3M
STE buffer containing 3H-dsRNA (2,700 count/min, 0.27 jig) and 32P-dsRNA (2,240 count/min, 0.01 jig).
Annealing of 32P-dsRNA alone was carried out in a
similar manner although without the addition of
3H-dsRNA.
J. VIROL.
@ .
03H S
.
32P
80
0 0
0
00
0.0 0 0
°
6fl ri e'sribm a O
Y
932p MIXED
A32P
9-4
/!
A~~~~~~~~~~~~~
i.8P~~~~~~~~~~h
,I,{
..z z
on November 11, 2019 by guest
http://jvi.asm.org/
[image:5.484.57.249.314.541.2]case with the32P-labeled dsRNA extracted from virions
(Fig. 6);
about40%
of the32P
was lost from the annealedproduct.
In contrast, the 3H contained in thereplicase
product
wastotally
retained after
annealing.
Annealing in the presence of an excess of plus-RNA proceeded
conspicuously
faster(Fig.
7) than that done in the absence of plus-RNA (Fig. 5) because ofthe
dependence
ofannealing
on concentration. Plateau levels of
100%
for 3H and 60%for32P wereachieved.Amajor
conclusion from thepreceding
experiments
is that thedsRNA synthesized in vitro
by
replicase
iscom-posed of an unlabeled
plus-RNA
strand and a nascent 3H-labeled minus-RNA strand.Pre-sumably,
therefore,
theinvitroreplicase
reaction proceedsunilaterally
be means ofthesynthesis
of the minus-RNA strand upon a
preexisting
RNA
template.
The
annealing
data also show that the 32p_ labeledplus-RNA inthe virion dsRNA was notcompletely
replaced by
the unlabeledplus-RNA
even in the presence ofa 400-fold excess of un-labeled
plus-RNA
(Fig.
6).
The reason for the approximately 10%~discrepancy
between the theoretical value of50%
reduction and the ob-served value of 40% is unknown. Thisphe-hLJ
N
03 x
5 l0 20 30
PLUS RNA ag
60
FIG. 6. Mixedannealing of the3H-labeledreplicase product and32P-labeled virion dsRNA in the presence
ofunlabeledplus-RNA. Tothe mixture of'H-dsRNA (3,076 count/min, 0.3 Mg) and 32P-dsRNA (1,600 count/min, 0.005 4g) wasadded theindicatedamount
of unlabeledplus-RNA, andthe mixturewas denatured inDMSO. Thedenatured RNA wasannealedin0.3 ml of 0.3 Mf STEbufferfor 20 hr.
a
2 60 ] -
---
4--0 40- l
20
2 3 4 5 6
HOURS
FIG. 7. Kinetics of mixed annealing of the 3H-labeledreplicaseproductand32P-labeledviriondsRNA
in the presence ofan excess of plus-RNA. Each
an-nealing mixture (0.3 ml) contained 3H-dsRNA and
3"P-dsRNA in the amounts given in the legend to
Fig. Sand, inaddition, 25lig of unlabeledplus-RNA.
nomenon, however, may be related to the fact
that a small portion of the
DMSO-denatured,
virion dsRNA anneals at a faster rate than the
major portion (13).
Ifso, the discrepancy does not essentially affect the above conclusion.DISCUSSION
Existence ofreplicaseas aseparate entity from
transcriptase. Evidence has been provided
that
the
replication
and transcription ofthereovirus genome are independent processes catalyzedby
two distinct enzyme-template complexes which are distinguishable by their buoyant densities in CsCl (Fig. 2). The reason for the heterogenous distribution of replicase in CsCl has not been studied, however.
It is possible that replicase and transcriptase are two individual proteins. Alternatively, replicase might be converted to transcriptase as theenzyme-template complexis
incorporated
into the subviral particles. The latter possibility is compatible with experimental evidence that transcriptase is detectable only in the subviral particles of infected cells and the virus core ofmaturevirions and that theseparticles apparently
lack replicase activity. The fact that dsRNA replicationininfectedcells disappears soon after protein synthesis is inhibited by an addition of cycloheximide (14) is also compatible with this hypothesis, since continued synthesisofreplicase
wouldberequired.
Examination of the in vitro replicase reaction
andproduct. The dsRNA synthesized invitro by replicase contained three major size-classes of
100- s _ 0
90-i
80- \
70- /m32p
60- _09_ _ _ _
60 ---.-.-0-
on November 11, 2019 by guest
http://jvi.asm.org/
[image:6.484.242.433.63.230.2] [image:6.484.34.227.353.567.2]dsRNA segments (12) and appeared to be
indistinguishable from virion dsRNA with
re-spect to double-strand characteristics (Fig. 3-5).
That the replicase product could be denatured
and annealed in the same manner as the virion
dsRNA (Fig. 4, 5) provided arationale for the
hybridization-competition experiment. This
ex-periment allowed the determination of whether
the in vitro dsRNA synthesis involved the
simultaneous synthesis of bothplus- and
minus-RNA strands. The results (Fig. 6, 7) indicated
that the dsRNA segments synthesized in vitro
were composed of a nascent 3H-labeled
minus-RNA strandand anunlabeledplus-RNA strand
whichwaspreexistentin theenzymepreparation.
The replicase preparation contained
tran-scriptase activity and incubation for the replicase
reaction stimulated synthesis of ssRNA in
addi-tion to the replicase product. This ssRNA
ap-pearedtobeindistinguishablefrom theplus-RNA
synthesized byreovirus-coretranscriptaseinthat
it also contained three size-classes of ssRNA
resembling the messenger RNA formed in
reovirus-infected cells (Fig. 2). The ssRNA did
not self-anneal or hybridize with plus-RNA
(unpublished data). These observations suggest
that no free minus-RNA was present in the
products synthesized bythe cell-freereplicaseand
transcriptase preparation. We believe, therefore,
that the in vitro replicase reaction proceeds by
the synthesis ofminus-RNA upon a plus-RNA
template, presumably of single-strand form,
rather than by replacement ofthe minus-strand
of dsRNA. The latter possibility has not been
completely ruled out. Our interpretation ofthe
datareportedhere is inagreementwith themodel
ofreovirus dsRNAreplication recently proposed
by Schonberg et al. (9). The possibilitythat the
replicase productistriple-stranded is unlikely in
view of the similarity of the product to virion
dsRNA in all physical properties determined.
Assumingthevalidityof the above
interpreta-tion, the fact thatthe replicase product did not
contain any detectable amounts of labeled
plus-RNA suggests that theplus-RNA synthesized by
the contaminating transcriptase did not
subse-quently serve asa template for furtherreplicase
activity. This failureto reinitiate thesynthesis of
minus-RNA by using the nascent plus-RNA as
template is consistent with the demonstration
that the in vitro replicase reaction slows down
rapidly (Fig. 1).
Itisinterestingthat the nascentplus-RNAdid
not readily serve as template in the cell-free
system. One hypothesisto explain this would be
that for the nascentplus-RNA to become
func-tional as a template for replicase action, a
con-comitant protein synthesis would be required.
The protein(s) couldbe eitherareplicaseorthose
proteins required for the formation ofan active
replicase-template complex. We have previously
shown that, in reovirus-infected cells,the
inhibi-tion of protein synthesis by cycloheximide leads
to a rapid cessation of viral dsRNA synthesis,
and the plus-RNA (mRNA) synthesized and
accumulated in the presence ofcycloheximide is
notlaterincorporated into dsRNAafter removal
of thedrug (14).
ACKNOWLEDGMENT
This investigation was supported by Public Health Service
researchgrantAl-02454 from the NationalInstitute ofAllergy andInfectiousDisease.
LITERATURE CITED
1. Banerjee, A. K., and A. J. Shatkin. 1970. Transcriptionin
vitro by reovirus-associated ribonucleic acid-dependent
polymerase. J. Virol. 6:1-ll.
2.Bellamy, A. R., and W. K.Joklik.1967. Studiesonthe A-rich RNAof reovirus. Proc. Nat.Acad.Sci. U.S.A.
58:1389-1395.
3. Borsa, J., J. Grover, and J. D. Chapman. 1970.Presenceof
nucleoside triphosphate phosphohydrolaseactivityin
puri-fiedvirions of reovirus. J. Virol. 6:295-302.
4. Gomatos, P. J. 1970. Comparison of virion polymerase of
reovirus with the enzyme purified from reovirus-infected cells. J. Virol. 6:610-620.
5. Levin, D. H., N. Mendelsohn, M. Schonberg, H.Klett, S.
Silverstein, A. M. Kapuler, and G. Acs. 1970.Properties of RNAtranscriptase in reovirus subviral particles.Proc. Nat.Acad.Sci. U.S.A. 66:890-897.
6. Lowry, 0. H., N. J. Rosebrough, A. L. Farr, and R. J.
Randall. 1951.Proteinmeasurementwiththe Folin phenol
reagent.J. Biol. Chem. 193:265-275.
7.Lozeron, H. A., and W. Szybalski. 1966. Suppressionof RNA precipitation duringCs2S04density gradient centrifugation. Biochem.Biophys. Res. Commun. 23:612-618.
8.Nonoyama, M., Y. Watanabe, andA.F. Graham. 1970. De-fectivevirionsof reovirus. J. Virol. 6:226-236.
9. Schonberg,M., S. C. Silverstein, D. H. Levin. and G. Acs. 1971. Asynchronous synthesis of thecomplementary strands ofthereovirusgenome. Proc. Nat. Acad. Sci.U.S.A. 68: 505-508.
10. Shatkin, A.J., and J.D.Sipe. 1968. RNA polymerase activity in purified reoviruses. Proc. Nat. Acad. Sci. U.S.A. 61: 1462-1469.
11. Skehel, J.J., and W. K. Joklik. 1969. Studiesonthe invitro transcriptionofreovirusRNAcatalyzed by reoviruscore.
Virology39:822-831.
12. Watanabe,Y.,C. J.Gauntt,and A. F.Graham. 1968. Reo-virus-inducedribonucleicacidpolymerase.J. Virol. 2:869-877.
13.Watanabe, Y.,and A. F.Graham. 1967.Structuralunits of reovirusribonucleicacid and theirpossiblefunctional sig-nificance.J. Virol. 1:665-677.
14. Watanabe, Y.,H.Kudo,andA.F.Graham. 1967.Selective inhibitionofreovirusribonucleic acidsynthesis by cyclo-heximide. J.Virol.1:36-44.
15.Watanabe, Y.,S.Millward,and A.F. Graham.1968. Regu-lationoftranscriptionof the reovirusgenome.J.Mol. Biol. 36:107-123.
J. VIROL.