0022-538X/07/$08.00⫹0 doi:10.1128/JVI.00445-07
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
MINIREVIEW
Epstein-Barr Virus Entry
䌤
Lindsey M. Hutt-Fletcher*
Department of Microbiology and Immunology, Center for Molecular and Tumor Virology and Feist-Weiller Cancer Center, Louisiana State University Health Sciences Center, Shreveport, Louisiana
One of the many conundrums of herpesvirology is why a herpesvirus is so profligate in its use of four or more envelope glycoproteins for entry into a cell when other viruses can man-age very well with only one or two. Profligacy does, however, have its advantages. Epstein-Barr virus (EBV), originally rec-ognized for its ability to infect and transform lymphocytes, is now clearly understood to infect epithelial cells as part of its normal cycle of persistence in a human host, and under some circumstances, the virus may infect T cells, natural killer cells, smooth muscle cells (47), and possibly monocytes as well (11, 50). Our understanding of how EBV enters each of these cell types is very incomplete, but some of the major players in-volved in B-cell and epithelial cell infections are being identi-fied, and they provide a window into the flexibility of tropism that the use of different combinations of virus and cell mem-brane proteins can provide. This review summarizes what we know about these players so far.
ATTACHMENT
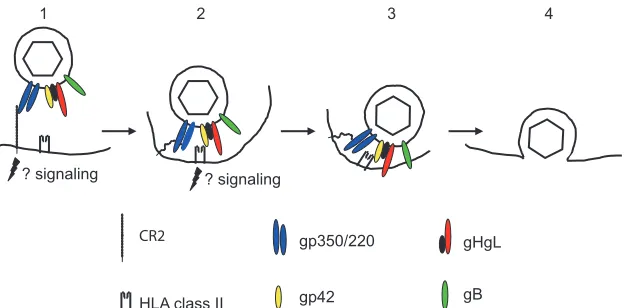
(i) B cells.Eight virus glycoproteins have been implicated in some way in EBV entry into either a B cell or an epithelial cell (Table 1). One of the most abundant of these in the virus envelope, gp350/220 (22), is responsible for attachment of the virus with high affinity (36) to the complement receptor type 2 (CR2) on B cells (7, 8, 40, 41, 60, 61). An EBV recombinant lacking gp350/220 can transform B cells with much-reduced efficiency (20), so CR2 is perhaps not the only portal by which the virus can access a B cell. It is, however, clearly the pre-dominant one (Fig. 1). Antibodies to gp350/220 that block virus binding neutralize B-cell infection, and soluble forms of either CR2 or gp350/220 can do the same (35, 61).
Glycoprotein gp350/220 is a highly glycosylated single-pass membrane protein, which as a result of alternative splicing is made in two forms, with approximate masses of 350 and 220 kDa (1, 17). Residues 500 to 757, which include three repeats of a 21-amino-acid motif with amphipathic characteristics, are lost from the full-length 907-amino-acid protein as a result of the splice. It does, however, maintain the reading frame, and both forms of the protein retain the CR2 binding domain at the amino terminus (61). Solution of the structure of an
unligan-ded form of gp350/220 has mapped this domain to a glycan-free surface (59) that includes a sequence with some similarity to the natural ligand of CR2, the C3dg fragment of comple-ment (26, 39, 61). The extracellular domain of CR2 is com-posed of tandem repeats of structural modules 60 to 75 amino acids in length known as short consensus repeats (SCR), and the binding site for gp350/220 has been genetically mapped to a region composed of residues from the membrane distal SCR1 and SCR2 (30). Attachment initially positions the virus at approximately 50 nm from the cell surface (38), but the segmental flexibility that the repeated SCRs may give to CR2 (68) and the possibility of an exchange in binding from gp350 to gp220 may contribute to moving the virus closer to the cell membrane.
Binding of gp350/220 also triggers capping of CR2 and en-docytosis of the virus (38, 60). CR2 can function as a signal transducer both independently and as part of a signal trans-duction complex that includes CD19 and CD35 (5). Cross-linking by gp350/220 activates NF-B (53, 58) and induces interleukin-6 via a protein kinase C pathway (4, 62). None of these signaling events may be critical to penetration of the cell membrane, but they clearly have potential consequences for downstream events in infection.
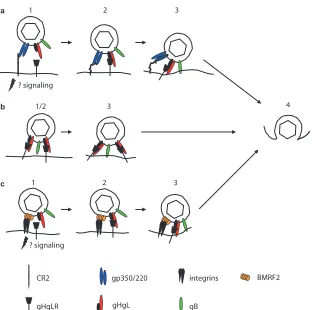
(ii) Epithelial cells.Attachment of the virus to an epithelial cell is a more-complicated affair (Fig. 2). CR2 is expressed at least at low levels on some epithelial cells in culture (6) in the absence of CD19 and CD35, and infection of an epithelial cell that has been engineered to express high levels of CR2 can be very efficient (3, 29). Notably, in this regard, EBV-mediated cross-linking of CR2 on epithelial cells, which lack the other components of the CR2 signaling complex found on B cells, stimulates relocalization and clustering of CR2 and the formin homolog overexpressed in spleen (FHOS/FHOD1). Formins are molecular scaffolds that nucleate actin, linking signal trans-duction to actin reorganization and gene transcription (10), and this may contribute to successful delivery of the virus to the nucleus of the cell. However, which, or even whether, epithelial cells normally express CR2 in vivo remains uncer-tain. Studies with monoclonal antibodies to CR2 were com-promised by the finding that the antibody most commonly used cross-reacted with an unrelated epithelial cell protein (73).
At least three other possible attachment mechanisms have been proposed that involve neither gp350/220 nor CR2. The first was a demonstration that virus coated with immunoglob-ulin A specific to gp350/220 can bind productively to the poly-meric immunoglobulin-A receptor (54). This may be particu-larly relevant to infection via the basolateral surface of an
* Mailing address: Department of Microbiology and Immunology, Louisiana State University Health Sciences Center, 1501 Kings Highway, Shreveport, LA 71130. Phone: (318) 675-4948. Fax: (318) 675-5764. E-mail: [email protected].
䌤Published ahead of print on 25 April 2007.
7825
on November 8, 2019 by guest
http://jvi.asm.org/
epithelial cell in an immune host, although, since in polarized cells virus was transported intact from the basolateral to the apical surface, it may be more relevant totransepithelial trans-port than direct infection (9). The second was a demonstration that in the absence of CR2 a complex of two additional glyco-proteins, gH and gL, can serve as epithelial ligands (34, 43). EBV gH, a type 1 membrane protein, is, like its homologs in other herpesviruses, misfolded and retained in the endoplas-mic reticulum in the absence of the much smaller type 2 pro-tein, gL (27, 72). No separate function in addition to facilitat-ing foldfacilitat-ing and transport of gH has been ascribed to gL, and the two proteins, which associate noncovalently, are usually referred to as a unit, the gHgL complex. EBV derived from a B cell can bind well to a CR2-negative epithelial cell, but recombinant viruses that lack gHgL lose this ability (34, 43). A soluble form of gHgL made in baculovirus can bind specifically to epithelial cells, but not B cells, and its binding can be reduced by a monoclonal antibody specific for the gHgL com-plex (3). The same antibody can also reduce virus binding (34). These observations have been interpreted to mean that there is an epithelial cell receptor for gHgL that can serve in attach-ment. The identity of the molecule is not yet known, but op-erationally it has been referred to as gHgLR.
Finally, and most intriguing, on polarized epithelial cells, which presumably resemble more closely the environment that
the virus encounters in vivo, an interaction between a multi-span virus membrane protein encoded by the BMRF2 open reading frame and integrins has been demonstrated (63). An-tibodies to integrins and to a BMRF2 fusion protein only partially block binding to polarized epithelial cells but have a significant impact on infection via the basolateral surface of the polarized monolayer. The RGD motif present in an exposed loop of BMRF2 has been shown to be a ligand for1,␣5,␣3, and␣v integrins (71). The protein is not required for cell-cell fusion (12, 31). However, whether the BMRF2 integrin inter-action is primarily responsible for attachment or whether it is most relevant to postattachment events to which signaling may make an important contribution is not yet clear. There is ap-parently very little BMRF2 protein in the virion (22).
FUSION AND PENETRATION
Fusion of the EBV envelope with both a B cell and an epithelial cell requires three glycoproteins, gHgL and gB, which are conserved throughout the herpesvirus family and which have been referred to as the core fusion machinery (55). However, how and where this machinery is activated differs significantly for each cell type.
(i) B cells.Fusion of the virus with a normal B cell (Fig. 1) requires endocytosis, which is triggered by the interaction
be-TABLE 1. EBV envelope glycoproteins with roles in entry into B cells or epithelial cells
Protein/genea Type Role in virus entry
gp350/BLLF1 Single pass type1 Attachment to B-cell receptor CR2/CD21
gH/BXLF2 Single pass type 1 Fusion; attachment to epithelial cell receptor/coreceptor
gL/BKRF2 Single pass type 2 Chaperone for gH
gp42/BZLF2 Single pass type 2 Fusion; interaction with B-cell coreceptor HLA class II
gB/BALF4 Single pass type 1 Fusion
gN/BLRF1 Single pass type 1 Codependent with gM for expression; possibly involved in postfusion events
gM/BBRF3 Multispanning
BMRF2 Multispanning Binds integrins; important for infection of polarized epithelial cells
[image:2.585.45.540.80.175.2]aEBV envelope proteins are conventionally named for their apparent masses (e.g., gp350); the alphabetical names of homologs in other herpesviruses, if they exist, (e.g., gH); or their gene names only (e.g., BMRF2).
FIG. 1. A putative model of the steps involved in entry of virus into a B lymphocyte. (1) The virus binds to CR2 via gp350, possibly initiating signaling events and triggering endocytosis. (2) CR2 may now bind to gp220 rather than gp350, and this, combined with the potential flexibility of CR2, may allow the virus to approach closer to the cell membrane, where gp42 can interact with HLA class II. (3) Interaction of gp42 with HLA class II triggers the interaction of the core fusion machinery, gHgL and gB, with the endosomal membrane and may also initiate further signaling events. (4) The virus and endosomal membranes fuse, allowing entry of the tegumented capsid into the cytoplasm. Modified from reference 18, with permission.
7826 MINIREVIEW J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:2.585.137.450.513.667.2]tween gp350/220 and CR2 and occurs in a low-pH compart-ment, although low pH itself is not required (32). The virus proteins that are both necessary and sufficient for efficient fusion are gHgL, gB, and gp42. A role for gH was first dem-onstrated when it was shown that virosomes made from pro-teins depleted of those that bound to an antibody to gH could attach to B cells but could not fuse (14). It was later confirmed by analysis of the phenotype of a recombinant gH-null virus (34). A role for EBV gB was not possible to evaluate geneti-cally, since, unlike its homologs in other herpesviruses, it is essential for virus morphogenesis and egress (16). However, when cell-cell fusion assays were developed as models of virus-cell fusion, it became apparent that its contribution is also very important (12, 31).
The fourth and final protein involved in fusion of EBV with a B cell, gp42, has homologs only among the lymphocryptovi-ruses (48, 49). It is another type 2 membrane protein, and it associates noncovalently with gHgL. The original observation was that gp42 resembles a C-type lectin and that a soluble form made by replacing the signal sequence with one that is effi-ciently cleaved can bind to HLA class II (56). Within the three-part complex of gHgLgp42 in the virus, this interaction is
now thought to provide the trigger for B-cell fusion. A mono-clonal antibody that was initially mapped to gH (57) but ulti-mately proved to react with gp42 (28) inhibited interaction with HLA class II (27) and also blocked virus-cell fusion (33). Reciprocally, a monoclonal antibody to HLA class II that blocked interaction with gp42 also inhibited infection. The soluble form of gp42 initially used to document HLA class II binding could inhibit infection by competing with gp42 in the virus, and B cells that lacked HLA class II could be efficiently infected only if HLA class II expression was restored (27). A recombinant gp42-null virus could infect a B cell only if the cells and the bound virus were treated with an exogenous fusogen such as polyethylene glycol (65) or if the soluble form of gp42, which retained a gHgL binding domain at its amino terminus, was added intrans to reform three-part complexes (66).
[image:3.585.138.450.63.373.2]Binding of gp42 to HLA class II shows some allelic speci-ficity in that nonfunctional HLA-DQ alleles have been identi-fied (13), but since all three alleles, HLA-DP, HLA-DQ, and HLA-DR, can be used, it is perhaps unlikely that this has a major impact in an outbred human population. A crystal struc-ture of the ectodomain of gp42 liganded to HLA-DR1 has
FIG. 2. Putative models of the steps involved in entry of virus into epithelial cells after attachment via different cell proteins. (a) Entry via CR2 and gHgLR. (1) The virus binds to CR2 via gp350; signaling may occur, but endocytosis is not triggered. (2) A switch to interaction with gp220, combined with the potential flexibility of CR2, may allow the virus to approach closer to the cell membrane, where gHgL can interact with gHgLR. (3) Interaction of gHgL with gHgLR triggers the interaction of the core fusion machinery, gHgL and gB, with the cell membrane. (b) Entry via gHgLR alone (which is less efficient). (1) There is no separate attachment event. (2) gHgL interacts directly with gHgLR. (3) Interaction of gHgL with gHgLR triggers the interaction of the core fusion machinery, gHgL and gB, with the cell membrane. (c) Entry via integrins and gHgLR. (1) BMRF2 interacts with␣51 integrins, possibly initiating signaling events. (2) gHgL interacts with gHgLR. (3) Interaction of gHgL with gHgLR triggers the interaction of the core fusion machinery, gHgL and gB, with the cell membrane. Step 4 is the same for all routes: the virus and cell membrane fuse, allowing entry of the tegumented capsid into the cytoplasm. Modified from reference 18, with permission.
on November 8, 2019 by guest
http://jvi.asm.org/
been solved (37). The protein is most closely related to natural killer cell receptors, such as the murine receptor LY49, which interact with major histocompatibility complex (MHC) class I molecules, but it does not use the canonical surface site which many C-type lectins use to bind to their ligands. Instead, this site forms a hydrophobic pocket that, it is suggested, may be used to accommodate another virus protein or another cell protein important for virus entry (37), perhaps after triggering by HLA class II. This is consistent with the observation that mutations in the hydrophobic pocket do not affect HLA class II binding but inhibit fusion (52). The contact region on HLA-DR1 is within the1 domain to one side of the peptide binding groove. It was thought on theoretical grounds to be likely to interfere sterically with the binding of T-cell receptors to the peptide MHC complex and has been shown experimentally to reduce T-helper-cell recognition of B cells (45, 46, 56). Both peptide-loaded and immature MHC class II proteins that have not yet trafficked to the peptide-loading compartment can in-teract with gp42 (46), and, as discussed below, this has proven to have an interesting impact on virus tropism. Whether or not additional signaling events may be initiated as a result of a gp42-HLA class II interaction is not yet known, but since signaling via HLA class II molecules is well documented under other circumstances, this remains a reasonable possibility (67).
(ii) Epithelial cells.Fusion with a normal epithelial cell, at least one that is nonpolarized, takes place at neutral pH and does not appear to require endocytosis (3, 32). Epithelial cells do not constitutively express HLA class II, and thus, not sur-prisingly, infection is not blocked by the monoclonal antibody that blocks gp42 and HLA class II interactions. A gp42-null virus can infect an epithelial cell as well as, or even better than, wild-type virus. However, not only is gp42 dispensable for epithelial cell infection, but its presence is actually inhibitory (66). Stoichiometric analysis of gHgL and gp42 in the virion indicated that wild-type EBV carries far more gHgL than gp42, implying that there are two kinds of gH complexes, one the three-part complex composed of gHgLgp42 and the other a two-part complex composed of only gHgL. Only three-part complexes can mediate B-cell infection, but conversion of all two-part complexes to three-part complexes by the addition of soluble gp42 intransblocks epithelial cell infection. This was interpreted to mean that fusion with an epithelial cell is trig-gered by a direct interaction between gHgL and a novel epi-thelial cell surface molecule. Three monoclonal antibodies that interact with gH or gHgL and have no effect on B-cell infection can also inhibit infection of epithelial cells, whether virus is bound via gp350/220 to CR2 or via gHgL to gHgLR. One of them blocks gHgL binding to gHgLR, suggesting that the cell molecule that triggers fusion might be the same protein, gHgLR, which can serve as an attachment receptor in the absence of CR2. Consistent with this is the observation that soluble gp42 also blocks binding of the virus to gHgLR. Use of gHgL for attachment as well as fusion does, however, seem to com-promise infection. The virus can bind efficiently to gHgLR on a CR2-negative epithelial cell, but infection levels are low, either because fusion becomes less efficient or perhaps because engagement of gHgLR does not induce a downstream event required for completion of transport of the virus genome to the nucleus (3).
Beyond this, epithelial cell penetration also requires higher
levels of gB than does B-cell penetration (42), presumably reflecting a need for more gB in fusion (31). However, the mechanics of fusion itself remain obscure, as they do for all herpesviruses. Mutations in an amino-terminal region of gH that is predicted to form a coiled coil significantly reduced fusion in a cell-cell fusion assay (44), and insertion or point mutations in a region close to the transmembrane domain can either differentially affect B-cell and epithelial cell fusion or abrogate fusion with either cell type (69, 70). However, some epithelial cell fusion can be mediated in the absence of gHgL by a gB construct with a mutation in the cytoplasmic tail. This has been interpreted to mean that gB may be the critical fusion protein of EBV. Certainly the crystal structure of the homolog of gB in herpes simplex virus shows structural homology with the vesicular stomatitis virus G protein, which is the virus fusogen (15). Both form trimers characterized by an alpha-helical coiled-coil core and extended-hairpins that are char-acteristic of class I and class II fusion proteins, respectively. At this point, however, perhaps the most that can be definitively said is that virus fusion with a B cell requires a trigger that is transmitted via gp42 to gHgL and gB, whereas fusion with an epithelial cell requires a trigger that is transmitted directly to gHgL. Whether there is a temporal cascade of events culmi-nating in fusion mediated by gB or whether gHgL and gB coordinately contribute to the process remains to be deter-mined.
TRANSFER OF VIRUS BETWEEN B CELLS AND EPITHELIAL CELLS
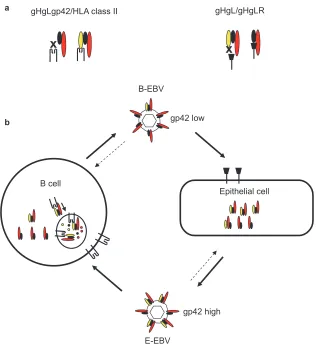
gp42 as a switch of tropism.The observation that two-part gHgL and three-part gHgLgp42 complexes are mutually exclu-sive for epithelial and B-cell entry suggested that the relative amounts of each complex in the virion might impact virus tropism. Analyses of viruses made in B cells and epithelial cells confirmed this. In a B cell, some three-part gHgLgp42 com-plexes prove to interact with immature MHC class II in the endoplasmic reticulum and are targeted to the HLA class II trafficking pathway, where they are vulnerable to degradation. This does not happen in an HLA class II-negative epithelial cell. Epithelial cell viruses are, as a result, rich in complexes containing gp42 and are as much as two orders of magnitude more infectious for a B cell than viruses produced by a B cell. B-cell viruses, lower in three-part complexes but rich in two-part complexes, are better able to infect an epithelial cell, although the phenotype is not nearly as striking. This suggested a model (Fig. 3) of persistent infection in which virus reacti-vating from a latently infected memory B cell as it terminally differentiates into a plasmablast (23) is equipped to infect an epithelial cell, whereas virus amplified in an epithelial cell is strongly B cell tropic (2).
EBV is an orally transmitted virus, and the switch in tropism effected by levels of gp42, which could impact transmission, raised the question of the cellular origins of viruses that are shed in saliva. Viruses made in a B cell, low in gp42, have been shown to be able to bind to gHgLR on an epithelial cell almost as well as they can bind CR2 on a B cell. In contrast, virus made in an epithelial cell, high in gp42, binds gHgLR very poorly (3). A comparison of the differential abilities of viruses from saliva and viruses from transformed B cells obtained from
7828 MINIREVIEW J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
a series of EBV-positive donors then facilitated the determi-nation that the majority of viruses shed in saliva are derived from HLA class II-negative cells (21). For the most-efficient transmission, this either means that the virus directly accesses a B cell in the oral mucosa or that transmission to an epithelial cell for amplification before accessing a B cell is influenced by other events. Two have been proposed, and both implicate the differential use of gp350/220.
gp350/220 as an impediment to epithelial infection. Anti-bodies to gp350/220, which inhibit infection of a B cell depen-dent on CR2 for entry, can enhance infection of an epithelial cell, which is not (64). The effect is not mediated by Fc receptor binding but is further increased by antibody cross-linking, which may patch gp350/220 in the virus envelope. Saliva from EBV-seropositive individuals has similar effects that can be reversed by depletion of antibodies. The interpretation of these results was that, if gp350/220 is not absolutely required, then its presence may impede access of other essential proteins to the cell surface. Antibodies to gp350/220, an abundant and immunogenic protein, can overcome this in the saliva of an
immune host to facilitate reinfection of an epithelial cell or to facilitate transmission through the epithelium in a new host.
[image:5.585.135.453.64.409.2]There are long-standing observations that epithelial infec-tion is more efficient if epithelial cells are cocultivated with B cells that are making the virus (19). A novel approach to investigation of the phenomenon demonstrated that binding EBV to a B cell before adding both to epithelial cells signifi-cantly enhanced infection of the epithelial cells, even in the absence of internalization and infection of the B cell (51). The mechanism of this B-cell transfer is uncertain but intriguing. The authors also suggested that gp350/220 is inhibitory to direct epithelial cell infection and proposed that the gp350/220 interaction with CR2 on the B cells unmasks envelope com-ponents necessary for epithelial cell infection. This parallels the hypothesis for enhanced infection in the presence of anti-bodies to gp350/220 in saliva. Neither study addressed what the envelope components necessary for efficient epithelial cell in-fection might be, but one likely candidate would be gB, the protein that is needed in higher amounts for epithelial than for B cell fusion.
FIG. 3. Glycoprotein gp42 as a switch of cell tropism. (a) B-cell infection requires an interaction between gHgLgp42 and HLA class II, whereas epithelial cell infection requires an interaction between gHgL and gHgLR. Complexes lacking gp42 cannot bind to HLA class II, and complexes containing gp42 cannot bind to gHgLR. (b) gHgLgp42 complexes can interact with HLA class II in the endoplasmic reticulum of a B cell. They may then be targeted to endosomal vesicles rich in proteases, where incoming proteins are digested into peptides for loading into the peptide binding groove of HLA class II before continuing on to the cell surface. The result is a loss of gHgLgp42 complexes to degradation and a relative decrease in gHgLgp42 complexes in viruses produced from B cells or plasmablasts (B-EBV). Complexes in epithelial cells are not lost to this degradative pathway, and viruses produced from epithelial cells (E-EBV) are richer in gp42. Epithelial cells high in gp42 are better able to infect B cells, and B cells low in gp42 are better able to infect epithelial cells. Modified from reference 18, with permission.
on November 8, 2019 by guest
http://jvi.asm.org/
POSTFUSION EVENTS
There is regrettably very little concrete information about what happens between fusion and the arrival of the virus ge-nome in the nucleus, only speculation based on what is known for other herpesviruses. Allusion has been made throughout this brief review to the likelihood that signaling events trig-gered by interactions between the virus envelope and cell pro-teins have important consequences for postfusion events, but how they may impact intracellular transport and uncoating of the virion is a subject that has not yet been substantively ad-dressed. Two glycoproteins in the virus envelope have, how-ever, also been tentatively implicated in what are presumably immediate postfusion events that may perhaps have no con-nection to cell signaling. These are glycoprotein gN, a small type I membrane protein, and glycoprotein gM. Glycoprotein gM is a multispan membrane protein with a long, highly charged cytoplasmic tail rich in prolines and replete with po-tential phosphorylation sites that might provide a mechanism for the regulation of protein-protein interactions. Glycopro-teins gN and gM are codependent for expression (24, 25) and play important roles in virus assembly and acquisition of the final virus envelope. Only very little enveloped virus is made by a recombinant EBV that lacks gN and gM. However, that enveloped virus which is made and is able to bind specifically to CR2 on a B cell is impaired in infection in a way that cannot be rescued by addition of exogenous fusogens such as polyeth-ylene glycol. One hypothesis is that a complex that is necessary for assembly of an enveloped particle when a tegumented capsid associates with a membrane is also essential to its dis-assembly when a tegumented capsid must disassociate from a membrane with which its envelope has fused.
SUMMARY
The major players involved in moving EBV through a B-cell membrane have been identified, at least on the part of the virus. The model emerging is that gp350/220 binds to CR2, gp42 binds to HLA class II, and fusion is mediated by gHgL and gB. The possibility clearly exists, however, that there are additional cell partners for gHgL and gB. Some of the players involved in moving EBV through an epithelial cell membrane have been identified. Fusion involves gHgL and gB, but attach-ment may involve gp350/220, gHgL, or BMRF2, or possibly, if the virus is transferred from the surface of a B cell, no attach-ment receptor specific for any viral protein at all. With the possible exception of CR2, no cell partners have yet been identified on an epithelial cell, though there is clear evidence that they exist. There is also no conceptual understanding of the mechanism of fusion and very little insight into events inside the cell that may be triggered by interactions occurring at the cell surface. Much is still to be learned.
Despite the incompleteness of our understanding, however, what we know already provides a fascinating glimpse into dif-ferential use of virus proteins and cell partners to manipulate virus tropism and influence virus trafficking between one cell compartment and another. EBV is associated with human ma-lignancies, and primary infection in a minority of individuals causes acute, but self-limiting, mononucleosis. The majority of humans, however, live without incident with a virus that
con-tinually infects their lymphocytes and epithelial cells. The strat-egies that have evolved to make this possible also continue to be remarkable.
ACKNOWLEDGMENTS
Research by L.M.H.-F. is supported by Public Health Service grants AI061017 from the National Institute of Allergy and Infectious Dis-eases and DE016669 from the National Institute of Dental and Cranio-facial Research.
REFERENCES
1.Beisel, C., J. Tanner, T. Matsuo, D. Thorley-Lawson, F. Kezdy, and E. Kieff. 1985. Two major outer envelope glycoproteins of Epstein-Barr virus are encoded by the same gene. J. Virol.54:665–674.
2.Borza, C. M., and L. M. Hutt-Fletcher.2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med.8:594– 599.
3.Borza, C. M., A. J. Morgan, S. M. Turk, and L. M. Hutt-Fletcher.2004. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol.78:5007–5014.
4.D’Addario, M., T. A. Libermann, J. Xu, A. Ahmad, and J. Menezes.2001. Epstein-Barr virus and its glycoprotein-350 upregulate IL-6 in human B cells via CD21, involving activation of NF-kB and different signaling pathways. J. Mol. Biol.308:501–514.
5.Fearon, D. T., and R. H. Carter.1995. The CD19/CR2/TAPA-1 complex of B lymphocytes: linking natural to acquired immunity. Annu. Rev. Immunol. 13:127–149.
6.Fingeroth, J. D., M. E. Diamond, D. R. Sage, J. Hayman, and J. L. Yates. 1999. CD-21 dependent infection of an epithelial cell line, 293, by Epstein-Barr virus. J. Virol.73:2115–2125.
7.Fingeroth, J. D., J. J. Weis, T. F. Tedder, J. L. Strominger, P. A. Biro, and D. T. Fearon.1984. Epstein-Barr virus receptor of human B lymphocytes is the C3d complement CR2. Proc. Natl. Acad. Sci. USA81:4510–4516. 8.Frade, R., M. Barel, B. Ehlin-Henricksson, and G. Klein.1985. Gp140, the
C3d receptor of human B lymphocytes, is also the Epstein-Barr virus recep-tor. Proc. Natl. Acad. Sci. USA82:1490–1493.
9.Gan, Y., J. Chodosh, A. Morgan, and J. W. Sixbey.1997. Epithelial cell polarization is a determinant in the infectious outcome of immunoglobulin A-mediated entry by Epstein-Barr virus. J. Virol.71:519–526.
10.Gill, M. B., J. Roecklein-Canfield, D. R. Sage, M. Zambela-Soediono, N. Longtine, M. Uknis, and J. D. Fingeroth.2004. EBV attachment stimulates FHOF/FHOD1 redistribution and co-aggregation with CD21:formin inter-actins with the cytoplasmic domain of human CD21. J. Cell Sci.117:2709– 2720.
11.Guerreiro-Cacais, A. O., L. Li, D. D. Donat, M. T. Bejarano, A. J. Morgan, M. Masucci, L. M. Hutt-Fletcher, and V. Levitsky.2004. The capacity of Epstein-Barr virus to infect monocytes and inhibit their development into dendritic cells is affected by the cell type supporting virus replication. J. Gen. Virol.85:2767–2778.
12.Haan, K. M., S. K. Lee, and R. Longnecker.2001. Different functional domains in the cytoplasmic tail of glycoprotein gB are involved in Epstein-Barr virus-induced membrane fusion. Virology290:106–114.
13.Haan, K. M., and R. Longnecker.2000. Coreceptor restriction within the HLA-DQ locus for Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 97:9252–9257.
14.Haddad, R. S., and L. M. Hutt-Fletcher.1989. Depletion of glycoprotein gp85 from virosomes made with Epstein-Barr virus proteins abolishes their ability to fuse with virus receptor-bearing cells. J. Virol.63:4998–5005. 15.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and
S. C. Harrison.2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science313:217–220.
16.Herrold, R. E., A. Marchini, S. Frueling, and R. Longnecker.1995. Glyco-protein 110, the Epstein-Barr virus homolog of herpes simplex virus glyco-protein B, is essential for Epstein-Barr virus replication in vivo. J. Virol. 70:2049–2054.
17.Hummel, M., D. Thorley-Lawson, and E. Kieff.1984. An Epstein-Barr virus DNA fragment encodes messages for the two major envelope glycoproteins (gp350/300 and gp220/200). J. Virol.49:413–417.
18.Hutt-Fletcher, L. M.2005. EBV entry and epithelial infection, p. 359–378.In
E. S. Robertson (ed.), Epstein-Barr virus. Caister Academic Press, Norfolk, England.
19.Imai, S., J. Nishikawa, and K. Takada.1998. Cell-to-cell contact as an efficient mode of Epstein-Barr virus infection of diverse human epithelial cells. J. Virol.72:4371–4378.
20.Janz, A., M. Oezel, C. Kurzeder, J. Mautner, D. Pich, M. Kost, W. Ham-merschmidt, and H. J. Delecluse.2000. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of addi-tional viral ligands. J. Virol.74:10142–10152.
21.Jiang, R., R. S. Scott, and L. M. Hutt-Fletcher.2006. Epstein-Barr virus shed in saliva is high in B-cell-tropic gp42. J. Virol.80:7281–7283.
7830 MINIREVIEW J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
22.Johannsen, E., M. Luftig, M. R. Chase, S. Weicksel, E. Cahir-McFarland, D. Illanes, D. Sarracino, and E. Kieff.2004. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA101:16286–16291.
23.Laichalk, L. L., and D. A. Thorley-Lawson.2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol.79:1296–1307.
24.Lake, C. M., and L. M. Hutt-Fletcher.2000. Epstein-Barr virus that lacks glycoprotein gN is impaired in assembly and infection. J. Virol.74:11162– 11172.
25.Lake, C. M., S. J. Molesworth, and L. M. Hutt-Fletcher.1998. The Epstein-Barr virus (EBV) gN homolog BLRF1 encodes a 15-kilodalton glycoprotein that cannot be authentically processed unless it is coexpressed with the EBV gM homolog BBRF3. J. Virol.72:5559–5564.
26.Lambris, J. D., V. S. Ganu, S. Hirani, and H. J. Muller-Eberhard.1985. Mapping of the C3d receptor (CR2) binding site and a neoantigenic site in the C3d domain of the third component of complement. Proc. Natl. Acad. Sci. USA82:4235–4239.
27.Li, Q. X., M. K. Spriggs, S. Kovats, S. M. Turk, M. R. Comeau, B. Nepom, and L. M. Hutt-Fletcher.1997. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol.71:4657–4662.
28.Li, Q. X., S. M. Turk, and L. M. Hutt-Fletcher.1995. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithe-lial cells. J. Virol.69:3987–3994.
29.Li, Q. X., L. S. Young, G. Niedobitek, C. W. Dawson, M. Birkenbach, F. Wang, and A. B. Rickinson.1992. Epstein-Barr virus infection and replica-tion in a human epithelial system. Nature356:347–350.
30.Martin, D. R., A. Yuryev, K. R. Kalli, D. T. Fearon, and J. M. Ahearn. 1991. Determination of the structural basis for selective binding of Ep-stein-Barr virus to human complement receptor type 2. J. Exp. Med. 174:1299–1311.
31.McShane, M. P., and R. Longnecker. 2004. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc. Natl. Acad. Sci. USA101:17474–17479. 32.Miller, N., and L. M. Hutt-Fletcher.1992. Epstein-Barr virus enters B cells
and epithelial cells by different routes. J. Virol.66:3409–3414.
33.Miller, N., and L. M. Hutt-Fletcher.1988. A monoclonal antibody to glyco-protein gp85 inhibits fusion but not attachment of Epstein-Barr virus. J. Vi-rol.62:2366–2372.
34.Molesworth, S. J., C. M. Lake, C. M. Borza, S. M. Turk, and L. M. Hutt-Fletcher.2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324–6332.
35.Moore, M. D., M. J. Cannon, A. Sewall, M. Finlayson, M. Okimoto, and G. R. Nemerow.1991. Inhibition of Epstein-Barr virus infection in vitro and in vivo by soluble CR2 (CD21) containing two short consensus repeats. J. Virol.65:3559–3565.
36.Moore, M. D., R. G. DiScipio, N. R. Cooper, and G. R. Nemerow.1989. Hydrodynamic, electron microscopic, and ligand-binding analysis of the Epstein-Barr virus/C3dg receptor (CR2). J. Biol. Chem.34:20576–20582. 37.Mullen, M. M., K. M. Haan, R. Longnecker, and T. S. Jardetzky.2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell9:375–385.
38.Nemerow, G. R., and N. R. Cooper.1984. Early events in the infection of human B lymphocytes by Epstein-Barr virus. Virology132:186–198. 39.Nemerow, G. R., R. A. Houghton, M. D. Moore, and N. R. Cooper.1989.
Identification of the epitope in the major envelope proteins of Epstein-Barr virus that mediates viral binding to the B lymphocyte EBV receptor (CR2). Cell56:369–377.
40.Nemerow, G. R., C. Mold, V. K. Schwend, V. Tollefson, and N. R. Cooper. 1987. Identification of gp350 as the viral glycoprotein mediating attachment of Epstein-Barr virus (EBV) to the EBV/C3d receptor of B cells: sequence homology of gp350 and C3 complement fragment C3d. J. Virol.61:1416– 1420.
41.Nemerow, G. R., R. Wolfert, M. McNaughton, and N. R. Cooper.1985. Identification and characterization of the Epstein-Barr virus receptor on human B lymphocytes and its relationship to the C3d complement receptor (CR2). J. Virol.55:347–351.
42.Neuhierl, B., R. Feederle, W. Hammerschmidt, and H. J. Delecluse.2002. Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and ef-ficiency of infection. Proc. Natl. Acad. Sci. USA99:15036–15041. 43.Oda, T., S. Imai, S. Chiba, and K. Takada.2000. Epstein-Barr virus lacking
glycoprotein gp85 cannot infect B cells and epithelial cells. Virology276: 52–58.
44.Omerovic, J., L. Lev, and R. Longnecker.2005. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with B cells and epithelial cells. J. Virol.79:12408–12415.
45.Ressing, M. E., D. van Leeuwen, F. A. W. Verreck, R. Gomez, B. Heemskerk, M. Toebes, M. M. Mullen, T. S. Jardetzky, R. Longnecker, M. W. Schilham, T. H. M. Ottenhoff, J. Neefjes, T. N. Schumacher, L. M. Hutt-Fletcher, and E. J. H. J. Wiertz.2003. Interference with T cell receptor-HLA-DR
inter-actions by Epstein-Barr virus gp42 results in reduced T helper cell recogni-tion. Proc. Natl. Acad. Sci. USA100:11583–11588.
46.Ressing, M. E., D. van Leeuwen, F. A. W. Verreck, S. Keating, R. Gomez, K. L. M. C. Franken, T. H. M. Ottenhoff, M. Spriggs, T. N. Schumacher, L. M. Hutt-Fletcher, M. Rowe, and E. J. H. J. Wiertz.2005. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that me-diates class II immune evasion. J. Virol.79:841–852.
47.Rickinson, A. B., and E. Kieff.2007. Epstein-Barr virus, p. 2655–2700.In
D. M. Knipe and P. M. Howley (ed.), Fields virology. Lippincott Williams & Wilkins, Philadelphia, PA.
48.Rivailler, P., Y.-G. Cho, and F. Wang.2002. Complete genomic sequence of an Epstein-Barr virus-related herpesvirus naturally infecting a New World primate: a defining point in the evolution of oncogenic lymphocryptoviruses. J. Virol.76:12055–12068.
49.Rivailler, P., H. Jiang, Y.-G. Cho, C. Quink, and F. Wang.2002. Complete nucleotide sequence of the rhesus lymphocryptovirus: genetic validation for an Epstein-Barr virus animal model. J. Virol.76:421–426.
50.Savard, M., C. Belanger, M. Tardif, P. Gourde, L. Flamand, and J. Gosselin. 2000. Infection of primary human monocytes by Epstein-Barr virus. J. Virol. 74:2612–2619.
51.Shannon-Lowe, C. D., B. Neuhierl, G. Baldwin, A. B. Rickinson, and H.-J. Delecluse.2006. Resting B cells as a transfer vehicle for Epstein-Barr virus infection of epithelial cells. Proc. Natl. Acad. Sci. USA103:7065– 7070.
52.Silva, A. L., J. Omerovic, T. S. Jardetzky, and R. Longnecker.2004. Muta-tional analysis of Epstein-Barr virus glycoprotein gp42 reveals funcMuta-tional domains not involved in receptor binding but required for membrane fusion. J. Virol.78:5946–5956.
53.Sinclair, A. J., and P. J. Farrell.1995. Host cell requirements for efficient infection of quiescent primary B lymphocytes by Epstein-Barr virus. J. Virol. 69:5461–5468.
54.Sixbey, J. W., and Q.-Y. Yao.1992. Immunoglobulin A-induced shift of Epstein-Barr virus tissue tropism. Science255:1578–1580.
55.Spear, P. G., and R. Longnecker.2003. Herpesvirus entry: an update. J. Vi-rol.77:10179–10185.
56.Spriggs, M. K., R. J. Armitage, M. R. Comeau, L. Strockbine, T. Farrah, B. MacDuff, D. Ulrich, M. R. Alderson, J. Mu¨llberg, and J. I. Cohen. 1996. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DRchain and inhibits antigen presentation. J. Virol. 70:5557–5563.
57.Strnad, B. C., T. Schuster, R. Klein, R. F. Hopkins III, T. Witmer, R. H. Neubauer, and H. Rabin.1982. Production and characterization of mono-clonal antibodies against the Epstein-Barr virus membrane antigen. J. Virol. 41:258–264.
58.Sugano, N., W. Chen, M. L. Roberts, and N. R. Cooper.1997. Epstein-Barr virus binding to CD21 activates the initial viral promoter via NF-B induc-tion. J. Exp. Med.186:731–737.
59.Szakonyi, G., M. G. Klein, J. P. Hannan, K. A. Young, R. Z. Ma, R. Asokan, V. M. Holers, and X. S. Chen.2006. Structure of the Epstein-Barr virus major envelope glycoprotein. Nat. Struct. Mol. Biol.13:996–1001. 60.Tanner, J., J. Weis, D. Fearon, Y. Whang, and E. Kieff.1987. Epstein-Barr
virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorp-tion, capping, and endocytosis. Cell50:203–213.
61.Tanner, J., Y. Whang, J. Sample, A. Sears, and E. Keiff.1988. Soluble gp350/220 and deletion mutant glycoproteins block Epstein-Barr virus ad-sorption to lymphocytes. J. Virol.62:4452–4464.
62.Tanner, J. E., C. Alfieri, T. A. Chatila, and F. Diaz-Mitoma.1996. Induction of interleukin-6 after stimulation of human B-cell CD21 by Epstein-Barr virus glycoproteins gp350 and gp220. J. Virol.70:570–575.
63.Tugizov, S. M., J. W. Berline, and J. M. Palefsky.2003. Epstein-Barr virus infection of polarized tongue and nasopharyngeal epithelial cells. Nat. Med. 9:307–314.
64.Turk, S. M., R. Jiang, L. S. Chesnokova, and L. M. Hutt-Fletcher.2006. Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J. Virol.80:9628–9633.
65.Wang, X., and L. M. Hutt-Fletcher. 1998. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 72:158–163.
66.Wang, X., W. J. Kenyon, Q. X. Li, J. Mullberg, and L. M. Hutt-Fletcher. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol.72:5552–5558. 67.Watt, T. H.1997. Signaling via MHC class II molecules, p. 141–161.InM.
Harnete and K. P. Rigley (ed.), Lymphocyte signaling: mechanisms, subver-sion and manipulation. John Wiley and Sons Ltd., New York, NY. 68.Weisman, H. F., T. Bartow, M. K. Leppo, H. C. Marsh, Jr., G. R. Carson,
M. F. Concino, M. P. Boyle, K. H. Roux, M. L. Weisdeldt, and D. T. Fearon. 1990. Soluble human complement receptor type 1: in vivo inhibitor of com-plement suppressing post-ischemic myocardial inflammation and necrosis. Science249:146–151.
69.Wu, L., C. M. Borza, and L. M. Hutt-Fletcher.2005. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J. Virol.79:10923–10930.
on November 8, 2019 by guest
http://jvi.asm.org/
70.Wu, L., and L. M. Hutt-Fletcher.20 February 2007. Point mutations in EBV gH that abrogate or differentially affect B cell and epithelial cell fusion. Virology363:148–155. [Epub ahead of print.]
71.Xiao, J., J. M. Palefsky, R. Herrera, and S. M. Tugizov.2007. Charac-terization of the Epstein-Barr virus glycoprotein BMRF2. Virology359: 382–396.
72.Yaswen, L. R., E. B. Stephens, L. C. Davenport, and L. M. Hutt-Fletcher.
1993. Epstein-Barr virus glycoprotein gp85 associates with the BKRF2 gene product and is incompletely processed as a recombinant protein. Virology 195:387–396.
73.Young, L. S., C. W. Dawson, K. W. Brown, and A. B. Rickinson.1989. Identification of a human epithelial cell surface protein sharing an epitope with the C3d/Epstein-Barr virus receptor molecule of B lymphocytes. Int. J. Cancer43:786–794.
7832 MINIREVIEW J. VIROL.