Phosphorylation of Human Metapneumovirus M2-1 Protein
Upregulates Viral Replication and Pathogenesis
Hui Cai,aYu Zhang,aMijia Lu,bXueya Liang,aRyan Jennings,aStefan Niewiesk,aJianrong Lia
Department of Veterinary Biosciences, College of Veterinary Medicine, The Ohio State University, Columbus, Ohio, USAa; Laboratory of New Drugs Safety Evaluation, Zhejiang Academy of Medical Sciences, Hangzhou, Zhejiang, People’s Republic of Chinab
ABSTRACT
Human metapneumovirus (hMPV) is a major causative agent of upper- and lower-respiratory-tract infections in infants, the elderly, and immunocompromised individuals worldwide. Like all pneumoviruses, hMPV encodes the zinc binding protein M2-1, which plays important regulatory roles in RNA synthesis. The M2-1 protein is phosphorylated, but the specific role(s) of the phosphorylation in viral replication and pathogenesis remains unknown. In this study, we found that hMPV M2-1 is phos-phorylated at amino acid residues S57 and S60. Subsequent mutagenesis found that phosphorylation is not essential for zinc binding activity and oligomerization, whereas inhibition of zinc binding activity abolished the phosphorylation and oligomer-ization of the M2-1 protein. Using a reverse genetics system, recombinant hMPVs (rhMPVs) lacking either one or both phos-phorylation sites in the M2-1 protein were recovered. These recombinant viruses had a significant decrease in both genomic RNA replication and mRNA transcription. In addition, these recombinant viruses were highly attenuated in cell culture and cot-ton rats. Importantly, rhMPVs lacking phosphorylation in the M2-1 protein triggered high levels of neutralizing antibody and provided complete protection against challenge with wild-type hMPV. Collectively, these data demonstrated that phosphoryla-tion of the M2-1 protein upregulates hMPV RNA synthesis, replicaphosphoryla-tion, and pathogenesisin vivo.
IMPORTANCE
The pneumoviruses include many important human and animal pathogens, such as human respiratory syncytial virus (hRSV), hMPV, bovine RSV, and avian metapneumovirus (aMPV). Among these viruses, hRSV and hMPV are the leading causes of acute respiratory tract infection in infants and children. Currently, there is no antiviral or vaccine to combat these diseases. All known pneumoviruses encode a zinc binding protein, M2-1, which is a transcriptional antitermination factor. In this work, we found that phosphorylation of M2-1 is essential for virus replication and pathogenesisin vivo. Recombinant hMPVs lacking phosphor-ylation in M2-1 exhibited limited replication in the upper and lower respiratory tract and triggered strong protective immunity in cotton rats. This work highlights the important role of M2-1 phosphorylation in viral replication and that inhibition of M2-1 phosphorylation may serve as a novel approach to develop live attenuated vaccines as well as antiviral drugs for pneumoviruses.
H
uman metapneumovirus (hMPV) is one of the leading causes of upper and lower respiratory tract infection in humans, particularly in infants, young children, the elderly, and immuno-compromised individuals. The virus was first identified in chil-dren suffering from respiratory illness in the Netherlands in 2001 (1). Subsequent epidemiological studies suggested that 5 to 15% of all respiratory tract infections in infants and young children were caused by hMPV, a proportion second only to that of human respiratory syncytial virus (hRSV) (2–5). hMPV belongs to the Metapneumovirusgenus within the Pneumovirinaesubfamily of the familyParamyxoviridae. Avian metapneumovirus (aMPV) is the only other member of theMetapneumovirusgenus. The Pneu-movirusgenus includes hRSV, the most common cause of pediat-ric respiratory infection, pneumonia virus of mice (PVM), and bovine RSV, which cause respiratory tract infections in animals. Currently, there is no FDA-approved vaccine for hMPV and hRSV.A unique characteristic of pneumovirus replication and tran-scription is the requirement of a cofactor, the M2-1 protein, for the RNA-dependent RNA polymerase (RdRp) complex, in addi-tion to the large (L) polymerase catalytic subunit and the accessory phosphoprotein (P) (6–8). All known pneumoviruses possess the M2 gene, which carries two overlapping open reading frames (ORFs), producing the M2-1 and M2-2 proteins, although the
location of the M2 gene in the genome varies among different pneumoviruses (1,6,9). The M2 gene of hMPV is located between the fusion protein (F) and the small hydrophobic protein (SH) genes, whereas the M2 gene of hRSV is located between the F and L genes. The functions of hRSV M2-1 are well characterized. HRSV M2-1 functions as a transcriptional elongation factor and antiterminator that enhances readthrough at intergenic junctions (10–13). Thus, M2-1 is essential for synthesis of full-length mR-NAs and polycistronic mRmR-NAs (10–13). HRSV M2-1 is an RNA binding protein, specifically interacting with viral mRNA (14,15). In addition, hRSV M2-1 was shown to interact with the P protein and colocalize with the N and L proteins (16,17), suggesting that M2-1 is a component of the RNP complex and plays an important
Received19 April 2016Accepted25 May 2016
Accepted manuscript posted online1 June 2016
CitationCai H, Zhang Y, Lu M, Liang X, Jennings R, Niewiesk S, Li J. 2016.
Phosphorylation of human metapneumovirus M2-1 protein upregulates viral replication and pathogenesis. J Virol 90:7323–7338.doi:10.1128/JVI.00755-16.
Editor:D. S. Lyles, Wake Forest University
Address correspondence to Jianrong Li, li.926@osu.edu.
H.C. and Y.Z. contributed equally to this work.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
on November 7, 2019 by guest
http://jvi.asm.org/
role in regulating RNA synthesis. Single-amino-acid mutations or deletions in the zinc binding motif of M2-1 were lethal to hRSV (6,
18,19), suggesting that the M2-1 protein is absolutely essential for the hRSV life cycle.
The functions of hMPV M2-1 have not been elucidated. How-ever, available evidence suggests that the M2-1 protein of hMPV plays a different role in the virus life cycle than hRSV. Unlike hRSV, the hMPV minigenome replicates efficiently without the M2-1 protein (20). Deletion of the M2-1 gene from the hMPV genome yielded a viable virus that was highly attenuated in both cell culture and animal models (20). This demonstrates that hMPV M2-1 was not required for virus replication, although it enhances infectivity. In contrast, deletion of M2-1 from the hRSV genome was lethal to the virus (6,18,19). Similar to hRSV M2-1, hMPV M2-1 incorporates zinc ions at a molecular ratio of 1:1 (21,
22). However, the role of individual amino acids in the zinc bind-ing motif (CCCH) differs between hMPV and hRSV M2-1. In hMPV M2-1, the third cysteine (C21) and the last histidine (H25) in the zinc binding motif (CCCH) were found to be essential for zinc binding activity, whereas the first two cysteines (C7 and C15) play only minor or redundant roles in zinc binding (21). In con-trast, in hRSV M2-1, all four residues in the CCCH motif were equally important for zinc binding activity (H. Cai and J. Li, un-published data). Finally, mutations to the zinc binding motif (CCCH) in hMPV M2-1 yielded viable recombinant viruses, dem-onstrating that zinc binding activity was not required for virus viability (21). In contrast, mutations to the zinc binding motif (CCCH) in hRSV M2-1 were lethal to the virus (19), suggesting that zinc binding is absolutely essential for the hRSV life cycle.
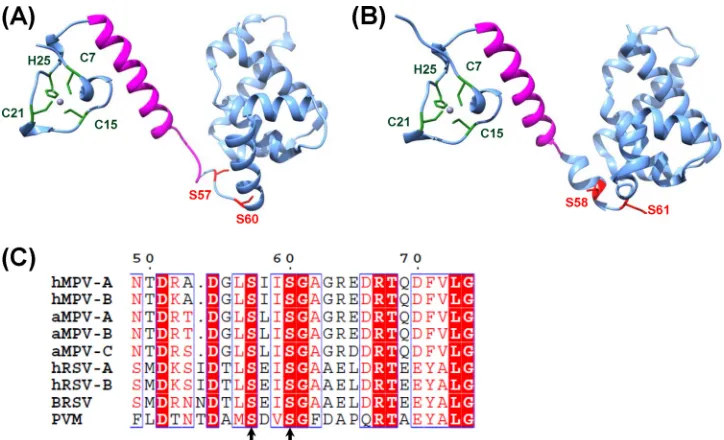
Significant differences were also observed in the crystal
struc-ture between the hRSV and hMPV M2-1 proteins. The hMPV M2-1 protein forms an asymmetric tetramer in which three of the protomers exhibit a closed conformation and one forms an open conformation (23). Each protomer is composed of an N-terminal zinc finger domain and an␣-helical tetramerization motif, form-ing a rigid unit, followed by a flexible linker and an␣-helical core domain (Fig. 1A). Interestingly, hMPV M2-1 exhibits a dynamic equilibrium between open and closed conformations in solution. The closed state of hMPV M2-1 is stabilized by RNA binding through simultaneous recognition of RNA by the zinc finger and core domains. In contrast to the hMPV M2-1 structure, the M2-1 protein of hRSV forms a disk-like symmetrical tetramer, which is driven by a long helix forming a four-helix bundle at its center, and is stabilized by contact between the zinc-binding domain and adjacent protomers (24). Each M2-1 monomer forms three dis-tinct regions, including the zinc binding domain, tetramerization helix, and the core domain, which are linked by unstructured or flexible sequences (Fig. 1B). The open and closed conformations of hMPV M2-1 were not observed for hRSV M2-1.
The M2-1 protein of hRSV was also found to be phosphory-lated (15,25). Initially, it was thought that residues T56 and S58 were the phosphorylation sites in hRSV M2-1, and phosphoryla-tion of M2-1 protein decreased its RNA binding capacity at least 5-to 10-fold (25). However, subsequent mass spectrometry (MS) and mutagenesis analysis found that S58 and S61, but not T56, are the phosphorylation sites in hRSV M2-1 protein (15). This is con-sistent with the fact that T56 is not conserved in M2-1 proteins of other pneumoviruses (Fig. 1C). Interestingly, phosphorylation of S58 creates a new casein kinase I (CKI) site at serine 61 in the hRSV M2-1 protein (15). Unphosphorylated M2-1 protein purified
FIG 1Location of phosphorylation sites and zinc binding sites in hMPV and RSV M2-1 proteins. (A) hMPV M2-1 monomer. The structure was generated using PDB entry4CS7. (B) hRSV M2-1 monomer. The structure was generated using PDB entry4C3D. The predicted oligomerization domain is highlighted in magenta. The phosphorylation sites are highlighted in red. The zinc binding motif is indicated by green. (C) Sequence alignment of pneumovirus M2-1 proteins (aa 49 to 74). Representative members of pneumoviruses include hMPV-A, human metapneumovirus subtype A (GenBank accession no.ACJ70116.1; gi 215794520); hMPV-B (accession no.FJ168778.1; gi 215794505); aMPV-A, avian metapneumovirus subtype A (accession no.AAT68647.1; gi 49823139); aMPV-B (accession no.BAJ23930.1; gi 310772463); aMPV-C (accession no.ACR23029.1; gi 237847064); hRSV-A, human respiratory virus type A (accession no.
M74568.1; gi 333959); hRSV-B (accession no.AAB82437.1; gi 2582031); BRSV, bovine respiratory syncytial virus (accession no.M82816.1; gi 210823); and PVM, pneumonia virus of mice (accession no.YP_173333.1; gi 56900724). Fully conserved residues are highlighted by red boxes, conserved substitutions are indicated by red letters, and black letters mean no match. Phosphorylation sites are highlighted by arrows.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.111.473.65.285.2]fromEscherichia colicould be phosphorylatedin vitroby casein kinase I. Removal of S58 prevented phosphorylation at S61, which subsequently abolished phosphorylation at both sites. Impor-tantly, phosphorylation is required for the efficient transcriptional activity of hRSV M2-1, as mutations of S58 and S61 resulted in a severe decrease in antitermination activity in a minigenome rep-lication assay (15). Unlike previous observations from Cuesta et al., it was found that maintenance of the zinc binding motif, but not phosphorylation, was required for interaction with viral mRNA (25). Given the fact that S58 and S61 are conserved in M2-1 proteins of all pneumoviruses (Fig. 1C), it is likely that M2-1 proteins of all pneumoviruses are also phosphorylated, although it has not been experimentally demonstrated. Currently, it is not known whether phosphorylation of M2-1 plays a role in regulat-ing the pneumovirus life cyclein vitroand viral pathogenesisin vivo. Filling this major gap will rely on the successful recovery of viable recombinant pneumoviruses that lack phosphorylation sites in M2-1.
In this study, we found that the M2-1 protein of hMPV is phosphorylated at residues S57 and S60, which is equivalent to residues S58 and S61 in the hRSV M2-1 protein. Subsequently, we revealed the relationship between M2-1 phosphorylation, zinc binding, and oligomerization. We found that M2-1 phosphoryla-tion was not required for zinc binding and oligomerizaphosphoryla-tion. Con-versely, we found the zinc binding activity of M2-1 was essential for phosphorylation and oligomerization. Using a reverse genetics system, we successfully recovered three recombinant hMPVs missing either one or both phosphorylation sites in M2-1. We found that recombinant hMPV lacking phosphorylation in the M2-1 protein decreased both genomic RNA replication and mRNA transcription. These recombinant viruses exhibited signif-icantly decreased viral replicationin vitroand pathogenesisin vivo. This work suggests that inhibition of M2-1 phosphorylation serves as a means to attenuate hMPV and perhaps other pneumo-viruses.
MATERIALS AND METHODS
Ethics statement.The animal study was conducted in strict accordance with theGuide for the Care and Use of Laboratory Animalsof the National Research Council (26) and was approved by The Ohio State University Institutional Animal Care and Use Committee (IACUC; animal protocol no. 2009A0221). The animal care facilities at The Ohio State University are AAALAC accredited. Every effort was made to minimize potential distress, pain, or discomfort to the animals throughout all experiments.
Cell lines.Vero E6 cells (ATCC CRL-1586) and BHK-SR19-T7 cells (kindly provided by Apath, LLC, Brooklyn, NY) were grown in Dulbec-co’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS). The medium for the BHK-SR19-T7 cells was supplemented with 10g/ml puromycin (Life Technologies) during every other passage to select for T7 polymerase-expressing cells.
Plasmids and site-directed mutagenesis. Plasmids carrying the hMPV minigenome or the full-length genomic cDNA of hMPV strain NL/1/00 and support plasmids expressing hMPV N protein (pCITE-N), P protein P), L protein L), and M2-1 protein (pCITE-M2-1) were kindly provided by R. A. M. Fouchier at the Department of Virology, Erasmus Medical Center, Rotterdam, The Netherlands (8). The F cleavage site in the genome of hMPV NL/1/00 was modified to a trypsin-independent F cleavage site, as described previously (27). The M2-1 mu-tants of hMPV were generated by site-directed mutagenesis using the QuikChange methodology (Stratagene, La Jolla, CA). All constructs were sequenced at The Ohio State University Plant Microbe Genetics Facility to confirm the presence of the introduced mutations.
Expression and purification of recombinant hMPV M2-1 protein fromE. coli.The hMPV M2-1 gene was PCR amplified from a cDNA clone of strain NL/1/00 and was inserted into anE. coliexpression system pGEX-4T-1 vector at BamHI and NotI sites (21). To facilitate protein purification, a glutathioneS-transferase (GST) fusion protein was fused to the N terminus of the M2-1 gene. The resulting plasmids were trans-formed intoE. coliRosetta (DE3) and grown at 37°C until the absorbance at 600 nm reached 0.6 to 0.8. The cells next were chilled on ice for 10 min, and protein expression was induced by the addition of 20M isopropyl--D-1-thiogalactopyranoside (IPTG) and 75M ZnSO4. The cells were
grown for an additional 20 h at 25°C and harvested by centrifugation at 5,000⫻gfor 10 min. Bacterial pellets were resuspended in lysis buffer (40 mM Tris-HCl [pH 7.4], 1.0 M NaCl, 0.5 mM dithiothreitol [DTT], and 20 M ZnSO4) supplemented with 1 mg/ml of lysozyme and protease
inhib-itor cocktail (Roche, Mannheim, Germany). After a 30-min incubation on ice, the cells were lysed using sonication, followed by centrifugation at 15,000⫻gat 4°C for 40 min. The supernatant was collected and loaded into a column containing 5 ml of glutathione HiCap matrix (Qiagen). The G resins were washed with 150 ml of resuspension buffer, followed by washing with 100 ml of the cleavage buffer (50 mM Tris-HCl [pH 7.6], 2.5 mM CaCl2, 150 mM NaCl, and 0.2 mM DTT). To isolate GST-free M2-1,
20 ml of the cleavage buffer containing 2 U/ml of thrombin (Sigma, St. Louis, MO) was loaded into the column and incubated at room temper-ature for 8 h. The reaction was stopped by adding 4-amidinophenylmeth-anesulfonyl fluoride hydrochloride (APMSF) (Sigma). The GST fusion hMPV M2-1 mutants S57A, S60A, S57A-S60A, C21S, and H25L were generated similarly. The purified hMPV M2-1 proteins were dialyzed against phosphate-buffered saline (PBS) buffer containing 300 mM NaCl and 10M ZnSO4. Protein concentration was determined by the Brad-ford assay (Sigma).
Expression and purification of recombinant hMPV M2-1 protein from insect cells.The hMPV M2-1 gene was cloned into the pFastBac Dual vector (Invitrogen) at EcoRI and HindIII sites (21). To facilitate protein purification, a 6⫻His tag was fused to the N terminus of the M2-1 protein. The resulting plasmid was transformed into DH 10Bac compe-tent cells (Invitrogen), and baculovirus expressing M2-1 protein was gen-erated by transfection of bacmids into SF9 cells. To express the M2-1 protein, SF9 cells were infected with baculovirus at a multiplicity of infec-tion (MOI) of 3.0. At 72 h postinfecinfec-tion, cells were harvested by centrif-ugation at 5,000⫻gfor 10 min, and the pellets were washed with cold PBS. M2-1 protein was purified using Ni bead columns as described pre-viously. Protein concentration was determined by Bradford assay.
Colorimetric determination of the zinc content.Purified hMPV M2-1 was dialyzed against 50 mM PBS (pH 7.0) containing 0.3 M NaCl at 4°C overnight. M2-1 proteins at a concentration of 2M in 1 ml of solution containing 100M 4-(2-pyridylazo) resorcinol (PAR) were in-cubated at 25°C for 20 min, and the absorbance at 500 nm was monitored for 5 min. Upon addition of 100M p-chloromercuriphenylsulfonic acid (PMPS), the release of strongly bound Zn2⫹was monitored. The amount of Zn2⫹bound to hMPV M2-1 was quantified by comparing the sample readings to a standard curve generated using 2M, 3M, and 4M ZnSO4(Sigma). Buffer containing 50 mM PBS (pH 7.0), 0.3 M NaCl, and
100M PAR was used as the blank control. The zinc ion contents of
GST-M2-1 and mutants were also measured at 2M and compared to
those of rM2-1 at the same protein concentration.
Glutaraldehyde cross-linking.A volume of 1.2g of rM2-1, GST-M2-1, or mutant protein was diluted in 20l of reaction buffer (50 mM PBS, pH 7.0, 300 mM NaCl) and incubated with increasing amounts of glutaraldehyde from 0 to 0.1% at 25°C for 30 s. The reaction was stopped by the addition of Tris-HCl (pH 7.4) at a final concentration of 50 mM. Subsequently, the cross-linked products were analyzed by 12% SDS-PAGE and the proteins were visualized by Coomassie blue staining.
MS.M2-1 proteins were separated by SDS-PAGE gel and digested with sequencing-grade trypsin (Promega, Madison, WI). Bands were briefly trimmed to minimize the polyacrylamide material. Gel pieces were then
on November 7, 2019 by guest
http://jvi.asm.org/
washed in 50% methanol–5% acetic acid for 1 h. Subsequently, the gels were washed three times with acetonitrile and ammonium bicarbonate (100 mM) for 5 min. The gels were then dried in a speed vacuum. The protease was then driven into the gel pieces by rehydrating them in 50l of sequencing-grade modified trypsin, chymotrypsin, or a combination of
both at 20g/ml in 50 mM ammonium bicarbonate for 10 min. An
aliquot of 20l of 50 mM ammonium bicarbonate was added to the gel bands, and the mixture was incubated at room temperature overnight. After the peptides were extracted from the polyacrylamide with 50% ace-tonitrile and 5% formic acid repeatedly and concentrated in a speed vac-uum to approximately 25l, the mass spectrometry (MS) analysis was immediately performed to ensure high-quality tryptic peptides with min-imal nonspecific cleavage. Each sample was injected into a trapping col-umn (LC-Packings) and desalted with 50 mM acetic acid for 10 min. Protein identification was performed on a Thermo Finnigan LTQ-XL Orbitrap mass spectrometer equipped with a nanospray source operated in positive ion mode. Samples were separated on a capillary column (0.2
by 150 mm; Magic C18AQ 3200A; Michrom Bioresources, Inc.,
Au-burn, CA) using an UltiMate 3000 high-performance liquid chromatog-raphy (HPLC) system from LC Packings-Dionex Co. (Sunnyvale, CA). The raw data files collected on the mass spectrometer were converted to mzXML and MGF files using MassMatrix data conversion tools (version 1.3;http://www.massmatrix.net). The fragment mass tolerance was set to 0.5 Da. Identifications of phosphorylated peptides were verified manually.
Recovery of rhMPVs from the full-length cDNA clones. Recombi-nant hMPVs (rhMPVs) were rescued using a reverse genetics system as described previously (8,21). Briefly, BHK-SR19-T7 cells (kindly provided by Apath LLC), which stably express T7 RNA polymerase, were trans-fected with 5.0g of plasmid phMPV carrying the full-length hMPV genome, 2.0g of pCITE-N, 2.0g of pCITE-P, 1.0g of pCITE-L, and 1.0g of pCITE-M2-1 using Lipofectamine 2000 (Life Technologies). At day 6 posttransfection, the cells were harvested using cell scrapers and were cocultured with Vero E6 cells at 50 to 60% confluence. When extensive cytopathic effects (CPE) were observed, the cells were sub-jected to three freeze-thaw cycles, followed by centrifugation at 3,000 ⫻gfor 10 min. The supernatant was subsequently used to infect new Vero E6 cells. The successful recovery of the rhMPVs was confirmed by immunostaining, agarose overlay plaque assay, and reverse transcrip-tion-PCR (RT-PCR).
Immunostaining plaque assay.Vero E6 cells were seeded in 24-well plates and infected with serial dilutions of rhMPV. At day 6 postinfection, the supernatant was removed and cells were fixed in a prechilled acetone-methanol solution at room temperature for 15 min. Cells were permeab-ilized in phosphate-buffered saline (PBS) containing 0.4% Triton X-100 at room temperature for 10 min and blocked at 37°C for 1 h using 1% bovine serum albumin (BSA) in PBS. The cells were then labeled with an anti-hMPV N-protein primary monoclonal antibody (Millipore, Bil-lerica, MA) at a dilution of 1:1,000, followed by incubation with horse-radish peroxidase (HRP)-labeled rabbit anti-mouse secondary antibody (Thermo Scientific, Waltham, MA) at a dilution of 1:5,000. After incuba-tion with 3-amino-9-ethylcarbazole (AEC) chromogen substrate (Sigma, St. Louis, MO), positive cells were visualized under a microscope. The viral titer was calculated as the number of PFU per milliliter. The diame-ters of the plaques were measured using ImageJ.
Viral replication kinetics in Vero E6 cells.Confluent Vero E6 cells in 35-mm dishes were infected with wild-type (wt) rhMPV or mutant rhMPV at an MOI of 0.01. After 1 h of adsorption, the inoculum was removed and the cells were washed three times with PBS. Fresh DMEM (supplemented with 2% FBS) was added and the infected cells were incu-bated at 37°C. At different time points postinfection, the supernatant and cells were harvested by three freeze-thaw cycles, followed by centrifuga-tion at 1,500⫻gat room temperature for 15 min. The virus titer was determined by an immunostaining assay in Vero E6 cells.
Genetic stability of rhMPV mutants in cell culture.Confluent Vero E6 cells in T-25 flasks were infected with each rhMPV mutant at an MOI
of 0.01. At day 5 postinfection, the cell culture supernatant was harvested and used for the next passage in Vero E6 cells at the same MOI. Using this method, each rhMPV mutant was repeatedly passaged 15 times in Vero E6 cells. At each passage, the M2-1 gene was amplified by RT-PCR and se-quenced. At passage 15, the entire genome of each recombinant virus was amplified by RT-PCR and sequenced.
Quantification of viral genomic RNA replication and mRNA synthe-sis by real-time RT-PCR.Confluent Vero E6 cells were infected with each rhMPV mutant at an MOI of 5.0. At 6, 12, and 24 h postinfection, total RNA was isolated from virus-infected cells using the TRIzol reagent (Life Technologies). Viral genomic RNA copies were quantified by real-time RT-PCR using two primers specifically targeting the hMPV leader
se-quence (5=-AAAACGCGTATAAATTAGATTCCAAAA-3=) and N gene
(5=-TGCAGTTGTTGTACCACATCTCTTT-3=). Poly(A)-containing
vi-ral mRNA was isolated from total RNA using a Dynabeads mRNA isola-tion kit (Life Technologies) according to the manufacturer’s recommen-dations. Using the viral mRNA as the template, the N-protein mRNA copies were quantified by real-time RT-PCR using two primers targeting
the viral N-protein gene (5=-CAAGGAAAACAATGGCAAC-3=and 5=-T
GTAGATGATGAGCCTAATGC-3=). Real-time PCR was performed on a
StepOne real-time PCR machine (Applied Biosystems, Foster City, CA). PCR and cycling parameters were set by following the manufacturer’s recommendations. For cycling parameters, a holding stage at 95°C was maintained for 2 min, prior to 40 cycles of 94°C for 15 s for denaturation, 55°C for 30 s for annealing, and 72°C for 15 s for extension. Standard curves and StepOne software, v2.1, were used to quantify genomic RNA
and mRNA copy numbers. Viral RNA was expressed as the mean log10
number of RNA copies per milliliter⫾standard deviations.
Replication and pathogenesis of rhMPV in cotton rats. Forty-four-week-old female specific-pathogen-free (SPF) cotton rats (Harlan Labo-ratories, Indianapolis, IN) were randomly divided into eight groups (5 cotton rats per group). Prior to virus inoculation, the cotton rats were anesthetized with isoflurane. The cotton rats in groups 1 to 3 were
inoc-ulated with 1.0⫻105PFU of wt rhMPV, S57A, and
rhMPV-S60A. The cotton rats in groups 4 to 7 were inoculated with 1.0⫻104PFU
of rhMPV, rhMPV-S57A, rhMPV-S60A, and rhMPV-S57A-S60A. The cotton rats in group 8 were mock infected with 0.1 ml of Opti-MEM medium and served as uninfected controls. Each cotton rat was inoculated intranasally with a volume of 100l. After inoculation, the animals were evaluated on a daily basis for mortality and the presence of any respiratory symptoms. At day 4 postinfection, the cotton rats were sacrificed, and lungs and nasal turbinates were collected for both virus isolation and histological analysis.
Immunogenicity of rhMPVs in cotton rats.Thirty cotton rats (Har-lan Laboratories, Indianapolis, IN) were randomly divided into six groups (5 cotton rats per group). The cotton rats in group 1 (DMEM) were mock immunized with DMEM, mock challenged with Opti-MEM medium, and used as an unvaccinated uninfected control. Cotton rats in group 2 (chal-lenge control) were mock immunized with DMEM, chal(chal-lenged with wt rhMPV, and used as an unvaccinated challenged control. Cotton rats in groups 3 to 6 were intranasally inoculated with 1.0⫻104PFU of wt
rhMPV, rhMPV-S57A, rhMPV-S60A, or rhMPV-S57A-S60A in 0.1 ml Opti-MEM medium. After immunization, the cotton rats were evaluated daily for mortality and the presence of any symptoms of hMPV infection. Blood samples were collected from each rat weekly by facial vein retro-orbital bleeding, and serum was isolated for neutralizing antibody detec-tion. At week 4 postimmunization, the cotton rats in groups 2 to 6 were challenged intranasally with wt rhMPV at a dose of 1.0⫻105PFU per
cotton rat. After challenge, the animals were evaluated twice every day for mortality and the presence of any symptoms of hMPV infection. At day 4 postchallenge, all cotton rats from each group were euthanized by CO2
asphyxiation. The lungs and nasal turbinates from each cotton rat were collected for virus isolation and histological evaluation. The immunoge-nicity of rhMPV mutants was evaluated using the following methods: (i) humoral immunity was determined by analysis of serum neutralization of
on November 7, 2019 by guest
http://jvi.asm.org/
virus using an endpoint dilution plaque reduction assay; (ii) viral titers in the nasal turbinates and lungs were determined by an immunostaining plaque assay; and (iii) pulmonary histopathology and viral antigen distri-bution were determined using the procedure described below. The pro-tection was evaluated with respect to viral replication, antigen distribu-tion, and pulmonary histopathology.
Pulmonary histology.After sacrifice, the right lung of each animal was removed, inflated, and fixed with 4% neutral buffered formaldehyde. Fixed tissues were embedded in paraffin and sectioned at 5m. Slides were then stained with hematoxylin-eosin (H&E) for the examination of histological changes by light microscopy. The pulmonary histopatholog-ical changes were reviewed by 2 to 3 independent pathologists who were blinded to the groups. Histopathological changes were scored based on the extent of interstitial inflammation, edema, and peribronchiolar in-flammation.
Immunohistochemical (IHC) staining.The right lung of each animal was fixed in 10% neutral buffered formaldehyde and embedded in paraf-fin. Five-micrometer sections were cut and placed onto positively charged slides. After deparaffinization, sections were incubated with target re-trieval solution (Dako, Carpinteria, CA) for antigen rere-trieval. After body block, a primary mouse hMPV matrix (M) monoclonal anti-body (Virostat, Portland, ME) was added for 30 min at room temperature, followed by incubation with a biotinylated horse anti-mouse secondary antibody (Vector Laboratories, Burlingame, CA). Slides were further in-cubated with ABC Elite complex to probe biotin (Vector Laboratories) and then developed using a 3,3=-diaminobenzidine (DAB) chromogen kit (Dako) and hematoxylin as a counterstain. Lung sections from hMPV-infected and unhMPV-infected samples were used as positive and negative con-trols, respectively.
Determination of hMPV-neutralizing antibody. hMPV-specific neutralizing antibody titers were determined using a plaque reduction neutralization assay (21,28). Briefly, cotton rat sera were collected by retro-orbital bleeding weekly until challenge. The serum samples were heat inactivated at 56°C for 30 min. Twofold dilutions of the serum sam-ples were mixed with an equal volume of DMEM containing approxi-mately 100 PFU/well hMPV NL/1/00 in a 96-well plate, and the plate was incubated at room temperature for 1 h with constant rotation. The mix-tures were then transferred to confluent Vero E6 cells in a 96-well plate in triplicate. After 1 h of incubation at 37°C, the virus-serum mixtures were removed and the cells were overlaid with 0.75% methylcellulose in DMEM and incubated for another 4 days before virus plaque titration. The plaques were counted and 50% plaque reduction titers were calcu-lated as the hMPV-specific neutralizing antibody titers.
Determination of viral titer in lung and nasal turbinate.The nasal turbinate and the left lung from each cotton rat were removed, weighed, and homogenized in 1 ml of PBS solution using a Precellys 24 tissue homogenizer (Bertin, MD) by following the manufacturer’s recommen-dations. The presence of infectious virus was determined by an immuno-staining plaque assay in Vero cells as described above.
Statistical analysis.Quantitative analysis was performed by either densitometric scanning of autoradiographs or by using a phosphorimager (Typhoon; GE Healthcare, Piscataway, NJ) and ImageQuant TL software (GE Healthcare, Piscataway, NJ). Statistical analysis was performed by one-way multiple comparisons using SPSS (version 8.0) statistical analy-sis software (SPSS Inc., Chicago, IL). APvalue of⬍0.05 was considered statistically significant.
RESULTS
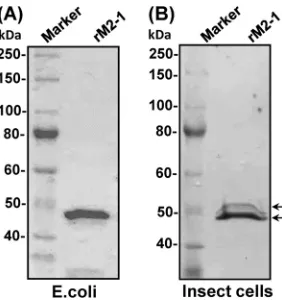
The M2-1 protein of hMPV is phosphorylated at amino acids S57 and S60.To determine the biological roles of M2-1 phosphor-ylation in the hMPV life cycle, we first mapped the phosphoryla-tion site in M2-1 protein. GST-tagged M2-1 protein was expressed in insect cells andE. coli, and the purified proteins were analyzed by SDS-PAGE. As shown inFig. 2A, a single protein band with a molecular mass of approximately 49 kDa was observed when
M2-1 was expressed in theE. coliexpression system. In contrast, at least two protein bands, with the lower band being dominant, were observed when M2-1 was expressed in insect cells (Fig. 2B). This suggests that the M2-1 protein underwent posttranscrip-tional modification in the eukaryotic expression system. To iden-tify the phosphorylation sites of hMPV M2-1, purified unphos-phorylated and phosunphos-phorylated M2-1 protein from insect cells were separated by SDS-PAGE and digested with trypsin, chymo-trypsin, or a combination of both. The digestion products were analyzed by liquid chromatography-tandem MS (LC-MS/MS). MS analysis showed that 95% of the amino acid sequence of M2-1 protein was covered and revealed that the phosphory-lated and unphosphoryphosphory-lated proteins differed in a unique pep-tide (53ADGLSIISGAGR64) (underlining indicates the phosphor-ylation sites of the peptide). The spectra of the digests were also examined for phosphorylation, which causes a mass shift of 80 Da to the native intact peptide. Masses of the unphosphorylated pro-tein (Fig. 3A) and 3 types of phosphorylated protein (Fig. 3BtoD) confirmed that hMPV M2-1 protein could be phosphorylated either at a single phosphorylation site at S57 (Fig. 3B) or S60 (Fig. 3C) or double phosphorylation sites at both S57 and S60 (Fig. 3D).
Mutations to the phosphorylation sites of M2-1 abolished phosphorylation.The two phosphorylation sites (S57 and S60) next were mutated to alanine, and two single mutants (S57A and S60A) and a double mutant (S57A-S60A) were purified either fromE. colior insect cells (Fig. 4A). Wild-type rM2-1 purified from insect cells had doublet bands, whereas all three M2-1 mu-tants had only a single protein band. This suggests that mutations to these two amino acid residues affected the phosphorylation of M2-1 protein. In contrast, both wild-type rM2-1 and M2-1 mu-tants had a single protein band when expressed inE. coli(data not shown), which is consistent with the fact that proteins produced by theE. coliexpression system lack posttranscriptional modifica-tion. To further confirm that mutation to one phosphorylation site affects the phosphorylation status of the other phosphoryla-tion site, insect cell-expressed rM2-1-S57A and rM2-1-S60A were
FIG 2Expression and purification of recombinant hMPV M2-1 protein. (A) M2-1 protein expressed inE. coli. The GST-tagged hMPV M2-1 protein was expressed inE. coliRosetta (DE3) and purified using a column containing glutathione HiCap matrix. (B) M2-1 protein expressed in insect cells. The GST-tagged hMPV M2-1 protein was cloned into the pFastBac Dual vector (Invitrogen), and baculovirus expressing M2-1 protein was generated by transfection of bacmids into SF9 cells. M2-1 proteins were purified using a column containing glutathione HiCap matrix. Doublet bands are indicated by arrows.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.351.492.65.215.2]sent for mass spectrometry analysis. As shown inFig. 4B, no phos-phorylation was detected at residue S57 in mutant rM2-1-S60A. Similarly, no phosphorylation was detected at residue S60 in mu-tant rM2-1-S57A (data not shown). Thus, these results demon-strated that mutations altering one phosphorylation site in the M2-1 protein also abolished phosphorylation of the other phos-phorylation site.
Phosphorylation of M2-1 is not essential for zinc binding activity.M2-1 is a zinc binding protein, and the zinc coordination pocket includes amino acid residues C7, C15, C21, and H25. Pre-viously, we have shown that mutations to C7 and C15 diminished approximately 40% of zinc binding activity, whereas mutations to C21 and H25 completely abolished zinc binding activity (21). We also showed that M2-1 protein purified fromE. coliwas capable of binding to zinc ions at a molecular ratio of 1:1 (21), suggesting that phosphorylation of M2-1 is not essential for zinc binding activity. To further determine the relationship between phosphorylation and zinc binding, the zinc binding activity of M2-1 mutants lack-ing phosphorylation sites was determined. As shown inFig. 5, M2-1 mutants lacking phosphorylation sites had no significant impairment of zinc binding activity. As controls, C21S and H25L mutants abolished zinc activity. Thus, phosphorylation of M2-1 is not essential for zinc binding activity.
Mutations in zinc binding motif affect the phosphorylation of M2-1.We also determined whether zinc binding activity is essential for phosphorylation of M2-1. To do this, M2-1 mu-tants (C21S and H25L) lacking zinc binding activity were ex-pressed and purified from insect cells. As shown inFig. 4A, zinc binding mutants had a single 50-kDa protein band, whereas wild-type rM2-1 had multiple protein bands in SDS-PAGE, suggesting that mutations to the zinc binding motif affected the phosphorylation of the M2-1 protein. Subsequently, mass spectrometry confirmed that these zinc binding mutant pro-teins (rM2-1-H25L) expressed from insect cells were not phos-phorylated at residues S57 and S60 (Fig. 4C). Mass spectrom-etry also confirmed that amino acid substitution H25L was present in rM2-1-H25L (Fig. 4D). Similarly, no phosphoryla-tion was detected at residues S57 and S60 in rM2-1-C21S (data not shown). Thus, these results demonstrated that the zinc binding activity of M2-1 is essential for phosphorylation.
Mutations in phosphorylation sites do not impair oligomer-ization of M2-1 protein.A recent crystal structure revealed that the hMPV M2-1 protein is a tetramer (23). We previously showed that mutations to the zinc binding motif of M2-1 abol-ished the oligomerization of M2-1 (21). Similarly, the effect of mutations in M2-1 phosphorylation sites on oligomerization
FIG 3Identification of phosphorylation sites in hMPV M2-1 by mass spectrometry. (A) Unphosphorylated hMPV M2-1; (B) phosphorylation site at residue S57; (C) phosphorylation site at residue S60; (D) phosphorylation sites at residues S57 and S60. M2-1 proteins were digested with trypsin, chymotrypsin, or a combination of the two. Peptides were isolated and analyzed by mass spectrometry. Protein identification was performed on a Thermo Finnigan LTQ-XL Orbitrap mass spectrometer. The raw data files collected on the mass spectrometer were converted to mzXML and MGF files using MassMatrix data conversion tools (version 1.3;http://www.massmatrix.net/download). Identified phosphorylated peptides were verified.
on November 7, 2019 by guest
http://jvi.asm.org/
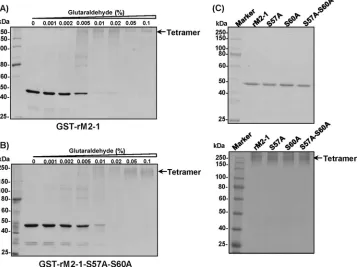
[image:6.585.41.543.62.390.2]was determined by a chemical cross-linking assay. A volume of 1.2g of GST-tagged rM2-1 protein was cross-linked by an increasing concentration (from 0 to 0.1%) of glutaraldehyde, and the resulting products were resolved by SDS-PAGE. For wild-type protein, the density of the M2-1 monomer (49 kDa) gradually decreased and the oligomerization product increased in the presence of increasing concentrations of glutaraldehyde. At 0.05% glutaraldehyde, a protein band with a molecular mass
of approximately 196 kDa was detected in the cross-linking experiment (Fig. 6A), which is consistent with the size of an M2-1 tetramer. Interestingly, rM2-1-S57A-S60A, which lacked two phosphorylation sites, formed a tetramer in a manner sim-ilar to that of rM2-1 (Fig. 6B). The oligomerization of the two single-point mutants rM2-1-S57A and rM2-1-S60A was tested at 0.1% glutaraldehyde. As shown in Fig. 6C, both mutants were able to form tetramers. Thus, mutations in M2-1
phos-FIG 4Zinc binding activity is essential for phosphorylation of M2-1. (A) Purification of recombinant M2-1 carrying mutations in phosphorylation sites or zinc binding sites. M2-1 mutations were generated by site-directed mutagenesis and cloned into the pFastBac Dual vector. M2-1 mutants were expressed in SF9 insect cells by baculovirus. Proteins were purified using a column containing glutathione HiCap matrix. Doublet bands are indicated by stars. (B) Analysis of phosphorylation of rM2-1-S60A by mass spectrometry. No phosphorylation was detected in S57 when one of the phosphorylation sites (S60) was mutated to A. (C) Analysis of phosphorylation of rM2-1-H25L by mass spectrometry. No phosphorylation was detected in S57 and S60 when one of the zinc binding sites (H25) was mutated to L. (D) Confirmation of the presence of the H25L mutation in rM2-1-H25L by mass spectrometry.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.46.545.59.553.2]phorylation sites did not impair the oligomerization of the hMPV M2-1 protein.
rhMPV carrying mutations in the phosphorylation site of M2-1 protein.In order to determine the effect of M2-1 phos-phorylation in viral replication and pathogenesis, we mutated the phosphorylation site of M2-1 in an infectious cDNA clone
of hMPV lineage A strain NL/1/00. Using a reverse genetics system, recombinant viruses with a single point mutation (rhMPV-S57A and rhMPV-S60A) and double mutations (rhMPV-S57A-S60A) were successfully recovered. The recom-binant viruses were first detected in Vero E6 cells by an immu-nostaining assay using a monoclonal antibody against N pro-tein. As shownFig. 7A, all rhMPV mutants were positive for viral N-protein expression at day 5 postinfection. The immu-nospots formed by these rhMPV mutants in Vero E6 cells were significantly smaller than those formed by wt rhMPV. The size of the immunospots can be ranked as rhMPV⬎ rhMPV-S57A ⫽ rhMPV-S60A⬎rhMPV-S57A-S60A. The recombinant viruses were also detected by an agarose overlay plaque assay in Vero E6 cells (Fig. 7B). After 8 days of incubation, the plaque sizes for rhMPV-S57A, rhMPV-S60A, and rhMPV-S57A-S60A were 0.47⫾0.23 mm, 0.42⫾ 0.51 mm, and 0.19⫾0.17 mm in diameter, respectively, which were all significantly smaller than that of wt rhMPV (2.35⫾0.74 mm) (P⬍0.05). This suggests that rhMPVs lacking phosphorylation sites in M2-1 had defects in cell-to-cell spread and/or viral repli-cation in cell culture. Using an agarose overlay plaque assay, all hMPV mutants were plaque purified. The plaque-purified viruses were further passaged three times in Vero E6 cells. The entire genome of each hMPV mutant was amplified and sequenced. All recombinant hMPVs retained the desired mutation in the M2-1 gene, and no additional mutations were found in the genome. Subsequently, these hMPV mutants were passed 15 times in Vero E6 cells, and sequencing found that all of the mutants retained the
FIG 5Phosphorylation of hMPV M2-1 is not essential for zinc binding activ-ity. Zinc binding activity was measured by a colorimetric assay. The standard curve was generated using 1M, 2M, 3M, and 4M ZnSO4. The amount of Zn2⫹bound to hMPV M2-1 protein was quantified by comparing the
sam-ple readings to a standard curve. The percentage of wild-type M2-1 binding for each M2-1 mutant is shown. Data are averages from three independent exper-iments⫾1 standard deviation. Statistical difference (P⬍0.01) is indicated.
FIG 6Phosphorylation of hMPV M2-1 does not impair oligomerization of M2-1 protein. (A) Glutaraldehyde cross-linking of GST-M2-1. GST-M2-1 protein (1.2g) was incubated with increasing amounts of glutaraldehyde from 0 to 0.1% at 25°C for 30 s. The reaction was stopped by adding Tris-HCl (pH 7.4) at a final concentration of 50 mM. The cross-linked products were analyzed by 12% SDS-PAGE, followed by Coomassie blue staining. The predicted tetramer is indicated. (B) Glutaraldehyde cross-linking of GST-M2-1-S57A. GST-M2-1-S57A protein (1.2g) was incubated with increasing amounts of glutaraldehyde from 0 to 0.1%. The cross-linked products were analyzed by SDS-PAGE. (C) Glutaraldehyde cross-linking of GST-M2-1 mutants. Each GST-M2-1 mutant (1.2 g) (upper) was incubated with 0.1% glutaraldehyde, and the products (lower) were analyzed by SDS-PAGE.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.76.248.64.207.2] [image:8.585.115.473.398.665.2]desired mutation in the M2-1 gene. At passage 15, the entire ge-nome was sequenced, and the results showed that no additional mutations were found except for the desired mutation in the M2-1 gene. This indicates that these rhMPV mutants were genetically stable in cell culture.
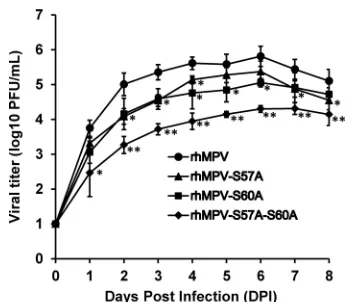
rhMPV lacking the phosphorylation site in M2-1 protein had defects in replication in cell culture.The replication kinetics of recombinant hMPV carrying mutations in the phosphoryla-tion site of the M2-1 protein were determined in Vero E6 cells (Fig. 8). Briefly, Vero E6 cells were infected with each recom-binant virus at an MOI of 0.01. At the indicated time points, viral titer in the supernatant was determined by an immuno-staining plaque assay. All rhMPV mutants had delayed cyto-pathic effects (CPE). wt rhMPV reached a peak titer of 105.8 PFU/ml at day 6 postinoculation. Recombinant rhMPV-S57A and rhMPV-S60A showed delayed replication kinetics com-pared to rhMPV and reached a peak titer of 105.4 and 105.1 PFU/ml, respectively, at day 6 postinoculation. The double mutant rhMPV-S57A-S60A was the most defective virus, ex-hibiting a 1.5-log reduction in peak titer compared to the wild-type virus. When Vero E6 cells were infected at a higher MOI (0.1), the highest titer of rhMPV, rhMPV-S57A, rhMPV-S60A, and rhMPV-S57A-S60A reached 106.7, 106.5, 106.3, and 105.8, respectively (data not shown). Although rhMPV-S57A and rhMPV-S60A exhibited a significant delay in viral replication, they grew to a titer comparable to that of wt rhMPV in cell culture. This result demonstrated that rhMPV lacking phos-phorylation in the M2-1 protein diminished viral replication in cell culture.
rhMPV lacking phosphorylation of M2-1 protein had defects in RNA synthesis but had no significant alteration in the balance between genome replication and mRNA transcription. One unique feature of RNA synthesis in nonsegmented negative-sense RNA viruses is that one polymerase complex carries out two
dis-FIG 7Recovery of recombinant hMPVs carrying mutations in the phosphorylation sites of M2-1. (A) Immunostaining spots formed by recombinant hMPVs. Vero E6 cells were infected with recombinant hMPV mutants and incubated at 37°C for 5 days. The cells were stained with an anti-hMPV N protein monoclonal antibody. (B) Plaque morphology of recombinant hMPVs. An agarose overlay plaque assay was performed on a monolayer of Vero E6 cells. Viral plaques were developed at day 8 postinfection. The plaque pictures were taken from different dilutions of each hMPV mutant. Data were averages from 20 plaques⫾1 standard deviation.
FIG 8Multistep growth curve of recombinant hMPVs carrying mutations in the phosphorylation site. Vero E6 cells in 35-mm dishes were infected with each recombinant hMPV at an MOI of 0.01. After adsorption for 1 h, the inocula were removed and the infected cells were washed 3 times with Opti-MEM. Fresh Opti-MEM containing 2% FBS then was added and cells were incubated at 37°C for various time periods. Aliquots of the cell culture fluid were removed at the indicated intervals. Viral titer was determined by an immunostaining assay in Vero E6 cells. Viral titers of each mutant at each time point were compared to those of rhMPV (*,P⬍0.05; **,P⬍0.01).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.105.480.63.348.2] [image:9.585.333.508.486.637.2]tinct RNA syntheses: replication and transcription (29). We next examined the genomic RNA replication and mRNA synthesis of these recombinant hMPVs in virus-infected cells. To do this, Vero E6 cells were infected by each recombinant virus at an MOI of 5.0. At 6, 12, and 24 h postinfection, total RNAs were isolated, and genomic RNA and N mRNA were separated by poly(A) binding beads and quantified by real-time RT-PCR. All three rhMPV mu-tants had a significant defect in both genomic RNA replication (Fig. 9A) and mRNA synthesis (Fig. 9B) compared to rhMPV (P⬍ 0.05). Recombinant rhMPV-S57A-S60A was the most defective mutant (P⬍0.01). However, when mRNAs were normalized to genomic RNA (Fig. 9C), no significant difference was observed compared to rhMPV (P⬎0.05). Thus, these data demonstrated that rhMPV mutants lacking phosphorylation of M2-1 protein had defects in both genomic RNA replication and mRNA synthe-sis but caused no significant alteration in the balance between replication and transcription.
rhMPV lacking phosphorylation of M2-1 protein replicated inefficiently in cotton rats.The replication and pathogenesis of rhMPVs carrying mutations in both M2-1 phosphorylation sites was determined in a cotton rat model. Briefly, 4-week-old SPF cotton rats were inoculated intranasally with two doses (1.0⫻105PFU or 1.0⫻104 PFU) of wt rhMPV or rhMPV mutants. At day 4 postinoculation, cotton rats were sacrificed, viral replication was determined in nasal turbinate and lungs, and pulmonary histology was evaluated (Table 1). At a dose of 1.0⫻105PFU, average viral titers in lungs and nasal turbinates of rhMPV-inoculated cotton rats reached 105.02 and 104.82 PFU/g tissue, respectively. Recombinant rhMPV-S57A and rhMPV-S60A had significant defects in viral replication in cot-ton rats. For the rhMPV-S57A group, only 3 out of 5 cotcot-ton rats had detectable hMPV in nasal turbinates and lungs, with aver-age titers of 102.38and 102.70PFU/g of tissue, respectively. For the rhMPV-S60A group, only 3 out of 5 cotton rats had infec-tious hMPV in nasal turbinate, with an average titer of 103.07 PFU/g tissue, and all 5 cotton rats had detectable virus in lungs, with a titer of 102.87PFU/g tissue, which was significantly lower than that of wild-type rhMPV (P⬍0.05). At a dose of 1.0⫻104 PFU, average viral titers in the lungs and nasal turbinates of rhMPV-inoculated cotton rats were 103.91 and 103.83 PFU/g tissue, respectively. For rhMPV-S57A, only 2 out of 5 cotton rats had hMPV in the nasal turbinates and lungs, with a titer of 101.68and 101.99 PFU/g tissue, respectively. For hMPV-S60A, two out of five cotton rats had a low level of infectious virus (101.48PFU/g tissue) in nasal turbinate, and no infectious virus was found in lungs. No infectious virus was detected in either nasal turbinate or lungs in rhMPV-S57A-S60A-infected cotton rats. Therefore, these results demonstrated that rhMPVs lack-ing phosphorylation of M2-1 protein were attenuated in repli-cation in both the upper and lower respiratory tracts of cotton rats.
The lungs of hMPV-infected cotton rats were examined histo-logically. Wild-type rhMPV infection caused moderate histologi-cal lesions, including interstitial pneumonia, peribronchial lym-phocytic infiltrates, mononuclear cell infiltrates, and edematous thickening of the bronchial submucosa (Fig. 10). Recombinant rhMPV-S57A, rhMPV-S60A, and rhMPV-S57A-S60A caused only mild pulmonary histological changes (Fig. 10). Immunohis-tochemical (IHC) analysis found a large amount of viral antigen in epithelial cells of lungs of cotton rats infected by rhMPV. In
con-FIG 9hMPVs carrying mutations in the phosphorylation sites of M2-1 had significant defects in genomic RNA replication and mRNA transcription. (A) Viral genomic RNA replication. Vero E6 cells were infected with wild-type rhMPV or rhMPV mutants at an MOI of 5.0. Total RNA was isolated using the TRIzol reagent at 6, 12, and 24 h postinfection, and genomic RNA was quantified by real-time RT-PCR using specific primers annealing to the hMPV leader sequence and N gene. (B) N mRNA synthesis. Viral mRNA was further purified using a Dynabeads mRNA isolation kit and subse-quently quantified by real-time PCR using primers annealing to the N gene. (c) Ratio between mRNA and genomic RNA. The ratio between N mRNA and genomic RNA was calculated for each recombinant hMPV. The results are the averages from three independent experiments and are expressed as mean standard errors of the numbers of copies of the transcribed N mRNA or viral genomic RNA. All three mutants had significant defects in mRNA synthesis and genome replication compared to rhMPV at three time points (*,P⬍0.05; **,P⬍0.01).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.337.507.65.525.2]trast, little or no viral antigen was found in the lungs of cotton rats infected by rhMPV mutants (Fig. 11). Thus, rhMPV lacking phos-phorylation of the M2-1 protein caused fewer histologic lesions in the lungs of cotton rats.
Vaccination with recombinant rhMPVs lacking phosphory-lation in the M2-1 protein protected cotton rats from wild-type hMPV infection.Since all three rhMPV mutants were attenuated in vitroandin vivo, the immunogenicity of these mutants was determined by vaccination of cotton rats, followed by challenge with wt rhMPV. After vaccination, serum antibody levels were determined weekly by a plaque reduction neutralization assay. wt rhMPV infection and reinfection was used as a control. An ideal live attenuated vaccine candidate should generate immunogenic-ity similar to or higher than that of wt hMPV. As shown inFig. 12, rhMPV-S57A and rhMPV-S60A triggered a neutralizing antibody titer comparable to that of wt rhMPV immunization at week 4 postinfection (P ⬎ 0.05). Antibody was detectable at week 1
postimmunization, and the levels gradually increased during weeks 2 to 4. However, the antibody titer of rhMPV-S57A-S60A was significantly lower than that of S57A and rhMPV-S60A (P⬍ 0.01), suggesting that rhMPV-S57A-S60A is overly attenuated. No hMPV-specific antibody was detected in the un-vaccinated control. At week 4 postimmunization, all un-vaccinated cotton rats were challenged with wt rhMPV at a dose of 105PFU, and cotton rats were sacrificed at day 4 postinfection. No infec-tious virus was detectable in either nasal turbinates or lungs from animals vaccinated with either rhMPV-S57A, rhMPV-S60A, or rhMPV-S57A-S60A following challenge with rhMPV (Table 2). Interestingly, rhMPV-S57A-S60A was capable of preventing hMPV replication in both nasal turbinates and lungs despite the fact that it triggered a low antibody titer.
To determine whether vaccination can protect the lung from damage after challenge, all lungs were examined histologically. The unvaccinated challenged control had moderate histologic le-sions characterized by interstitial pneumonia, mononuclear cell infiltration, and edematous thickening of the bronchial submu-cosa. In contrast, no or only mild histological changes were found in the lungs of cotton rats vaccinated with S57A, rhMPV-S60A, and rhMPV-S57A-S60A (Table 2). Furthermore, hMPV antigen expression in lung tissue was examined by IHC. A large number of hMPV antigens was found at the luminal surface of the bronchial epithelial cells in lung tissues from the unvaccinated challenged controls (Fig. 13). In contrast, no or little antigen was found on the luminal surface of bronchial epithelial cells in the rhMPV-S57A, rhMPV-S60A, and rhMPV-S57A-S60A groups (Fig. 13). Therefore, these results demonstrate that rhMPVs lack-ing phosphorylation sites in M2-1 are capable of triggerlack-ing high levels of neutralizing antibody and provide complete protection against hMPV infection.
DISCUSSION
[image:11.585.41.545.78.217.2]Numerous prokaryotic and eukaryotic transcription factors use phosphorylation as a means to positively or negatively control activity (30,31). The M2-1 protein of pneumoviruses is a typical
TABLE 1Replication of rhMPV carrying M2-1 phosphorylation site mutations in cotton rats
Mutanta Dose
Viral replication in:
Lung histologyc
Lung IHCd
Nasal turbinateb Lung
% of infected animals
Mean titer, log10 (PFU/g)
% of infected animals
Mean titer, log10 (PFU/g)
rhMPV 105 100 4.82⫾0.21a 100 5.02⫾0.23a 2.5a 3.0a
rhMPV-S57A 105 60 2.38⫾0.26b 60 2.70⫾0.27c 0.8b 0.5b
rhMPV-S60A 105 60 3.07⫾0.54b 100 2.87⫾0.54c 0.8b 0.8b
rhMPV 104 100 3.91⫾0.19a 100 3.83⫾0.51b 1.8a 2.5a
rhMPV-S57A 104 40 1.68⫾0.26c 40 1.99⫾0.74c 0.6b 0.5b
rhMPV-S60A 104 40 1.48⫾0.07c 0 ND 0.8b 0.5b
rhMPV-S57A-S60A 104 0 ND 0 ND 0.6b 0c
DMEM 0 0 ND 0 ND 0c 0c
aCotton rats were inoculated intranasally with DMEM or with 1.0⫻105PFU or 1.0⫻104wild-type rhMPV or rhMPV mutants. DMEM indicates an unvaccinated unchallenged
control. At day 4 postimmunization animals were euthanized. Viral titers were determined in nasal turbinate and lung tissues, and lung sections were prepared for histological examination.
b
ND indicates that infectious virus was not detectable. Values within a column followed by different lowercase letters (a, b, and c) are significantly different (P⬍0.05). cThe severity of histological changes in lung tissues was scored for each individual animal. The values represent the mean score for each group. 0, no change; 1, mild change; 2,
moderate change; 3, severe change.
dhMPV antigen expression in the lung was determined by IHC, and the quantity of antigen present was scored on a subjective scale. The values represent the mean score for each
group. 0, no antigen; 1, small amount; 2, moderate amount; 3, large amount.
FIG 10Lung histological changes in cotton rats infected by rhMPVs. The right lung from each cotton rat was preserved in 4% (vol/vol) phosphate-buffered paraformaldehyde. Fixed tissues were embedded in paraffin, sec-tioned at 5m, and stained with hematoxylin-eosin (HE) for the examination of histological changes by light microscopy.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.41.287.522.674.2]zinc binding protein and forms tetramers in solution (17,21–23). Previously, we showed that the zinc binding activity of hMPV M2-1 is essential for viral replicationin vitroandin vivo(21). In this study, we mapped the phosphorylation sites in the hMPV M2-1 protein to residues S57 and S60 and found that phosphor-ylation of M2-1 is not essential for zinc binding and oligomeriza-tion. However, zinc binding is essential for phosphorylation of M2-1 and oligomerization. Furthermore, we found that rhMPV lacking phosphorylation in M2-1 significantly decreased viral RNA synthesis, replication, and pathogenesis. Thus, phosphory-lation of M2-1 positively regulates the replication of hMPV.
All nonsegmented negative-sense (NNS) RNA viruses share a similar strategy for replication and gene expression (29). The
ge-nome of NNS RNA viruses is encapsidated with the N protein to form a nuclease-resistant helical N-RNA complex that is the func-tional template for mRNA synthesis as well as genomic RNA rep-lication. The N-RNA complex is tightly associated with the viral RNA-dependent RNA polymerase (RdRp), which is comprised of the L protein catalytic subunit and the P protein cofactor and results in the assembly of a viral RNP complex. This structure contains the essential virally encoded components of the RNA synthesis machinery (29). The RdRp of thePneumovirinae sub-family of the sub-familyParamyxoviridaerequires an additional cofac-tor, the M2-1 protein (7), whereas the polymerase of Ebola virus (Filoviridaefamily) requires VP30 as an additional cofactor (32– 34). Interestingly, M2-1 shares structural and functional similarity with Ebola virus VP30 (24). Both M2-1 and VP30 are phosphor-ylated, possess RNA binding and zinc binding activity, interact with the nucleoprotein, and are thought to play regulatory roles in RNA synthesis (10,15,24,33). The M2-1 protein of hRSV is an elongation and antitermination factor (7,10–12). However, this function is not observed for hMPV M2-1 and filovirus VP30 (8,
20,34–36). Deletion of M2-1 from the hRSV genome is lethal to virus, whereas deletion of hMPV M2-1 yielded a recombinant virus that was highly attenuatedin vitroandin vivo(18–20). De-letion of the VP30 gene from the Ebola virus genome resulted in a replication-deficient Ebola virus (Ebola⌬VP30), which can repli-cate efficiently only if the VP30 protein is provided intrans(35,
36). Thus, it appears that hMPV M2-1 shares more functional similarities with filovirus VP30 than hRSV M2-1.
The hMPV M2-1 protein is a basic protein of 187 amino acids (aa). The N terminus of M2-1 includes three functional sites (do-mains): the zinc binding domain (aa 7 to 31) and the oligomer-ization domain (aa 32 to 53), followed by two phosphorylation sites (S57 and S60). Prior to this study, the relationship between these functional sites was not known. We found that phosphory-lation of M2-1 did not affect zinc binding and oligomerization of M2-1, whereas zinc binding activity abolished phosphorylation of M2-1 and oligomerization. The thermostability of GST–M2-1
FIG 11IHC staining of lungs from cotton rats infected by rhMPVs. Lung tissues were fixed in 4% (vol/vol) phosphate-buffered paraformaldehyde. Deparaf-finized sections were stained with monoclonal antibody against hMPV matrix protein (Virostat, Portland, ME) to determine the distribution of viral antigen.
FIG 12Recombinant hMPVs triggered a high level of neutralizing antibody titer in cotton rats. Cotton rats were immunized with each recombinant hMPV intranasally at a dose of 1.0⫻104PFU per rat. Blood samples were collected from each rat weekly by retro-orbital bleeding. The hMPV neutralizing anti-body was determined using a plaque reduction neutralization assay as de-scribed in Materials and Methods. Data were the average titers from four cotton rats⫾1 standard deviation. Antibody titers of each mutant at each time point were compared to those of rhMPV (*,P⬍0.05; **,P⬍0.01).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:12.585.114.470.65.281.2] [image:12.585.41.233.490.647.2]and mutants was measured by differential scanning fluorimetry (DSF). It was found that the melting point (Tm) of the
phosphor-ylation site mutants (S57A, S60A, and S57A-S60A) was not signif-icantly altered (data not shown). However, theTmof the zinc
binding mutants (C21S and H25L) decreased by 3 to 5°C. This suggests that mutations to the zinc binding motif significantly altered the local and global structure of the M2-1 protein (data not shown). Our findings are in good agreement with the crystal struc-ture. Based on the crystal structure, the zinc binding residues C7, C15, C21, and H25 all are located at a coiled-coil structure (23). With the help of zinc ion coordination, these coils form stable subdomains required for correct folding of M2-1. Thus, abolish-ing the zinc bindabolish-ing ability would disturb M2-1 foldabolish-ing and cause a global tertiary structure change. In contrast, the
phosphoryla-tion sites are located at a short linker region between two stable␣ helices (23), so mutations to the phosphorylation sites may not alter the structure of M2-1 and thus not affect other functions, such as zinc binding and oligomerization.
[image:13.585.39.547.77.195.2]Phosphorylation of the components of the RNP complex has been shown to play many crucial roles in transcription, replica-tion, and genome stability (33,37–39). To date, N/NP, P, and M2-1/VP30 in the RNP complex have been shown to be phos-phorylated (33,37,39–42). For example, phosphorylation of the NP protein of measles virus (38), rabies virus (37), and Nipah virus (39) has been shown to upregulate transcriptional activity. In contrast, phosphorylation of mumps virus NP downregulates viral RNA synthesis (43). The P proteins of all NNS RNA viruses are heavily phosphorylated during the viral life cycle. Recombi-nant vesicular stomatitis virus (VSV) P protein expressed from Escherichia colilacking phosphorylation was unable to supportin vitrotranscription, demonstrating that P protein phosphorylation is essential for viral mRNA transcription (40, 41). Mutation of phosphorylation sites Ser60, Thr62, and Ser64 in the N-terminal domain of P inhibited transcription by 50 to 98%; however, ge-nome RNA replication was largely unaffected (42). Interestingly, phosphorylation of Tyr14 in the N-terminal region of P protein is essential for both viral mRNA transcription and genome replica-tion (44). It appears that phosphorylation of P controls the bal-ance between transcription and replication (42, 44). For N/NP and P proteins, it is difficult to silence all phosphorylation sites in an infectious virus due to multiple phosphorylation sites. How-ever, it is generally accepted that phosphorylation of RNP compo-nents can both up- and downregulate activity and that these dif-ferences may also depend on the site that is being phosphorylated. It is relatively simple to determine the role of M2-1 phosphor-ylation in the hMPV life cycle, because there are only two phos-phorylation residues in M2-1 protein. We found that mutations to these two phosphorylation sites resulted in significant decreases in genomic RNA and mRNA synthesis compared to rhMPV. How-ever, there was no significant difference in the mRNA/genomic RNA ratio between any of the rhMPV mutants and wild-type virus (P⬎0.05). This suggests that mutations to the M2-1 phosphory-lation sites did not significantly alter the balance between genome
TABLE 2Immunogenicity of rhMPV carrying M2-1 phosphorylation site mutations in cotton rats
Groupa
No. of cotton rats per group
Viral replication in:
Lung histologyc
Lung IHCd
Nasal turbinateb Lung
% of infected animals
Mean titer, log (PFU/g)
% of infected animals
Mean titer, log (PFU/g)
DMEM 5 0 ND 0 ND 0b 0b
Challenge control 5 100 3.96⫾0.40 100 4.04⫾0.29 2.5a 3.0a
rhMPV 5 0 ND 0 ND 1.0c 0.5c
rhMPV-S57A 5 0 ND 0 ND 0.5c 0.5c
rhMPV-S60A 5 0 ND 0 ND 0.8c 0.5c
rhMPV-S57A-S60A 5 0 ND 0 ND 0.5c 0.5c
aAnimals were immunized intranasally with DMEM or 1.0⫻104PFU wild-type rhMPV or rhMPV mutants. At day 28 postimmunization, animals were challenged with 105PFU
wild-type rhMPV. DMEM indicates an unvaccinated unchallenged control. Challenge control indicates unvaccinated challenged control. rhMPV indicates that cotton rats were vaccinated with rhMPV and challenged with rhMPV.
b
ND indicates infectious virus was not detectable.
cThe severity of lung histology was scored for each lung tissue. The average score for each group is shown. 0, no change; 1, mild change; 2, moderate change; 3, severe change.
Values within a column followed by different lowercase letters (a, b, and c) are significantly different (P⬍0.05).
dhMPV antigen expression in the lung was determined by IHC, and the quantity of antigen present was scored on a subjective scale. The values represent the mean score for each
group. 0, no antigen; 1, small amount; 2, moderate amount; 3, large amount.
FIG 13IHC staining of lungs from cotton rats vaccinated by rhMPV mutants followed by rhMPV challenge. Lung tissues were fixed in 4% (vol/vol) phos-phate-buffered paraformaldehyde. Deparaffinized sections were stained with monoclonal antibody against hMPV matrix protein (Virostat, Portland, ME) to determine the distribution of viral antigen.