The Glycoprotein B Cytoplasmic Domain
Lysine Cluster Is Critical for
Varicella-Zoster Virus Cell-Cell Fusion Regulation
and Infection
Edward Yang, Ann M. Arvin, Stefan L. Oliver
Departments of Pediatrics and Microbiology & Immunology, Stanford University School of Medicine, Stanford, California, USA
ABSTRACT The conserved glycoproteins gB and gH-gL are essential for herpesvirus entry and cell-cell fusion induced syncytium formation, a characteristic of varicella-zoster virus (VZV) pathology in skin and sensory ganglia. VZV syncytium formation, which has been implicated in the painful condition of postherpetic neuralgia, is reg-ulated by the cytoplasmic domains of gB (gBcyt) via an immunoreceptor tyrosine-based inhibition motif (ITIM) and gH (gHcyt). A lysine cluster (K894, K897, K898, and K900) in the VZV gBcyt was identified by sequence alignment to be conserved among alphaherpesviruses, suggesting a functional role. Alanine and arginine substi-tutions were used to determine if the positive charge and susceptibility to posttrans-lational modifications of these lysines contributed to gB/gH-gL cell-cell fusion. Criti-cally, the positive charge of the lysine residues was necessary for fusion regulation, as alanine substitutions induced a 440% increase in fusion compared to that of the
wild-type gBcyt while arginine substitutions had wild-type-like fusion levels in an in
vitro gB/gH-gL cell fusion assay. Consistent with these results, the alanine substitu-tions in the viral genome caused exaggerated syncytium formation, reduced VZV
ti-ters (⫺1.5 log10), and smaller plaques than with the parental Oka (pOka) strain. In
contrast, arginine substitutions resulted in syncytia with only 2-fold more nuclei,
a ⫺0.5-log10 reduction in titers, and pOka-like plaques. VZV mutants with both an
ITIM mutation and either alanine or arginine substitutions had reduced titers and small plaques but differed in syncytium morphology. Thus, effective VZV propaga-tion is dependent on cell-cell fusion regulapropaga-tion by the conserved gBcyt lysine clus-ter, in addition to the gBcyt ITIM and the gHcyt.
IMPORTANCE Varicella-zoster virus (VZV) is a ubiquitous pathogen that causes chickenpox and shingles. Individuals afflicted with shingles risk developing the painful condition of postherpetic neuralgia (PHN), which has been difficult to treat because the underlying cause is not well understood. Additional therapies are needed, as the current vaccine is not recommended for immunocompro-mised individuals and its efficacy decreases with the age of the recipient. VZV is known to induce the formation of multinuclear cells in neuronal tissue, which has been proposed to be a factor contributing to PHN. This study examines the role of a lysine cluster in the cytoplasmic domain of the VZV fusion protein, gB, in the formation of VZV induced multinuclear cells and in virus replication kinet-ics and spread. The findings further elucidate how VZV self-regulates multinu-clear cell formation and may provide insight into the development of new PHN therapies.
KEYWORDS cell-cell fusion, glycoproteins, human herpesviruses, varicella-zoster virus
Received25 August 2016Accepted18
October 2016
Accepted manuscript posted online26
October 2016
CitationYang E, Arvin AM, Oliver SL. 2017. The
glycoprotein B cytoplasmic domain lysine cluster is critical for varicella-zoster virus cell-cell fusion regulation and infection. J Virol 91:e01707-16. https://doi.org/10.1128/ JVI.01707-16.
EditorKlaus Frueh, Oregon Health & Science
University
Copyright© 2016 American Society for
Microbiology. All Rights Reserved.
Address correspondence to Edward Yang, edwyang@stanford.edu.
A.M.A. and S.L.O. contributed equally to this article.
crossm
on November 7, 2019 by guest
http://jvi.asm.org/
V
aricella-zoster virus (VZV), a member of the subfamily Alphaherpesvirinae, is a human pathogen that causes varicella (chickenpox) during primary infection and herpes zoster (shingles) upon reactivation from latently infected sensory neurons (1, 2). The current vaccination program of children in the United States has been successful in reducing the incidence of varicella by 90% (3). However, the current FDA-approvedzoster vaccine has been shown to be less efficacious for vaccine recipients aged⬎60
years, with vaccine effectiveness decreasing from 69% in the first year to 4.2% in the eighth year (4). The waning protection of the zoster vaccine is a significant medical concern, as patients afflicted with herpes zoster have a 27 to 74% risk of developing the painful complication of postherpetic neuralgia (PHN) depending on their age (5). PHN can last from weeks to months after the acute zoster rash has healed, with treatment limited to pain management, as the cause of the complication is not well understood (6).
Multinuclear cells called syncytia are a hallmark of VZV infection resulting from the fusion of infected cells (1). They have been observed in skin lesions and trigeminal ganglia taken at autopsy from individuals afflicted with zoster at time of death (7, 8). The fusion of infected satellite cells and neurons observed in the cadaver ganglia and in the human dorsal root ganglia xenograft model has implicated the damage to the fused cells as a potential factor contributing to PHN after VZV reactivation in the sensory nerve ganglia (9, 10). Understanding and defining the mechanisms that govern VZV syncytium formation could lead to the development of effective PHN treatments. VZV cell-cell fusion is mediated by the conserved herpesvirus fusion machinery of glycoproteins gB and gH-gL, which are expressed on the surface of infected cells (11). The three glycoproteins are also present on the alphaherpesvirus particles and are required for fusion of the virion envelope and cell membrane during entry (11, 12). Transient expression of VZV gB and gH-gL has been shown to be necessary and
sufficient to induce membrane fusion usingin vitrocell-cell fusion assays (13–15). In
contrast, other human herpesviruses require additional viral accessory proteins for membrane fusion, including glycoprotein D (gD) for herpes simplex virus 1 (HSV-1) and gp42 for Epstein-Barr virus (EBV) for specific cell types (16, 17). Defining the individual roles of the glycoproteins involved in membrane fusion has been critical in under-standing the mechanism of VZV syncytium formation and its relationship with patho-genesis.
VZV gB is a type 1 transmembrane protein, encoded by open reading frame 31 (ORF31), that has been shown to be essential for infection based on a deletion mutagenesis study (18). After translation, gB is exported from the endoplasmic retic-ulum (ER), processed in the Golgi apparatus, trafficked to the cell surface, endocytosed,
and then returned to thetrans-Golgi network (TGN) for insertion into the viral envelope
(12, 19, 20). The protein forms a trimer, with each 931-amino-acid (aa) monomer having an ectodomain, transmembrane domain, and a 124-aa cytoplasmic domain (21, 22). VZV gB is presumed to be the fusogen based on structural homology modeling with the solved crystal structure of HSV-1 gB (22, 23). The glycoprotein has also been shown to associate with a proposed entry receptor, myelin-associated glycoprotein (MAG), via attached sialic acids to facilitate fusion and entry (24). However, MAG is unlikely to be the sole receptor, as MAG is absent on cells susceptible to VZV infection.
Cell-cell fusion mediated by VZV gB/gH-gL has previously been shown to be regulated by a functional immunoreceptor tyrosine-based inhibition motif (ITIM) in the gB cytoplasmic domain (gBcyt) (22). ITIM-containing proteins have various functions, including negatively regulating immune cell responses, promoting cell proliferation, and blocking apoptosis (25). Substituting an alanine (Y881F) or tryptophan (Y881W) for
the tyrosine residue in the gBcyt ITIM sequence,879IKYMTL884, induced a hyperfusion
phenotype in anin vitrocell fusion assay. This fusion dysregulation caused exaggerated
syncytium formation in melanoma cells infected with the Y881F mutant virus, which resulted in reduced replication kinetics and propagation compared to those of the parental Oka (pOka) strain. VZV skin pathogenesis was also impaired by the
on November 7, 2019 by guest
http://jvi.asm.org/
ated syncytium formation in infected human skin xenografts implanted in severe combined immunodeficiency (SCID) mice.
Further emphasizing the importance of gBcyt fusion regulation, exaggerated syn-cytium formation has also been observed in melanoma cells infected with the gB-36 VZV mutant virus, which lacks amino acids 896 to 931 of the gBcyt (26). The truncation
of the gBcyt preserved the ITIM sequence but removed the YXX⌽motif,920YSRV923,
which has been shown to have a role in gB trafficking and processing (27). This motif was unlikely to be responsible for the exaggerated syncytium formation, because
disrupting the YXX⌽ motif with a Y920F substitution reduces rather than increases
gB/gH-gL-mediated cell-cell fusion (22). This suggests the presence of an additional fusion regulatory element in the terminal 36 amino acids of the gBcyt.
In the present study, the last 36 amino acids of the gBcyt were examined for an additional fusion regulatory element. A lysine cluster in the VZV gBcyt was identified to be conserved for alphaherpesviruses by sequence alignment and postulated to con-tribute to VZV gB/gH-gL-mediated cell-cell fusion regulation. This concept was sup-ported by a previous study in which disrupting an HSV-1 gBcyt lysine cluster induced hyperfusion in a virus-free cell-cell fusion assay (28). However, the role of the lysine cluster in the context of herpesvirus infection has not been previously examined. To characterize the VZV lysine cluster, mutant gB constructs were generated with alanine or arginine substitutions of the lysine cluster and evaluated in the recently developed gB/gH-gL stable reporter fusion assay (SRFA) (15). The role of the lysine cluster in VZV syncytium formation, replication kinetics, and spread was examined by transferring the mutations into recombinant VZV. The data derived demonstrated the positive charge of the gBcyt lysine cluster to be critical for cell-cell fusion regulation and effective VZV propagation.
RESULTS
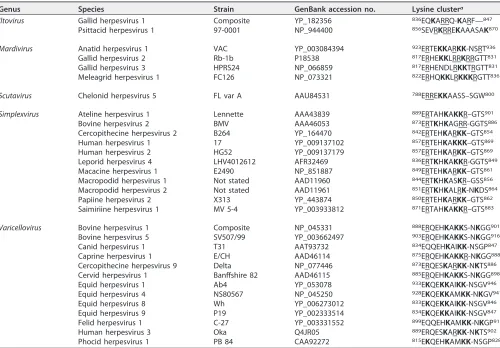
Positive charge of the VZV gBcyt lysine cluster contributes to VZV gB/gH-gL-mediated cell-cell fusion regulation. To identify a conserved sequence within the terminal 36 amino acids of the gBcyt likely responsible for fusion regulation, the predicted cytoplasmic domains of all known alphaherpesvirus gB homologues were aligned and compared to that of VZV gB (Table 1). The lysine at position 894 (K894) of VZV gB was conserved for 29 of the 32 alphaherpesviruses. The three lysines at position 894 (K894), 897 (K897), and 898 (K898) of VZV gB were present in 10 species in the
genus Varicellovirus and in five species in the genus Simplexvirus, including herpes
simplex viruses 1 and 2. A fourth lysine residue at position 900 (K900) of VZV gB was
conserved among seven members of the genus Varicellovirus. All four lysines of the
cluster (K894, K897, K898, and K900) were conserved for VZV, simian varicella virus (cercopithecine herpesvirus 9), equid herpesvirus 4, and felid herpesvirus 1. In addition, the arginine at position 896 was conserved for VZV, cercopithecine herpesvirus 9, papiine herpesvirus 2, macacine herpesvirus 1, human herpesvirus 2, cercopithecine herpesvirus 2, meleagrid herpesvirus 1, gallid herpesvirus 3, gallid herpesvirus 2, and anatid herpesvirus 1. The conserved nature of the lysines among members of the
alphaherpesvirinaeand the absence of the K897, K898, and K900 residues in the gB-36 virus examined by Heineman and Hall (26) suggested that the gBcyt lysine cluster might be a potential fusion regulatory element.
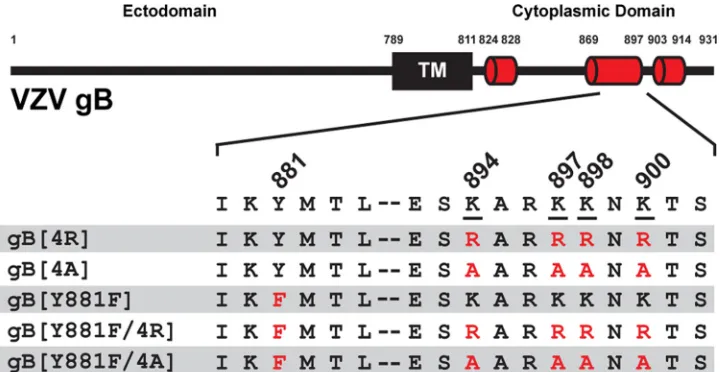
To determine the role of the gBcyt lysine cluster in VZV gB/gH-gL-mediated cell-cell fusion, gB mutants were generated and evaluated in the stable reporter fusion assay (SRFA), a quantitative cell-cell fusion assay (15). Lysines can be positively charged at physiological pH and are susceptible to posttranslational modifications (PTMs), includ-ing ubiquitination and acetylation (29, 30). To examine the effects of these lysine properties on gB/gH-gL-mediated cell-cell fusion, two mutants were generated that replaced all four lysine residues with either arginines (gB[4R]), to preserve the positive charge while removing any potential lysine-specific PTMs, or alanines (gB[4A]), to remove the positive charge at the positions (Fig. 1). The gB[4R] mutant had gB/gH-gL-mediated cell-cell fusion levels comparable to those with wild-type gB (gB[WT])
on November 7, 2019 by guest
http://jvi.asm.org/
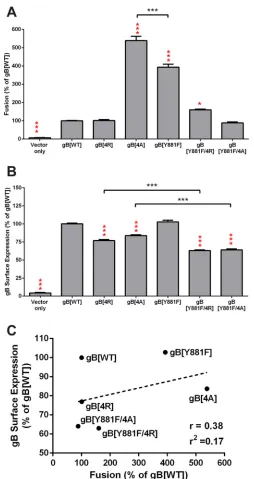
(Fig. 2A). In contrast, the gB[4A] mutant had a 440% increase in fusion compared to the level of gB[WT], indicating that the alanine substitution resulted in a loss of fusion regulation. gB[4A] was more hyperfusogenic than gB[Y881F], which had only a 300% increase in fusion compared to that with gB[WT]. Thus, the positive charge of the gBcyt lysine cluster contributes to the regulation of gB/gH-gL-mediated cell-cell fusion when expressed in the absence of other VZV proteins.
To ascertain if disrupting both the lysine cluster and the ITIM in the gBcyt would result in an additive increase in gB/gH-gL-mediated cell-cell fusion, two gB double mutants were generated with the Y881F mutation and either the arginine (gB[Y881F/ 4R]) or alanine (gB[Y881F/4A]) substitution of the lysine cluster (Fig. 1). The gB[Y881F/ 4R] mutant had a 60% increase in fusion compared to gB[WT], while the gB[Y881F/4A] mutant had gB[WT]-like fusion levels (Fig. 2A). Thus, substitutions in the gBcyt lysine cluster do not further enhance the Y881F hyperfusion phenotype.
[image:4.585.43.543.91.439.2]Whether the hyperfusion phenotype was a result of increased gB surface expression was determined by flow cytometry analysis of CHO cells transiently expressing gB[WT] or gBcyt mutants (Fig. 2B). gB[4R] and gB[4A] exhibited surface expression levels that were 23% and 16% lower than that of gB[WT], respectively. The double mutants gB[Y881F/4R] and gB[Y881F/4A] had reductions in surface expression of 36% and 37% compared with gB[WT], respectively. Furthermore, the surface expression levels of the double mutants were also significantly lower than those of their respective single mutants. The lower surface expression of the double mutants compared to that of the
TABLE 1Amino acid sequences that align with the VZV gBcyt lysine cluster for all species in the five genera of the subfamily Alphaherpesvirinae
Genus Species Strain GenBank accession no. Lysine clustera
Iltovirus Gallid herpesvirus 1 Composite YP_182356 836EQKARRQ-KARF—847
Psittacid herpesvirus 1 97-0001 NP_944400 856SEVRKRREKAAASAK870
Mardivirus Anatid herpesvirus 1 VAC YP_003084394 923ERTEKKARKK-NSRT936
Gallid herpesvirus 2 Rb-1b P18538 817ERHEKKLRRKRRGTT831 Gallid herpesvirus 3 HPRS24 NP_066859 817ERHENDLRKKTRGTT831 Meleagrid herpesvirus 1 FC126 NP_073321 822ERHQKKLRKKKRGTT836
Scutavirus Chelonid herpesvirus 5 FL var A AAU84531 788ERREKKAASS–SGW800
Simplexvirus Ateline herpesvirus 1 Lennette AAA43839 889ERTAHKAKKR–GTS901
Bovine herpesvirus 2 BMV AAA46053 873ERTKHKAGRR-GGTS886 Cercopithecine herpesvirus 2 B264 YP_164470 842ERTEHKARKK–GTS854 Human herpesvirus 1 17 YP_009137102 857ERTEHKAKKK–GTS869 Human herpesvirus 2 HG52 YP_009137179 857ERTEHKARKK–GTS869 Leporid herpesvirus 4 LHV4012612 AFR32469 836ERTKHKAKKR-GGTS849 Macacine herpesvirus 1 E2490 NP_851887 849ERTEHKARKK–GTS861 Macropodid herpesvirus 1 Not stated AAD11960 844ERTKHKASKR–GSS856 Macropodid herpesvirus 2 Not stated AAD11961 851ERTKHKALRK-NKDS864 Papiine herpesvirus 2 X313 YP_443874 850ERTEHKARKK–GTS862 Saimiriine herpesvirus 1 MV 5-4 YP_003933812 871ERTAHKAKKR–GTS883
Varicellovirus Bovine herpesvirus 1 Composite NP_045331 888ERQEHKAKKS-NKGG901 Bovine herpesvirus 5 SV507/99 YP_003662497 903ERQEHKAKKS-NKGG916 Canid herpesvirus 1 T31 AAT93732 834EQQEHKAIKK-NSGP847 Caprine herpesvirus 1 E/CH AAD46114 875ERQEHKAKKR-NKGG888 Cercopithecine herpesvirus 9 Delta NP_077446 873ERQESKARKK-NKTS886 Cervid herpesvirus 1 Banffshire 82 AAD46115 885ERQEHKAKKS-NKGG898 Equid herpesvirus 1 Ab4 YP_053078 933EKQEKKAIKK-NSGV946 Equid herpesvirus 4 NS80567 NP_045250 928EKQEKKAMKK-NKGV941 Equid herpesvirus 8 Wh YP_006273012 833EKQEKKAIKK-NSGV846 Equid herpesvirus 9 P19 YP_002333514 834EKQEKKAIKK-NSGV847 Felid herpesvirus 1 C-27 YP_003331552 899EQQEHKAMKK-NKGP912 Human herpesvirus 3 Oka Q4JR05 889ERQESKARKK-NKTS902 Phocid herpesvirus 1 PB 84 CAA92272 815EKQEHKAMKK-NSGP828
aThe amino acid sequences of known alphaherpesviruses gB homologues were aligned, and the sequences of the conserved lysine cluster and its flanking regions are shown. The lysines in the sequence are in bold, and the arginines are underlined.
on November 7, 2019 by guest
http://jvi.asm.org/
single mutants indicated that combining both ITIM and lysine cluster mutations in gBcyt further disrupted gB function and interfered with gB trafficking to the cell surface. Consistent with previous observations, gB[Y881F] had surface expression levels com-parable to those of gB[WT] (22). To determine if there was a correlation between surface expression and cell fusion levels, the mean levels of surface expression of gB[WT] and the gBcyt mutants were plotted against their cell fusion levels on a scatter plot (Fig. 2C).
A Pearson r value of 0.38 and r2 value of 0.17 were observed, indicating that no
significant correlation was found between surface expression of gB at the levels observed with the gBcyt mutants and the hyperfusion phenotype. Both the gB[4R] and gB[Y881F/4A] mutants were also implicated to be hyperfusogenic, as less gB surface expression was required to induce fusion levels equivalent to those with gB[WT] (Table 2). Thus, while the lysine cluster contributes to gB surface expression, the hyperfusion phenotype of gB[4A] is not a result of an increase in gB surface expression.
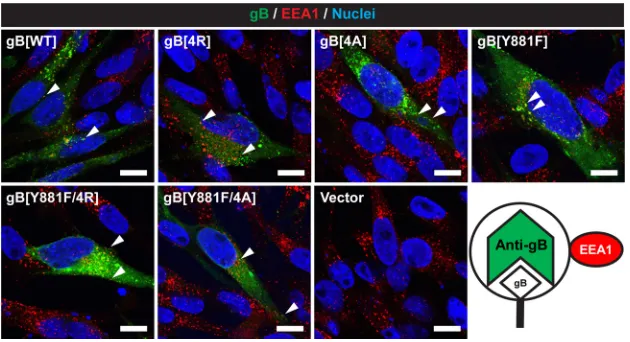
Trafficking of gB to endocytic vesicles is not inhibited by the gBcyt lysine cluster substitutions. To assess if the changes in gB/gH-gL-mediated cell fusion associated with the mutations to the gBcyt lysine cluster were a result of altered gB endocytosis, the subcellular localization of gB was examined in transfected melanoma cells by confocal microscopy (Fig. 3). Consistent with past observations, gB[WT] and gB[Y881F] had punctate localization in the cytoplasm that colocalized with early endosomes, detected with anti-early endosome antigen 1 antibody (EEA-1) (22). Similar colocalization of gB and early endosomes was observed for the gB[4R], gB[4A], gB[Y881F/4R], and gB[Y881F/4A] mutants. Thus, the mutations to the gBcyt lysine cluster do not abolish gB endocytosis.
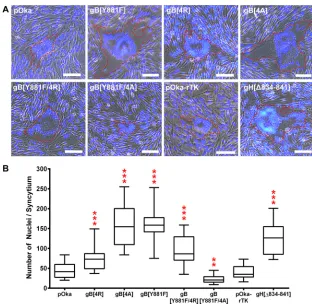
The gBcyt lysine cluster alanine substitutions induce exaggerated syncytium formation in infected melanoma cells.The effect of the gBcyt lysine mutations on VZV-induced syncytium formation was examined by generating four recombinant viruses with either the 4R, 4A, Y881F/4R, or Y881F/4A substitution into the parental Oka (pOka) bacterial artificial chromosome (BAC) with ORF36, which encodes the thymidine kinase, tagged with red fluorescent protein (RFP) at the C terminus (pOka-rTK). Infec-tious virus was recovered from all of the recombinant mutant pOka BACs. At 36 h postinfection, syncytia were observed in infected melanoma cells for all of the gBcyt mutant viruses, pOka-gB[4R], pOka-gB[4A], pOka-gB[Y881F/4R], and pOka-gB[Y881F/
FIG 1Mutagenesis of the varicella-zoster virus gBcyt lysine cluster. VZV gB constructs with substitution mutations of the tyrosine residue of the immunoreceptor tyrosine-based inhibition motif (ITIM) at position 881 (Y881F) and the lysine cluster, K894, K897, K898, and K900 are shown. Lysines were replaced with either arginine, which maintains the positive charge at the position but removes potential lysine specific post translational modifications, or alanine, which removes the positive charge at the position. On the black bar representing the full-length gB, the transmembrane domain is indicated with a black box (TM), and predicated alpha-helices in the cytoplasmic domain are represented by red cylinders. The numbers immediately above the black bar indicate position numbers. Amino acid residues 879 to 902 are presented with the lysines underlined and mutations indicated in red.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.43.403.71.257.2]4A] (Fig. 4A). The RFP tag on TK did not affect syncytium formation, as pOka-rTK-induced syncytia were similar to pOka-pOka-rTK-induced syncytia, with a median value of 35 nuclei/syncytium, compared to 40 nuclei/syncytium (Fig. 4B). pOka-gB[4R] had signifi-cantly enlarged syncytia, with a median value of 73 nuclei/syncytium. The increased
FIG 2Alanine substitutions of the gBcyt cluster enhance VZV gB-gH/gL-mediated cell-cell fusion. (A) The frequency of cell-cell fusion events of wild-type gB (gB[WT]) or the gB cytoplasmic domain (gBcyt) mutants, gB[4R], gB[4A], gB[Y881F], gB[Y881F/4R], and gB[Y881/4A], quantified by the VZV gB/gH-gL stable reporter fusion assay and flow cytometry. Fusion is represented as a percentage relative to the fusion events of gB[WT]. (B) Cell surface expression of gB on CHO-DSP1 cells transfected with plasmids expressing gB[WT] or the gBcyt mutants quantified by nonpermeabilized staining for gB and flow cytometry. gB surface expression is represented as a percentage relative to that of gB[WT]. For both assays, empty vector (vector only) was used as a negative control, and statistical differences were evaluated between either gB[WT] and the gBcyt mutants (red) or between the indicated gBcyt mutants (black) by ANOVA (*,P⬍0.05;***,P⬍0.001). Data represent the means of, at minimum, duplicate experiments, with bars indicating the standard errors of the means (SEM). (C) gB mutants were examined on a scatter plot comparing their mean surface expression and mean fusion levels relative to those of gB[WT]. The dotted line represents the trend line, with Pearsonrandr2values noted.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.79.333.67.546.2]number of nuclei of pOka-gB[4R] indicated that the substitution induced a hyperfusion
phenotype which corroborated the interpretation of the reduced in vitro surface
expression and gB[WT]-like cell-cell fusion data for gB[4R]. Exaggerated syncytium formation was observed for both pOka-gB[4A] and pOka-gB[Y881F], with median values of 154 and 158 nuclei/syncytium, respectively, which correlated with the hyperfusion phenotype observed in the cell-cell fusion assay. Consistent with the moderate hyper-fusion phenotype observed in the cell-cell hyper-fusion assay, pOka-gB[Y881F/4R] induced syncytia larger than pOka (median value of 87 nuclei/syncytium) but smaller than both gB[4A] and gB[Y881F]. In contrast to the other gBcyt mutants, pOka-gB[Y881F/4A] induced smaller syncytia than pOka, with a median value of 20 nuclei/ syncytium. This provided further evidence that combining the ITIM and lysine cluster mutations further disrupts gB function. The pOka-gH[Δ834-841] virus, which has the last eight amino acids of the gH cytoplasmic domain (gHcyt) deleted, also exhibited the exaggerated syncytium formation phenotype, with a median value of 126 nuclei/ syncytium, consistent with past observations (31). Thus, the positive charge of the gBcyt lysine cluster is critical for the regulation of cell-cell fusion in infected melanoma cells.
[image:7.585.41.546.84.177.2]VZV replication kinetics and spread are impaired by the gBcyt lysine cluster alanine substitutions.To determine if the gBcyt mutations inhibited propagation of the virus, the replication kinetics of the mutant viruses were compared to that of pOka in melanoma cells infected with cell-associated inoculum over 6 days. Correlating with the pOka-like syncytia morphology, the pOka-rTK virus had viral titers similar to those
TABLE 2Comparison of the gBcyt lysine cluster mutant phenotypes
gB construct
Cell-cell fusion (% of gB[WT])
In vitrosurface expression (% of gB[WT])
Cell-cell fusion phenotype
Syncytium formation
(median no. of nuclei/syncytium)
Viral titer at dpi 3 (log10)
Median plaque size (mm2)
gB[WT] 100 100 Wild type Wild type (40) 5.9 1.16
gB[4R] 100 77 Hyperfusion Exaggerated (73) 5.4 1.36
gB[4A] 538 84 Hyperfusion Exaggerated (154) 4.6 0.48
gB[Y881F] 393 103 Hyperfusion Exaggerated (158) 5.2 0.82 gB[Y881F/4R] 160 63 Hyperfusion Exaggerated (87) 5.1 0.63
gB[Y881F/4A] 88 64 Hyperfusion? Reduced (20) 4.9 0.4
FIG 3Substitutions to the gBcyt lysine cluster do not inhibit colocalization of gB with early endosomes in vitro. Shown are confocal microscopy images of melanoma cells transiently expressing wild-type gB (gB[WT]), gB[4R], gB[4A], gB[Y881F], gB[Y881F/4R], or gB[Y881/4A] at 24 h posttransfection. Cells were stained for gB (green), early endosome antigen (EEA-1; red), and nuclei (Hoechst 33342; blue). Arrow-heads indicate overlapping or adjacent staining of gB and EEA-1, highlighting representative endocytic vesicles containing gB. Cells transfected with empty vector (Vector) were used as a negative control. The scale bars represent 20m. The image in the right bottom corner demonstrates that the epitopes of the gB and EEA-1 antibodies are in the lumen and outside the endosome, respectively.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.49.364.491.662.2]of pOka from 2 to 5 days postinfection (dpi), demonstrating that the RFP-tagged TK did not interfere with virus replication in melanoma cells (Fig. 5C). The decrease in titer of
pOka at 6 dpi was a result of a ⬎95% infection of the monolayer. A significant
(0.5-log10) decrease in viral titers was observed for pOka-gB[4R] at 3 dpi compared to
the value for pOka (Fig. 5A). The hyperfusogenic pOka-gB[4A] had a significant (0.5- to
1.4-log10) reduction in viral titers compared to the value for pOka from 1 to 6 dpi, while
the less hyperfusogenic pOka-gB[Y881F] had a significant (0.3- to 0.7-log10) decrease in
viral titers only from 1 to 5 dpi. Both double mutant viruses, pOka-gB[Y881F/4R] and
pOkay-gB[Y881F/4A], had significant (0.5- to 1-log10) reductions in viral titers compared
to the values for pOka from 1 to 5 dpi (Fig. 5B). A 0.5-log10decrease in viral titers was
also observed for pOka-gH[Δ834-841] at 3 dpi, which correlated with previous obser-vations (31). Thus, the positive charge of the gBcyt lysine cluster contributes to the cell-cell fusion regulation required for effective VZV propagation.
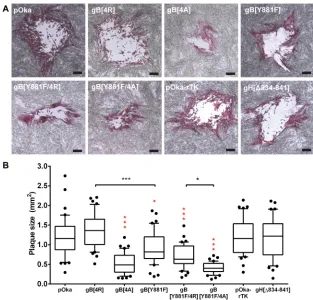
The reduced replication kinetics of mutant VZV with dysregulated cell-cell fusion has been attributed to the limited VZV spread caused by the exaggerated syncytium formation phenotype (31). To ascertain if the gBcyt mutations affected VZV spread in melanoma cells, the plaque sizes of the mutant viruses were compared to pOka at 4 dpi (Fig. 6A). The pOka-rTK, pOka-gB[4R], and pOka-gH[Δ834-841] viruses had plaques that were similar in size to those of pOka (Fig. 6B). In contrast, pOka-gB[4A], pOka-gB[Y881F], pOka-gB[Y881F/4R] and pOka-gB[Y881F/4A] had reduced plaque sizes compared to
FIG 4Alanine substitutions of the gBcyt lysine cluster induce exaggerated syncytium formation in infected melanoma cells. (A) Merged phase-contrast/fluorescence microscopy images of melanoma cells infected with either wild-type VZV (pOka), VZV with ORF36 tagged with RFP (pOka-rTK), gB mutant viruses on the pOka-rTK background (pOka-gB[4R], pOka-gB[4A], pOka-gB[Y881F], pOka-gB[Y881F/4R], and pOka-gB[Y881F/4A]), or gH mutant virus with aa 834 to 841 deleted (pOka-gH[Δ834-841]) at 36 h postinfection. The cells were stained for nuclei (blue), and representative syncytia are outlined in red. The scale bars represent 100m. (B) Box-and-whisker plot of the number of nuclei per syncytium on a single plane for 20 randomly selected syncytia for each virus. The whiskers indicate minimum and maximum values, with the 25th and 75th percentiles represented by the ends of the box. The median is indicated by the line within the box. Statistical differences between pOka and the mutants were evaluated by ANOVA (**,P⬍0.01;***,P⬍0.001).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.47.359.68.372.2]those of pOka, indicating impaired virus spread. Furthermore, the pOka-gB[4A] plaques were significantly smaller than those of pOka-gB[Y881F], which correlated with their differences in viral titers. While the double mutants had reduced replication kinetics similar to those of pOka, pOka-gB[Y881F/4R] had significantly larger plaques than
FIG 5Alanine substitutions of the gBcyt lysine cluster reduce VZV replication kinetics in melanoma cells. (A) Replication kinetics of wild-type pOka (black), pOka-gB[4R] (blue), pOka-gB[4A] (red), and pOka-gB[Y881F] (green) in melanoma cells over 6 days. (B) Replication kinetics of pOka (black), pOka-gB[Y881F/4R] (light blue), and pOka-gB[Y881F/4A] (light red) in melanoma cells over 6 days. (C) Replication kinetics of pOka (black), pOka-rTK (gray), and pOka-gH[Δ834-841] (orange) in melanoma cells over 6 days. Individual points represent means of harvested infected cells titrated in triplicate in melanoma cells, including the titrated-cell-associated inoculum at day zero. The titers were measured in log10PFU per milliliter. The bars indicate SEM.
Statistical differences between pOka and the mutant viruses were evaluated by two-way ANOVA (*,P⬍0.05;**,P⬍0.01;***, P⬍0.001).
FIG 6Alanine substitutions of the gBcyt lysine cluster reduce VZV spread in infected melanoma cells. Shown is a box-and-whisker plot of plaque sizes measured (n⫽40) in melanoma cells infected with wild-type VZV (pOka), rTK, gB[4R], gB[4A], gB[Y881F], gB[Y881F/4R], pOka-gB[Y881F/4A], or pOka-gH[Δ834-841] at 4 days postinfection. The area of the plaques was measured in square millimeters. The boxes represent the 10th to 90th percentile, with the median indicated by the line within the box and the outliers (⬍10th or⬎90th percentile) represented as individual black dots. Statistical differences were evaluated between pOka and the mutant viruses (red) or between the indicated mutant viruses (black) by ANOVA (*,P⬍0.05;***,P⬍0.001).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.44.442.71.198.2] [image:9.585.47.360.361.661.2]pOka-gB[Y881F/4A]. Thus, loss of the positive charge of the gBcyt lysine cluster limits VZV spread. In addition, the data further demonstrated that the increases in cell-cell fusion and dysregulated syncytium formation resulted in greater reductions in VZV propagation and spread.
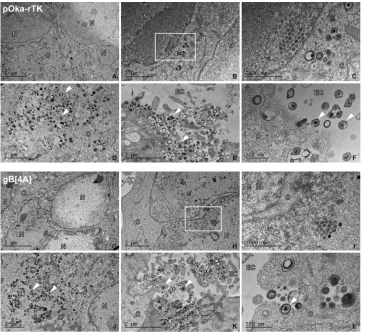
Viral particle formation and egress are not impaired by the gBcyt lysine cluster alanine substitutions.To elucidate if viral particle formation and egress were affected by the alanine substitutions in the gBcyt lysine cluster, electron micrographs of melanoma cells infected with either pOka-rTK, the control virus, or pOka-gB[4A] were captured at 48 h postinfection (hpi). Nucleocapsids in the nucleus and virus particles in the cytoplasm of similar size and morphology were observed in infected cells (Fig. 7A and G). Crystalline arrays of nucleocapsids were observed in the nuclei of pOka-gB[4A]-infected cells that were similar to those seen in pOka-rTK-pOka-gB[4A]-infected cells, indicating that the alanine substitution did not impair capsid formation (Fig. 7H and I and 7B and C). Enveloped viral particles and nucleocapsids were present in the cytoplasm of both pOka-rTK- and pOka-gB[4A]-infected cells, indicating that nuclear egress was not inhibited by the 4A mutation (Fig. 7D and J). At the surface of pOka-gB[4A]-infected cells, enveloped viral particles attached to the membrane were observed that were indistinguishable from those on the surface of pOka-rTK (Fig. 7K and L and 7E and F). Thus, the alanine substitutions in the gBcyt lysine cluster do not prevent viral particle formation and egress at 48 h postinfection.
FIG 7Alanine substitutions of the gBcyt lysine cluster do not inhibit VZV particle formation or egress in infected melanoma cells. Shown are electron micrographs of melanoma cells infected with pOka-rTK (A to F) or pOka-gB[4A] (G to L) at 48 h postinfection. The panels are representative images of nuclei (A and F); crystalline arrays of nucleocapids (B and H), with magnified images of the boxed area (C and I); vesicles containing viral particles (D and J); and viral particles on the cell surface (E, F, K, and L). C, N, and EC, cytoplasmic, nuclear, and extracellular space, respectively, with representative viral particles indicated with arrowheads. Scale bars are noted in the bottom left corner of each micrograph.
on November 7, 2019 by guest
http://jvi.asm.org/
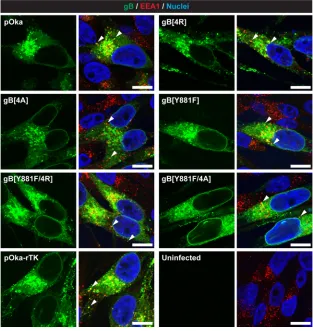
[image:10.585.41.406.71.404.2]Trafficking of gB to endocytic vesicles and gB processing during infection are not inhibited by the gBcyt lysine cluster substitutions.The role of the gBcyt lysine cluster in the trafficking of gB to endocytic vesicles during infection was evaluated by confocal micrographs taken of melanoma cells infected with the gBcyt mutant viruses at 24 h postinfection (Fig. 8). In pOka-infected cells, gB had nuclear rim localization, which was consistent with previous observations (27). Colocalization of gB and EEA-1 in the pOka-infected cells was also observed, which correlated with the data from melanoma cells transiently expressing gB (Fig. 3 and 8). All of the gBcyt mutant viruses and pOka-rTK had gB localization and colocalization with EEA-1 comparable to those of pOka. Thus, the gBcyt mutations did not impair trafficking of gB to endocytic vesicles. Proteolytic cleavage at the furin recognition motif in the ectodomain of gB
(491RSRR494) has been shown to have a functional role in VZV skin pathogenesis despite
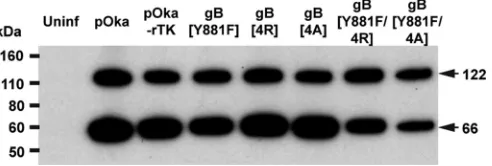
being dispensable for infection in melanoma cells (32). Cleavage of the immature full-length protein (124 kDa) produces an N-terminal cleavage product and C-terminal cleavage product of 66 kDa. To determine if the gBcyt mutations affected processing of gB during infection, gB was immunoprecipitated and probed using an antibody that recognized an epitope in the gBcyt. For pOka, pOka-rTK, and all of the gBcyt mutants, both the 124-kDa immature full-length protein and the 66-kDa cleaved product were detected by Western blotting (Fig. 9). Thus, the processing of gB is not dependent on either the ITIM or the lysine cluster of the gBcyt.
FIG 8Substitutions to the gBcyt lysine cluster do not inhibit localization of gB with early endosomes or the nuclear rim during infection. Shown are confocal microscopy images of melanoma cells infected with pOka, pOka-gB[4R], pOka-gB[4A], pOka-gB[Y881F], pOka-gB[Y881F/4R], or pOka-gB[Y881/4A], pOka-rTK, and pOka-gH[Δ834-841] at 24 h postinfection. Cells were stained for gB (green), early endosome antigen (EEA-1; red), and nuclei (Hoechst 33342; blue). For each virus, the left image is of gB alone and the right image is a merged image of gB with EEA-1 and nuclei. Arrowheads indicate colocalization of gB and EEA1, highlighting representative endocytic vesicles containing gB. Uninfected cells were used as a negative control. The scale bars represent 20m.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.49.362.73.404.2]DISCUSSION
Past efforts to elucidate the role of gB in herpesvirus fusion have focused primarily on its ectodomain (33–35). However, there is growing evidence that the gB cytoplasmic domain is critical for membrane fusion (22, 28, 36). The conserved gBcyt lysine cluster
was first recognized to have a role in membrane fusion in anin vitrocell-free cell-cell
fusion assay study involving HSV-1 gBcyt (28). The current study is the first to examine the function of a conserved alphaherpesvirus gBcyt lysine cluster in the context of infection. The VZV gBcyt lysine cluster was identified to be critical for the regulation of
in vitro cell-cell fusion, with arginine substitutions causing a moderate hyperfusion phenotype, as less gB surface expression was required for gB[WT]-like fusion. Further-more, the alanine substitutions induced an extreme hyperfusion phenotype and re-duced gB surface expression. The hyperfusion phenotype caused exaggerated syncy-tium formation and reduced virus propagation and spread in melanoma cells infected with the mutant viruses containing either substitutions. Correlating with the degree of hyperfusogenicity observed in the cell-cell fusion assay, larger syncytia and greater reduction in virus propagation and spread were observed with the alanine substitutions than with arginine substitutions. Thus, effective propagation and spread of VZV are dependent upon the strict regulation of cell-cell fusion by the gBcyt lysine cluster and ITIM.
The limiting effect of exaggerated cell-cell fusion on VZV propagation has also been observed in previous studies characterizing the role of the VZV gH cytoplasmic domains (gHcyt) in fusion regulation. Truncation of the terminal 8 aa of the 18-aa cytoplasmic domain of gH (gH[Δ834-841]) induced a hyperfusion phenotype that was also observed
in anin vitrocell-cell fusion assay (31). The gHcyt fusion regulation was identified to be
dependent upon the length rather than the sequence of the domain. The length of the HSV-1 gHcyt has also been shown to contribute to gB/gH-gL/gD cell-cell fusion (28). Similar to VZV gB[Y881F], the gH[Δ834-841] virus also caused exaggerated syncytium formation that translated into reduced replication kinetics of VZV in cultured melanoma cells and limited skin pathogenesis (31).
VZV syncytia have a disrupted intercellular environment, with rearrangements of
early endosomes and the trans-Golgi network (TGN) to the center of the syncytia
surrounded by a spheroid of nuclei (22). This suggests that VZV propagation is limited by the syncytium formation during the later stage of canonical VZV infection. Rear-rangement of early endosomes could limit trafficking of viral glycoproteins to the TGN, resulting in either an impairment of secondary envelopment or generation of nonin-fectious viral particles. Organelle rearrangements could also prevent trafficking of viral proteins required for capsid formation. This is supported by the exaggerated syncytium formation in the absence of viral particle formation observed in cells transfected with a mutant VZV BAC containing both the hyperfusogenic gB[Y881F] and gH[Δ834-841] mutations (31). Actin and microtubule cytoskeleton reorganization have also been observed in syncytia formed by cells transiently expressing influenza virus hemagglu-tinin (37). While the cytoskeleton has not been extensively studied in VZV syncytia, actin and microtubule rearrangements could interfere with egress of viral particles.
FIG 9Substitutions to the gBcyt lysine cluster do not impair proteolytic cleavage of gB during infection. Shown are results of immunoprecipitation and Western blotting of gB from melanoma cells either mock infected (uninfected) or infected with pOka, pOka-gB[4R], pOka-gB[4A], pOka-gB[Y881F], pOka-gB[Y881F/ 4R], or pOka-gB[Y881/4A], pOka-rTK, and pOka-gH[Δ834-841]. Arrows indicate the uncleaved (122-kDa) and the N-terminally cleaved (66-kDa) forms of gB. The molecular mass standard is indicated on the left.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:12.585.85.331.73.156.2]The exaggerated syncytium formation and reduced virus propagation caused by disrupting the VZV gBcyt lysine cluster are consistent with the observations of the gB-36 virus, which contained the ITIM but lacked the K897, K898, and K900 residues (26). The positive charge of the gBcyt lysine cluster was demonstrated to have a role in cell-cell fusion regulation by the hyperfusion phenotype of gB[4A]. A similar increase in fusion has been observed in an HSV-1 gB/gH-gL/gD cell-cell fusion assay when the corresponding lysines in the conserved lysine cluster of the HSV-1 gBcyt were replaced with the polar but not positively charged serines (K864S/K865S/K866S) (28). In contrast to the reduced surface expression of gB[4A] compared to gB[WT], the HSV-1 gBcyt lysine mutant had wild-type-like surface expression in cells transiently expressing HSV-1 gB, indicating that the conserved lysine cluster may have multiple roles in gB function or trafficking depending on the virus. However, the HSV-1 gBcyt lysine substitutions were not examined in the context of infection. Thus, the hyperfusion phenotype observed for both the HSV and VZV gBcyt lysine mutants was a result of an increase of gB fusogenicity and not overexpression of gB.
The-amino group of the lysine also had a role in VZV gB/gH-gL-mediated cell-cell
fusion and infection, as the arginine substitution of the lysine cluster increased syncy-tium size and reduced virus propagation in comparison to those of pOka. Similar to the single mutants, differences in phenotype were also observed with the gB[Y881F/4R] and gB[Y881F/4A] double mutants (Table 2). The gB[Y881F/4R] mutant was more
hyperfusogenic than gB[Y881F/4A], while it had similar surface expression in vitro.
While the two double mutant viruses had similarly reduced replication kinetics, differ-ences were observed in syncytium morphology and plaque size. The reduced surface expression of transiently expressed gB[Y881F/4A] compared to both the gB[WT] and single mutants coupled with the small syncytia observed in melanoma cells infected with the double mutant virus indicated that disrupting the ITIM and the positive charge of the lysine cluster further impaired gB function. Thus, the lysine cluster positive charge is not the only factor contributing to the regulation of cell-cell fusion by the gBcyt. It is uncertain if this is a result of an absence of a posttranslation modification or the introduction of the guanidino group of the arginine at one of the lysine positions. It is also possible that fusion regulation may be dependent upon a specific lysine residue or a combination of lysine residues in the cluster.
The mechanism by which the conserved lysine cluster contributes to VZV gB/gH-gL-mediated cell-cell fusion regulation is still not clear, although it has been suggested to have a role in maintaining the structure of the HSV-1 gBcyt (28). Currently, structural details of the gBcyt are limited, as all crystal structure data for herpesvirus gB lack both the transmembrane and cytoplasmic domains (23, 38, 39). The HSV-1 gBcyt has been shown to form trimers in the absence of the transmembrane and ectodomain, sug-gesting that the VZV gBcyt might also be trimeric (40). Both the HSV-1 and VZV gBcyt are predicted to have at minimum three alpha-helices, with the VZV gBcyt alpha-helices at positions 824 to 828, 869 to 897, and 903 to 914 (22, 40). The predicted alpha-helix at positions 869 to 897 in the VZV gBcyt, which contains the lysine cluster, has also been predicted in the HSV-1 gBcyt and supported by circular-dichroism spectrum analysis (40). An electrostatic interaction of the positive charged K864/K865/K866 residues of the second alpha-helix of the HSV-1 gBcyt and the negatively charged plasma membrane has been postulated (28). While this interaction of the lysine residues in the HSV-1 gBcyt has not been evaluated, purified truncated HSV-1 gBcyt lacking the second and third alpha-helices reduces binding of the gBcyt to anionic liposomes (40). The interaction of the lysine cluster and the cell membrane is also supported by data showing that residues of the second alpha-helix, including the lysines, are protected from proteolytic cleavage when soluble HSV-1 gBcyt in the presence of liposomes was treated with different proteases (41). How the association of the alpha-helix and the plasma membrane contributes to herpesvirus fusion regulation is still not clear. However, it has been proposed that the interaction of the alpha-helices in the HIV fusion protein, gp41, with the membrane serves to stabilize the trimeric fusion protein in its prefusion form (42, 43). Disrupting the positive charge of the gBcyt
on November 7, 2019 by guest
http://jvi.asm.org/
lysine cluster might weaken the association of the second alpha-helix with the mem-brane, leading to decreased stability of gB in its prefusion form and increased propen-sity of gB to induce fusion. While the VZV gB Y881 residue has been demonstrated to be phosphorylated, specific kinases or phosphatases have not yet been identified (22). The lysine cluster might also act to modulate the accessibility or binding of the VZV gBcyt ITIM to kinases or phosphatases through either its positive charge or posttrans-lational modifications, such as acetylation, which has been shown to have an important role in controlling phosphorylation of protein Akt (44).
In summary, strict regulation of VZV gB/gH-gL-mediated cell-cell fusion by the gBcyt and gHcyt is required for effective virus propagation. During VZV infection, cell-cell fusion regulation by the gHcyt and gBcyt is postulated to be maintained during the early stages of infection but lost as the infection progresses resulting in the canonical syncytium formation observed in infected skin and neuronal tissue. The identification of the lysine cluster role in cell-cell fusion regulation refines the knowledge of the molecular mechanisms that govern VZV syncytium formation during infection.
MATERIALS AND METHODS
Cells and viruses.CHO-DSP1 cells were propagated using F-12K nutrient mixture with Kaighn’s modification (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and penicillin (100 U/ml; Invitrogen) and maintained under puromycin selection (8g/ml; Invitrogen) (15). MeWo (human melanoma cells) (HTB-65; ATCC) and Mel-DSP2 cells were propagated in minimal essential medium (Invitrogen) supplemented with 10% FBS, nonessential amino acids (100M; Omega Scientific), and antibiotics (penicillin, 100 U/ml; streptomycin, 100g/ml; Invitrogen), with the latter cell line maintained under puromycin selection (5g/ml) (15). The parental Oka strain of VZV (pOka) was derived from a self-excisable BAC (pOka-DX) and used throughout the study (45). The recombinant virus pOka-TK-RFP (pOka-rTK) was generated in a previous study (46).
Vectors. The vectors gB[4R], gB[4A], gB[Y881F/4R], and pCAGGs-gB[Y881F/4A] were generated by site-directed mutagenesis using PCR. To generate pCAGGS-gB[4R] and pCAGGS-gB[4A], a PCR fragment was amplified using primer pair pCAGGS-gB-Age1 and [P]pCAGGS-gB892E and a second fragment was amplified using the primer pair of either [P]pCAGGS-gB-4R or [P]pCAGGS-gB-4A and pCAGGS-gB3Asc1, using pCAGGs-gB (a gift from Tadahiro Suenaga and Hisashi Arase, Osaka University, Osaka, Japan) (13) as the template (AccuPrime Taq; Invitrogen) (Table 3). The PCR fragments were separated on a 2% agarose gel, purified by gel extraction (Qiagen), digested with either Age1 or Asc1 restriction enzyme (New England BioLabs), and then purified using a PCR purification kit (Qiagen). The pCAGGS-gB vector was digested with Age1 and Asc1, dephosphorylated with antarctic phosphatase (Invitrogen), separated on a 0.8% agarose gel, and purified by gel extraction. The purified products were ligated together (T4 ligase; Invitrogen) and transformed into TOP10 electrocompetent cells (Invitrogen). The transformed cells were plated on LB agar plates supplemented with ampicillin (100
g/ml; Sigma) antibiotics. The clones were confirmed by Age1 and Asc1 digestion and sequencing. The vectors pCAGGs-gB[Y881F/4R] and pCAGGs-gB[Y881F/4A] were constructed using the pCAGGs-gB-Y881F vector generated by Oliver et al. for the template (22).
[image:14.585.42.373.82.185.2]The gB-Kan vectors, gB-Kan-4R, gB-Kan-4A, gB-Kan-Y881F/4R, and gB-Kan-Y881F/4A, were generated to introduce mutations of the gBcyt into the pOka BAC. To generate the vectors, a PCR fragment was amplified using primer pair pCAGGS-gB-Age1 and [P]pCAGGS-gB892E and a second fragment was amplified using the primer pair of either [P]pCAGGS-gB-4R or [P]pCAGGS-gB-4A and M13R using the gB-Kan or pCAGGS-gB-Y881F vectors generated by Oliver et al. for the template (22). The PCR fragments were separated on a 2% agarose gel, purified by gel extraction, digested with either Age1 or Spe1 restriction enzyme (New England BioLabs), and purified using a PCR purification kit. The gB-Kan vector was digested with Age1 and Spe11, dephosphorylated with antarctic phosphatase, separated on a 0.8% agarose gel, and purified by gel extraction. The purified products were ligated together and transformed into TOP10 cells. The transformed cells were plated on LB agar plates supplemented with ampicillin antibiotics. The clones were confirmed by restriction enzyme digestion and sequencing.
TABLE 3Primers used for gB mutagenesis
Primer name Purpose Sequence
pCAGGs-gB-Age1 Cloning CTTTTTTGCGTACCGGTACGTGC [P]pCAGGs-gB892E Cloning TTCTTGGCGCTCAGCCGCAGATA
[P]pCAGGs-gB-4R Cloning TCTCGAGCCCGCCGACGAAATCGGACTAGCGCCCTTTTAACTTC [P]pCAGGs-gB-4A Cloning TCTGCAGCCCGCGCAGCAAATGCGACTAGCGCCCTTTTAACTTC pCAGGs-gB3Asc1 Cloning AATGGCGCGCCCTGTGATGCGTAATGGAGACAC
M13R Cloning CAGGAAACAGCTATGAC gBseq009 Sequencing GCCACGGGAGCGCTGCTTTCC [31]F56625-56645 Cloning AGGTATAGGCAGTTCCCACGG [31]R59697-59717 Cloning TTTCATTGAGACTTGAAGCGC
on November 7, 2019 by guest
http://jvi.asm.org/
Stable reporter fusion assay of VZV gB/gH-gL-mediated fusion.Quantification of fusion events was performed as previously described (15, 47). Briefly, CHO-DSP1 cells were transfected with equimolar amounts of pcDNA3.1-gL, pME18s-gH-TL, and pCAGGs-gB plasmids (gifts from Tadahiro Suenaga and Hisashi Arase, Osaka University, Osaka, Japan) (13) or pCAGGs vector expressing mutant gB using Lipofectamine 2000 (Invitrogen). Transfected cells were harvested at 6 h posttransfection and cocultured with Mel-DSP2 cells. The cocultures were harvested after an additional 36 h, and the frequency of green fluorescent protein (GFP)-positive cells, indicating fusion events, was quantified using a FACSCalibur controlled by CellQuest Pro (BD Bioscience) and analyzed with FlowJo (TreeStar). A negative control was performed with pME18s- and pcDNA-empty vectors to establish background levels of GFP expression. Experiments were performed at minimum in duplicate.
Glycoprotein B surface expression.Surface expression of gB was analyzed as previously described (22). Briefly, CHO-DSP1 cells were transfected with either pCAGGs-gB or pCAGGS-gB mutant constructs. At 24 h posttransfection, cells were dislodged with enzyme-free cell dissociation buffer (Thermo Fisher Scientific), fixed, stained with anti-gB monoclonal antibody (Mab) SG2-2E6 (mouse; Meridian Life Sci-ences), and detected with anti-mouse Alexa Fluor 488 antibody (Thermo Fisher Scientific). Analysis was performed on a FACSCalibur controlled by CellQuest Pro (BD Bioscience), and gB surface expression was quantified with FlowJo (TreeStar). Experiments were performed at minimum in triplicate.
Mutagenesis of ORF31 (gB) in pOka-DX bacterial artificial chromosome (BAC) and generation of recombinant viruses. The mutant gB[4R], gB[4A], gB[Y881F/4R], and pOka-gB[Y881F/4A] BACs were generated as previously described (22). Briefly, the gB-Kan-4R, gB-Kan-4A, gB-Kan-Y881F/4R, and gB-Kan-Y881F/4A vectors were digested with Nae1 and BstZ171 (New England BioLabs), the fragments were separated on a 1% agarose gel, and the fragment of interest was gel purified. The fragment was inserted into the previously generated pOka-TK-RFP-ΔORF31 BAC, which lacks ORF31, by lambda Red recombination (46). Fragment insertion was confirmed by PCR using primers [31]F56625-56645 and [31]R59697-59717. To confirm the absence of spurious recombination, the BACs were digested with HindIII restriction enzyme (New England BioLabs), separated by gel electrophoresis, and compared to the pOka-DX BAC. The mutations in the BAC were confirmed by sequencing. Recombinant virus was generated by transfecting melanoma cells using Lipofectamine 2000.
Syncytium morphology assay.Melanoma cells in a six-well plate were infected with 250 PFU per well of pOka or gB mutant viruses. At 36 h postinfection, cells were fixed with 4% paraformaldehyde (PFA) and stained with Hoechst 33342 (Invitrogen) for visualization of nuclei. Phase-contrast and fluorescence microscopy images of 20 randomly selected syncytia were captured using an AX10 microscope (Zeiss) equipped with an X-Cite series 120 fluorescence excitation light source (Lumen Dynamics). The images were aligned and processed in Photoshop. The nuclei per syncytium were enumerated on a single plane to determine the size of the syncytium.
Replication kinetics and plaque size assay.Replication kinetics and virus spread of gB mutant viruses in melanoma cells were evaluated as previously described (22). Briefly, melanoma cells were infected with 1,000 PFU of either pOka or gB mutant viruses and harvested at 24-h intervals to examine replication kinetics. Viral titrations were performed by 10-fold dilutions on melanoma cells in triplicate. To measure virus spread, a plaque size assay was performed by capturing images of 40 randomly selected, PFA-fixed and stained plaques at 4 days postinfection using an AX10 microscope. The area of the plaques was calculated using ImageJ, as described previously (22).
TEM of VZV-infected melanoma cells.Samples for transmission electron microscopy (TEM) were prepared as previously described (31). Briefly, 12-mm glass coverslips seeded with 3⫻105melanoma
cells were infected with 1,000 PFU of pOka-rTK or pOka-gB[4A] virus. At 48 h postinfection, samples were fixed, dehydrated with ethanol and acetonitrile, and infiltrated with Epon. After Epon polymerization, the glass coverslip was removed from the block by three cycles of freezing in liquid nitrogen and thawing in boiling water. Sections were prepared and placed on grids that were then visualized using a JEOL 1400 transmission electron microscope at 80 kV at the Stanford Cell Sciences Imaging Facility. Electron micrographs were captured using a Gatan Multiscan 701 digital camera.
Confocal microscopy.Melanoma cells transfected with either pCAGGs-gB expression constructs using Lipofectamine 2000 or infected with 500 PFU of pOka, gB, or gH mutant viruses were fixed with 4% formaldehyde and permeabilized with 0.1% Triton-X after 24 h. Cells were stained with anti-gB MAb SG2-2E6 (mouse; Meridian Life Sciences) and EEA-1 (rabbit; Novus Biologicals). The primary anti-bodies were detected with anti-mouse Alexa Fluor 488 or anti-rabbit Alexa Fluor 555 secondary antibodies (Invitrogen). The nuclei were stained with Hoechst 33342 (Invitrogen). Images were captured with an inverted Zeiss LSM 780 multiphoton laser scanning confocal microscope at the Stanford Cell Sciences Imaging Facility. Image processing was performed using Zen 2.1 (black) software (Zeiss).
Immunoprecipitation and Western blotting.Immunoprecipitation of gB from infected lysate was performed as previously described (22). Briefly, lysate from melanoma cells mock infected or infected with pOka or gB mutant viruses were harvested at 72 h postinfection. Immunoprecipitation and Western blotting of gB was performed using anti-gB MAb SG2-2E6 and a rabbit antibody against the gBcyt epitope833PEGMDPFAEKPNAT846(32), respectively. The primary antibody was detected using secondary
horseradish peroxidase-conjugated antibodies to anti-rabbit and the ECL Plus detection kit (GE Health-care Bio-Sciences).
Sequence alignment.The amino acid sequence of alphaherpesvirus gB homologues were aligned using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/).
Statistical analysis.All quantitative results were analyzed by either one-way analysis of variance (ANOVA) or two-way ANOVA to determine the statistical significance using Prism (GraphPad Software).
on November 7, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
We acknowledge John Mulholland, Kitty Lee, Cedric Espenel, and John Perrino of the Stanford Cell Sciences Imaging Facility (NIH grant SIG 1S10RR02678001) for their technical assistance. We also acknowledge the following colleagues for providing reagents: Tadahiro Suenaga (Osaka University), Hisashi Arase (Osaka University), and Yasuko Mori (National Institute of Biomedical Innovation, Osaka and Kobe University Graduate School of Medicine).
Funding was provided by NIH grant AI102546.
REFERENCES
1. Arvin AM, Gilden D. 2013. Varicella-zoster virus, p 2015–2057.InHowley DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 2. Lippincott Williams & Wilkins, Philadelphia, PA.
2. Zerboni L, Sen N, Oliver SL, Arvin AM. 2014. Molecular mechanisms of varicella zoster virus pathogenesis. Nat Rev Microbiol 12:197–210. https://doi.org/10.1038/nrmicro3215.
3. Wooten KG, Kolasa M, Singleton MS, Shefer A. 2010. National, state, and local area vaccination coverage among children age 19 –35 months— United States, 2009. MMWR Morb Mortal Wkly Rep 59:1171–1177. 4. Tseng HF, Harpaz R, Luo Y, Hales CM, Sy LS, Tartof SY, Bialek S, Hechter
RC, Jacobsen SJ. 2016. Declining effectiveness of herpes zoster vaccine in adults agedⱖ60 years. J Infect Dis 213:1872–1875. https://doi.org/ 10.1093/infdis/jiw047.
5. Kost RG, Straus SE. 1996. Postherpetic neuralgia—pathogenesis, treat-ment, and prevention. N Engl J Med 335:32– 42. https://doi.org/10.1056/ NEJM199607043350107.
6. Kennedy PG. 2016. Issues in the treatment of neurological conditions caused by reactivation of varicella zoster virus (VZV). Neurotherapeutics 13:509 –513. https://doi.org/10.1007/s13311-016-0430-x.
7. Cheatham WJ, Dolan TF, Jr, Dower JC, Weller TH. 1956. Varicella: report of two fatal cases with necropsy, virus isolation, and serologic studies. Am J Pathol 32:1015–1035.
8. Esiri MM, Tomlinson AH. 1972. Herpes zoster. demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy. J Neurol Sci 15:35– 48.
9. Reichelt M, Zerboni L, Arvin AM. 2008. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia. J Virol 82: 3971–3983. https://doi.org/10.1128/JVI.02592-07.
10. Lungu O, Annunziato PW, Gershon A, Staugaitis SM, Josefson D, LaRussa P, Silverstein SJ. 1995. Reactivated and latent varicella-zoster virus in human dorsal root ganglia. Proc Natl Acad Sci U S A 92:10980 –10984. https://doi.org/10.1073/pnas.92.24.10980.
11. Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800 – 832. https://doi.org/10.3390/v4050800. 12. Maresova L, Pasieka TJ, Homan E, Gerday E, Grose C. 2005. Incorporation
of three endocytosed varicella-zoster virus glycoproteins, gE, gH, and gB, into the virion envelope. J Virol 79:997–1007. https://doi.org/10.1128/ JVI.79.2.997-1007.2005.
13. Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107: 866 – 871. https://doi.org/10.1073/pnas.0913351107.
14. Vleck SE, Oliver SL, Brady JJ, Blau HM, Rajamani J, Sommer MH, Arvin AM. 2011. Structure-function analysis of varicella-zoster virus glyco-protein H identifies domain-specific roles for fusion and skin tropism. Proc Natl Acad Sci U S A 108:18412–18417. https://doi.org/10.1073/ pnas.1111333108.
15. Yang E, Arvin AM, Oliver SL. 2016. Role for the alphaV integrin subunit in varicella-zoster virus-mediated fusion and infection. J Virol 90: 7567–7578. https://doi.org/10.1128/JVI.00792-16.
16. Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284: 17370 –17382. https://doi.org/10.1074/jbc.M109.005728.
17. Wang X, Kenyon WJ, Li Q, Mullberg J, Hutt-Fletcher LM. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J Virol 72:5552–5558.
18. Zhang Z, Selariu A, Warden C, Huang G, Huang Y, Zaccheus O, Cheng T, Xia N, Zhu H. 2010. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin-tropic factor. PLoS Pathog 6:e1000971. https://doi.org/ 10.1371/journal.ppat.1000971.
19. Wang Z, Gershon MD, Lungu O, Panagiotidis CA, Zhu Z, Hao Y, Gershon AA. 1998. Intracellular transport of varicella-zoster glycoproteins. J Infect Dis 178(Suppl 1):S7–S12.
20. Heineman TC, Connolly P, Hall SL, Assefa D. 2004. Conserved cytoplas-mic domain sequences mediate the ER export of VZV, HSV-1, and HCMV gB. Virology 328:131–141. https://doi.org/10.1016/j.virol.2004.07.011. 21. Maresova L, Pasieka T, Wagenaar T, Jackson W, Grose C. 2003.
Identifi-cation of the authentic varicella-zoster virus gB (gene 31) initiating methionine overlapping the 3=end of gene 30. J Med Virol 70(Suppl 1):S64 –S70. https://doi.org/10.1002/jmv.10324.
22. Oliver SL, Brady JJ, Sommer MH, Reichelt M, Sung P, Blau HM, Arvin AM. 2013. An immunoreceptor tyrosine-based inhibition motif in varicella-zoster virus glycoprotein B regulates cell fusion and skin pathogenesis. Proc Natl Acad Sci U S A 110:1911–1916. https://doi.org/10.1073/ pnas.1216985110.
23. Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. https://doi.org/10.1126/science.1126548. 24. Suenaga T, Matsumoto M, Arisawa F, Kohyama M, Hirayasu K, Mori Y,
Arase H. 2015. Sialic acids on varicella-zoster virus glycoprotein B are required for cell-cell fusion. J Biol Chem 290:19833–19843. https:// doi.org/10.1074/jbc.M114.635508.
25. Daëron M, Jaeger S, Du Pasquier L, Vivier E. 2008. Immunoreceptor tyrosine-based inhibition motifs: a quest in the past and future. Immunol Rev 224:11– 43. https://doi.org/10.1111/j.1600-065X.2008.00666.x. 26. Heineman TC, Hall SL. 2002. Role of the varicella-zoster virus gB
cyto-plasmic domain in gB transport and viral egress. J Virol 76:591–599. https://doi.org/10.1128/JVI.76.2.591-599.2002.
27. Heineman TC, Krudwig N, Hall SL. 2000. Cytoplasmic domain signal sequences that mediate transport of varicella-zoster virus gB from the endoplasmic reticulum to the Golgi. J Virol 74:9421–9430. https:// doi.org/10.1128/JVI.74.20.9421-9430.2000.
28. Rogalin HB, Heldwein EE. 2015. Interplay between the herpes simplex virus 1 gB cytodomain and the gH cytotail during cell-cell fusion. J Virol 89:12262–12272. https://doi.org/10.1128/JVI.02391-15.
29. Sokalingam S, Raghunathan G, Soundrarajan N, Lee SG. 2012. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS One 7:e40410. https://doi.org/10.1371/journal.pone.0040410.
30. Zee BM, Garcia BA. 2012. Discovery of lysine post-translational modifi-cations through mass spectrometric detection. Essays Biochem 52: 147–163. https://doi.org/10.1042/bse0520147.
31. Yang E, Arvin AM, Oliver SL. 2014. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytia formation and skin patho-genesis. PLoS Pathog 10:e1004173. https://doi.org/10.1371/journal .ppat.1004173.
32. Oliver SL, Sommer M, Zerboni L, Rajamani J, Grose C, Arvin AM. 2009. Mutagenesis of varicella-zoster virus glycoprotein B: putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J Virol 83:7495–7506. https://doi.org/10.1128/JVI.00400-09.
33. Vitu E, Sharma S, Stampfer SD, Heldwein EE. 2013. Extensive mutagen-esis of the HSV-1 gB ectodomain reveals remarkable stability of its postfusion form. J Mol Biol 425:2056 –2071. https://doi.org/10.1016/ j.jmb.2013.03.001.
on November 7, 2019 by guest
http://jvi.asm.org/
34. Connolly SA, Longnecker R. 2012. Residues within the C-terminal arm of the herpes simplex virus 1 glycoprotein B ectodomain contribute to its refolding during the fusion step of virus entry. J Virol 86:6386 – 6393. https://doi.org/10.1128/JVI.00104-12.
35. Reimer JJ, Backovic M, Deshpande CG, Jardetzky T, Longnecker R. 2009. Analysis of Epstein-Barr virus glycoprotein B functional domains via linker insertion mutagenesis. J Virol 83:734 –747. https://doi.org/ 10.1128/JVI.01817-08.
36. Chen J, Zhang X, Jardetzky TS, Longnecker R. 2014. The Epstein-Barr virus (EBV) glycoprotein B cytoplasmic C-terminal tail domain regulates the energy requirement for EBV-induced membrane fusion. J Virol 88: 11686 –11695. https://doi.org/10.1128/JVI.01349-14.
37. Richard JP, Leikina E, Chernomordik LV. 2009. Cytoskeleton reorganiza-tion in influenza hemagglutinin-initiated syncytium formareorganiza-tion. Biochim Biophys Acta 1788:450 – 457. https://doi.org/10.1016/j.bbamem.2008 .09.014.
38. Burke HG, Heldwein EE. 2015. Crystal structure of the human cytomeg-alovirus glycoprotein B. PLoS Pathog 11:e1005227. https://doi.org/ 10.1371/journal.ppat.1005227.
39. Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880 –2885. https://doi.org/10.1073/pnas.0810530106.
40. Chowdary TK, Heldwein EE. 2010. Syncytial phenotype of C-terminally truncated herpes simplex virus type 1 gB is associated with diminished membrane interactions. J Virol 84:4923– 4935. https://doi.org/10.1128/ JVI.00206-10.
41. Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane re-quirement for folding of the herpes simplex virus 1 gB cytodomain
suggests a unique mechanism of fusion regulation. J Virol 86: 8171– 8184. https://doi.org/10.1128/JVI.00932-12.
42. Chen SS, Lee SF, Wang CT. 2001. Cellular membrane-binding ability of the C-terminal cytoplasmic domain of human immunodeficiency virus type 1 envelope transmembrane protein gp41. J Virol 75:9925–9938. https://doi.org/10.1128/JVI.75.20.9925-9938.2001.
43. Postler TS, Desrosiers RC. 2013. The tale of the long tail: the cyto-plasmic domain of HIV-1 gp41. J Virol 87:2–15. https://doi.org/ 10.1128/JVI.02053-12.
44. Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. 2011. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal 4:ra46.
45. Tischer BK, Kaufer BB, Sommer M, Wussow F, Arvin AM, Osterrieder N. 2007. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J Virol 81:13200 –13208. https://doi.org/10.1128/ JVI.01148-07.
46. Oliver SL, Yang E, Arvin AM. 2016. Dysregulated glycoprotein B-mediated cell-cell fusion disrupts varicella-zoster virus and host gene transcription during infection. J Virol. 91:e01613-16. https://doi.org/ 10.1128/JVI.01613-16.
47. Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng Des Sel 25:813– 820. https://doi.org/10.1093/protein/gzs051.