Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Involvement of Cellular Double-Stranded DNA Break Binding
Proteins in Processing of the Recombinant Adeno-Associated
Virus Genome
LORENA ZENTILIN,1ALESSANDRO MARCELLO,1ANDMAURO GIACCA1,2*
Molecular Medicine Laboratory, International Centre for Genetic Engineering and Biotechnology, 34012 Trieste,1
and Laboratorio di Biologia Molecolare, Scuola Normale Superiore, 56126 Pisa,2Italy
Received 2 July 2001/Accepted 29 August 2001
Unlike postmitotic tissues in vivo, transduction of cultured cells is poor with recombinant adeno-associated virus (rAAV). The ability of rAAV to transduce cells is greatly enhanced by a variety of agents that induce DNA damage and is elevated in cells defective in the ataxia telangiectasia gene product (ATM), showing increased genomic instability. Here we show that DNA double-stranded break (DSB) repair pathways are involved in the regulation of rAAV transduction efficiency. By quantitative chromatin immunoprecipitation, we found that Ku86 and Rad52 proteins associate with viral DNA inside transduced cells. Both proteins are known to competitively recognize hairpin structures and DNA termini and to promote repair of DSBs, the former by facilitating nonhomologous end joining and the latter by initiating homologous recombination. We found that rAAV transduction is increased in Ku86-defective cells while it is inhibited in Rad52 knockout cells. These results suggest that binding of Rad52 to the rAAV genome might be involved in processing of the vector genome through a homologous recombination pathway.
Adeno-associated virus type 2 (AAV-2) is a defective human parvovirus presenting a biphasic life cycle, with productive replication occurring only in the presence of a coinfecting helper virus (reviewed in reference 8). In the absence of helper functions, the virus persists in infected cells as a latent viral genome (33). The characteristics of the AAV life cycle and its ability to infect several nondividing cell types, including mus-cle, liver, and brain cells, in the absence of host inflammatory or immune responses render this virus an excellent tool for gene transfer for human gene therapy (21, 32, 36, 57–59).
Despite the increasing popularity of recombinant AAV (rAAV) vectors and the successful applications of AAV-me-diated gene transfer in preclinical and clinical studies (3, 14, 31), the molecular mechanisms responsible for efficient rAAV transduction are still poorly defined. Recombinant AAVs are able to efficiently bind and enter a large number of cells by using widely expressed molecules as receptors (41, 51, 52). After internalization, however, functional rAAV transduction appears to be limited at different steps, including escape from endosomes and nuclear transport (19, 27), uncoating (6), and conversion from single-stranded to double-stranded DNA (48, 56). Even in highly permissive tissues, transgene expression takes several weeks to reach its maximal level and is often preceded by a lag period (58, 59).
In cultured cells, overexpression of adenovirus E4ORF6 in-creases the efficiency of rAAV transduction, and comparable enhancement can be obtained by treating cells with agents that affect genomic DNA integrity or metabolism (4, 20, 43). Both of these effects correlate with an improved conversion of the vector genome into double-stranded DNA (20, 21). These
ob-servations lead to the possibility that permissivity for AAV transduction could be linked to the induction of DNA damage checkpoints or DNA repair mechanisms that mediate replica-tion of single-stranded DNA AAV genomes. The proteins and the molecular pathways involved in such responses are still largely unexplored. In this respect, the phosphorylated form of an unknown cellular single-stranded DNA-binding protein has been shown to negatively regulate viral second-strand DNA synthesis by its association with the AAV genome termini (40). Here we investigate the fate of rAAV genomes in normal cells and in cells with defective DNA repair pathways and describe the direct molecular interaction between AAV DNA and proteins involved in double-stranded DNA break repair and recombination.
MATERIALS AND METHODS
Cell cultures.Cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum. Chinese hamster ovary (CHO-K1) cell lines were maintained in high-glucose (4.5 g/ml) DMEM.
To obtain the K8 hamster cell clone, we produced the cDNA coding for the wild-type Chinese hamster Ku86 protein by reverse transcription (RT)-PCR amplification of CHO-K1 total RNA. The cDNA was inserted into the expres-sion vector pcDNA3 (Clontech, Palo Alto, Calif.), and its nucleotide sequence was determined. This plasmid was transfected in the V15B, Ku86-deficient and radiation-sensitive, hamster cell line, and individual G418-resistant clones were selected and analyzed for restoration of resistance to gamma radiation. The K8 clone displayed response to gamma radiation similar to wild-type cells and was used in the subsequent experiments.
Rad52⫹/⫹and Rad52⫺/⫺primary fibroblast cell cultures were established from skin explants of newborn wild-type and Rad52⫺/⫺derivative C57BL/6 mice strains. The heterozygote Rad52⫹/⫺mouse strain was obtained from A. Pastink (Leiden University Medical Center, The Netherlands). Individual mice from heterozygote breeding progeny were genotyped for MmRAD52 by PCR analysis of DNA isolated from tail tips as described (42). Stable Rad52⫹/⫹and Rad52⫺/⫺ cell lines were obtained by spontaneous transformation of continuously in vitro-cultured primary fibroblasts.
Immortalized xeroderma pigmentosum fibroblasts cell lines XP3BRSV and XP12ROSV and Cockaine’s syndrome cell line CS1AN 5392 were kindly do-nated by M. Stefanini (Consiglio Nazionale delle Ricerche, Pavia, Italy).
* Corresponding author. Mailing address: Molecular Medicine Lab-oratory, International Centre for Genetic Engineering and Biotech-nology, Padriciano 99, 34012 Trieste, Italy. Phone: 39-040-3757.324. Fax: 39-040-226555. E-mail: [email protected].
12279
on November 8, 2019 by guest
http://jvi.asm.org/
The MRC5CVI and AT5BIVA cell lines were obtained from F. d’Adda di Fagagna (The Wellcome/CRC Institute, Cambridge, United Kingdom). The AT1 BR, AT3 BR, and HCT116 cell lines were obtained from the European Collection of Cell Cultures (Wiltshire, United Kingdom). All other cell lines were purchased from the American Type Culture Collection (Rockville, Md.)
Antibodies.A polyclonal antiserum was raised against recombinant human Rad52 (hRad52). The His-tagged hRad52 protein was overexpressed in Esche-richia coliusing the expression plasmid pFB581, kindly provided by S. West (Imperial Cancer Research Fund, Clare Hall Laboratories, Hertfordshire, United Kingdom). The protein was purified by affinity chromatography on Ni-nitrilotriacetic acid -agarose (Qiagen GmbH, Hilden, Germany) as previously described (7) and used for rabbit immunization. The serum was reactive against human Rad52 in Western blot and immunoprecipitation. Anti-Ku86 polyclonal serum was obtained as already described (15). Anti-USF-1 rabbit polyclonal antibody (C-20) was purchased from Santa Cruz Biotechnology, Santa Cruz, Calif.
rAAV vector preparation and characterization.rAAV-green fluorescent pro-tein (GFP) vector was prepared from plasmid pUF-5, kindly provided by N. Muzyczka (University of Florida, Gainesville) (61). Virus stocks were generated in 293 cells, cultured in 150-mm-diameter petri dishes, by cotransfecting each plate with 15g of the vector plasmid together with an equimolar amount of the
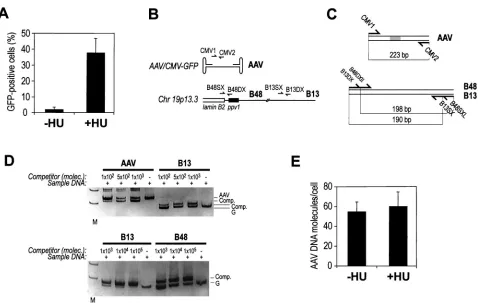
packaging/helper plasmid pDG (kindly provided by J. A. Kleinschmidt [26]), containing AAV and adenovirus helper functions. Forty-eight hours posttrans-fection, rAAV virions were released from the cells by three freeze-thaw cycles and purified by ammonium sulfate fractionation and CsCl2gradient centrifuga-tion as described (49). The rAAV titer was determined using pooled, dialyzed gradient fractions by a competitive PCR procedure (16). For this purpose, one pair of 20-bp primers were selected in the AAV-GFP genome, complementary to sequences in the cytomegalovirus (CMV) promoter and amplifying a segment of 243 bp (CMV1, 5⬘-ACGGTAAACTGCCCACTTGG-3⬘, and CMV2, 5⬘-CTTG GAAACCCCGTGAGTC-3⬘) (Fig. 1C). A competitor DNA fragment with the same sequence as the target amplification fragment but containing a 20-bp internal deletion was constructed according to an already described procedure (13). This fragment (223 bp) was purified, cloned into pGEM-T Easy cloning vector (Promega), and quantified.
To measure the number of viral genomes in vector stocks, 5l of each fraction was sequentially digested with 10 U of DNase I (Boehringer Mannheim) per ml for 30 min at 37°C, followed by 5 min of inactivation at 95°C. Proteinase K (Boehringer Mannheim, Mannheim, Germany) was sequentially added at 100
g/ml and incubated for 2 h at 56°C. Samples were then diluted and amplified in the presence of scalar amounts of competitor molecules (PCR conditions: 94°C, 30 s; 62°C, 30 s; and 72°C, 30 s for 30 cycles). A detailed description of the
FIG. 1. HU induces permissivity to AAV transduction through a postentry mechanism. (A) Human MRC fibroblasts were transduced with AAV-GFP either without or after overnight incubation in 1 mM HU. After 48 h, cells were analyzed for GFP fluorescence by flow cytometry. The graph shows the average results and standard deviations for 15 experiments of rAAV-GFP transduction of MRC fibroblasts. (B) To quantitatively detect AAV DNA in transduced cells, three primer pairs were selected, corresponding to DNA sequences in the AAV-GFP vector (primers CMV1and CMV2, mapping in the CMV promoter controlling GFP expression) and the human lamin B2 locus (primer pair B48SX and B48DX, mapping to a DNA replication origin [1, 23], and primer pair B13SX and B13DX, corresponding to a control region located⬇5 kb away). (C) The competitor for competitive PCR quantification of AAV DNA was a DNA fragment corresponding to the AAV vector PCR product with an internal 20-bp deletion. The competitor for quantification of B13 and B48 in the lamin B2 locus contains the B48 and B13 primers arranged to amplify DNA segments of different lengths than the respective genomic targets. (D) Fixed amounts of lysates from AAV-transduced cells were mixed with scalar amounts of competitor DNA and PCR amplified with the different primer pairs. After amplification, gels were stained with ethidium bromide, and the competitor (Comp.) and AAV or genomic DNA (G) bands were quantified. According to the principles of competitive PCR (22), the ratio between the amplification products in each reaction is linearly correlated with the input DNA amounts for the two species. Lanes M, molecular size markers. Molec., DNA molecules. (E) The number of AAV DNA molecules per cellular genome molecule (as evaluated by B48 and B13 quantification) was calculated in MRC5 cells either untreated or treated with HU and transduced with AAV-GFP. Quantification was performed at 48 h after transduction.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:2.612.64.542.74.377.2]principles of the competitive PCR technique has been provided elsewhere (16, 22, 53). The titers obtained for the several preparations used in this work ranged between 5⫻1010and 5⫻1012genomes per ml.
Transduction and analysis of cell cultures.Cells were seeded in 30-mm-diameter dishes (3⫻105cells) 24 h before infection. AAV-GFP was added to the cultures at a multiplicity of infection of 102particles per cell, as estimated from the number of AAV DNA genomes, in 2 ml of DMEM–10% FCS and left overnight. When indicated, cells were preincubated for 16 h with 1 mM hy-droxyurea (HU) (Calbiochem-Novabiochem Corporation, Darmstadt, Germany) from 1 M stocks dissolved in water; with 1M camptothecin (Calbiochem) from 1 mM stocks dissolved in dimethyl sulfoxide; with 0.15 U of bleomycin (Calbio-chem) per ml from 15-U/ml stocks dissolved in water; and with mitomycin C at 10⫺7M (Kyowa Italiana Farmaceutici, Milan, Italy) from 10⫺5M stocks dis-solved in water. After chemical treatment, cell cultures were washed with fresh medium immediately before incubation with AAV-GFP. Cells were exposed to UV radiation (254 nm) in a UV Stratalinker (Stratagene, La Jolla, Calif.) im-mediately before vector addition. After an additional 48 h, cells were examined for GFP expression by fluorescence microscopy or flow cytometry.
AAV-GFP DNA present in transduced cells was quantified by quantitative PCR. Cells were washed extensively with 2 M NaCl and incubated with trypsin (0.5 g/liter) for 2 to 3 min to strip any residual virus attached to the cell surface. In a control experiment, we observed that such treatment significantly reduced (over 50-fold) the amount of vector genomes adsorbed to the cell membrane but not yet internalized (data not shown). A fixed amount of cells was subsequently dissolved in lysis buffer (10 mM Tris HCl [pH 8.0], 0.05% NP-40, 0.05% Tween 100) and treated with DNase I (10 U/ml) for 30 min at 37°C and with proteinase K (100g/ml) for 2 h at 56°C. A fixed amount of diluted lysate was mixed with scalar amounts of competitor, and PCR was performed as described above.
Quantitative ChIP.Ten million cells (293, MRC5, or AT-5), either treated or not treated with 1 mM HU, were infected with AAV-GFP as described above. After 48 h of incubation, cells were fixed with formaldehyde for 5 min and treated as described by Orlando et al. (38). Quantitative chromatin immunopre-cipitation (ChIP) was performed as already described (34) with minor modifi-cations. Chromatin pellets were resuspended in 1 ml of RIPA lysis buffer 50 (50 mM Tris-HCl [pH 7.5], 50 mM NaCl, 1% sodium deoxycholate, 1% Nonidet P-40, 0.1% sodium dodecyl sulfate [SDS], 2 mM EDTA) with protease inhibitors (500M phenylmethylsulfonyl fluoride, 1M leupeptin, 1M pepstatin; Phar-macia, Uppsala, Sweden). Each sample was sonicated in Eppendorf tubes with 25 cycles of 10 s each at maximum power in ice. Sonicated chromatin was centri-fuged to spin down cell debris and incubated with the appropriate antibodies overnight at 4°C in order to immunoprecipitate protein-DNA cross-linked com-plexes.
After incubation, 40l of a 50% suspension of protein A-Sepharose CL-4B beads (Pharmacia) in TE buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA) was added. After a 1-h incubation at 4°C, beads were pelletted and washed three times with 1 ml of RIPA buffer 150 (RIPA lysis buffer with 150 mM NaCl). Pellets were then resuspended in 100l of TE buffer and digested with 5 U of DNase-free RNase (Boehringer Mannheim) for 30 min at 37°C. The protein-bound immunoprecipitated DNA was then sequentially digested with proteinase K (300g/ml) (Sigma Chemical Co., St. Louis, Mo.) in 0.5% SDS–100 mM NaCl for 6 h at 65°C to revert cross-links. DNA was extracted with phenol-chloroform-isoamyl alcohol, precipitated with ethanol, and resuspended in 50l of distilled water for competitive PCR quantification.
Primer sequences and amplification conditions for the B48 and B13 DNA segments in the lamin B2 genomic region have already been described (23). The multicompetitor molecule containing B48 and B13 primer recognition sequences (34) is depicted in Fig. 1C. Values obtained from the quantification of the B13 region were used as an internal control to normalize the total DNA concentra-tion recovered in the different samples. Results of the AAV genome quantifica-tion were expressed as fold enrichment with respect to values obtained using the anti-USF control antibody. Since the anti-USF antibody immunoprecipitates cross-linked B48 but not the adjacent B13 genomic region, the ratio between these two values was used as a positive ChIP control in each experiment.
RESULTS
Efficiency of AAV transduction is enhanced by genotoxic
agents.To gain insights into the molecular mechanisms that
govern intracellular permissivity to transduction with AAV vectors, we studied the properties of an AAV construct con-taining a CMV-GFP cassette (61). Transduction of human
MRC5 fibroblasts with this viral preparation at a multiplicity of infection of 102particles per cell resulted in few fluorescent
cells after 48 h. However, when cells were treated by overnight incubation with 1 mM HU, the efficiency of transduction rose to more than 40% GFP-positive cells, in agreement with pub-lished results (4, 43, 47) (Fig. 1A).
To verify that the effect of HU was related to molecular events occurring after virus internalization, we developed a competitive PCR procedure to quantitatively assay the amount of cell-associated AAV DNA with respect to cell genomic DNA. For this purpose, we designed one primer pair in the AAV-GFP genome and two primer pairs in a single-copy genomic region in human chromosome 19p13.3, corresponding to the lamin B2 genomic locus (23) (Fig. 1B). For each of these primer pairs, competitor DNA fragments were constructed that contain the respective primer recognition sites at a dis-tance which is different from that of the target DNA (Fig. 1C). Competitors were constructed according to an already de-scribed recombinant PCR procedure (22).
AAV DNA quantitation experiments were performed by lysing AAV-transduced cells and mixing fixed amounts of the cell lysates with known amounts of competitor DNAs. Figure 1D shows two representative competitive PCR gels for the quantification of DNA segments in the AAV genome (AAV) and in the human genome at the B48 and B13 segments of the lamin B2 locus. In each experiment, quantification was carried out by mixing a fixed amount of cell lysate DNA with scalar concentrations of competitor, followed by amplification with the respective primers. According to the principles of compet-itive PCR, the ratio between target and competitor DNA am-plification products exactly reflects the input ratio of the two DNA species, independent of amplification efficiency, mainte-nance of the exponential phase of the reaction, or presence of nonspecific amplification products (22).
By using this procedure, we analyzed the number of AAV DNA genomes inside the cells at 48 h after transduction, after cell treatment to release any residual virus associated with the cell. We consistently found that 50 to 70% of input virus DNA was recovered inside the cells in both untreated and HU-treated cells (Fig. 1E). Thus, HU acts at a step that is subse-quent to virus entry inside the cells. In keeping with similar experiments (46), by Southern analysis of Hirt extracts ob-tained from transduced cells which were not treated with HU, we found that most of the viral genome is maintained in a single-stranded form (not shown). This DNA is clearly unavail-able for transcription.
AAV transduction in cell lines with altered DNA repair
pathways.HU is known to generate DNA damage in cellular
DNA by blocking progression of DNA replication forks and to induce a checkpoint response (39). Induction of AAV trans-duction in human MRC fibroblasts as well as in human epi-thelial 293 and HeLa cells and hamster epiepi-thelial CHO cells was not restricted to HU but was also observed as a conse-quence of cell treatment with other agents known to damage DNA, such as UV radiation (254 nm), mitomycin C (10⫺7M),
camptothecin (1M), and bleomycin (0.15 U/ml) (see Fig. 3 and data not shown), in agreement with previous findings (4). It has been proposed that AAV DNA processing might be induced by activation of DNA repair mechanisms that follow DNA damage recognition inside the cells (4, 20, 21). Therefore
on November 8, 2019 by guest
http://jvi.asm.org/
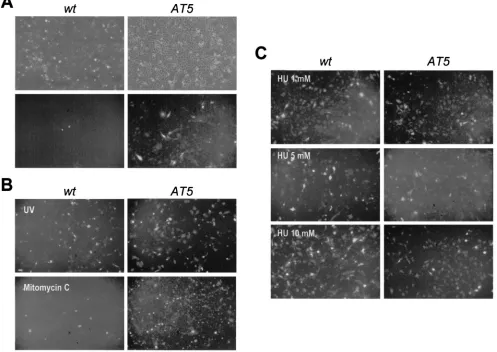
we set out to study the efficiency of AAV-GFP transduction in cell lines which are defective in different pathways of DNA repair. These included human immortalized xeroderma pig-mentosum fibroblasts XP3BRSV (xeroderma pigpig-mentosum group G) and XP12ROSV (xeroderma pigmentosum group A), which are defective in nucleotide excision repair; the Coc-kaine’s syndrome cell line CS1AN 5392 (CocCoc-kaine’s syndrome group B), defective in transcription-coupled nucleotide exci-sion repair; and an hMLH1-defective colorectal carcinoma cell line, HCT116, defective in mismatch repair. Analysis of the percentage of GFP-positive cells 48 h after transduction showed similar results for all these cell lines, with values that were in the same range of those of wild-type cells (Fig. 2A). Moreover, in all these cells, transduction efficiency was in-creased to the same extent as in wild-type cells by treatment with HU (Fig. 2A), camptothecin, or UV (not shown). These results clearly rule out a major role of these repair pathways in regulating rAAV transduction efficiency.
We then investigated cells defective in the ATM protein, a serine protein kinase belonging to a family of large proteins with a phosphatidylinositol 3-kinase domain that plays a cen-tral role in cellular signaling in response to DNA DSBs (5, 12). Three immortalized ataxia telangiectasia (AT) human fibro-blast cell lines from patients affected by the autosomal reces-sive disorder AT were used to study AAV-GFP infection. As shown in Fig. 2B, all these cell lines showed increased
permis-sivity, with numbers of GFP-positive cells three to eight times higher than in wild-type cells. This finding is also visually evi-dent from the panels of Fig. 3A, showing increased basal per-missivity of one of the AT clones (AT5). This result is in agreement with similar findings reported by J. Engelhart and collaborators using different AT cells (45). Incubation of the AT gene product ATM-defective cells with a genotoxic agent such as HU, UV, or mitomycin C before infection had a lim-ited effect on AAV transduction, indicating that the efficiency of AAV DNA molecular processing is already at nearly max-imal levels in these cells (Fig. 2B, 3B, and 3C).
Proteins involved in DSBs repair bind to AAV genome.
Un-der normal growth conditions, AT cells retain higher levels of unrepaired chromosome breaks and intramolecular recombi-nation than normal cells (35). Thus, a likely possibility is that enhanced AAV vector transduction in these cells is consequent to activation of proteins which participate in DSB recognition and repair. Two factors involved in these processes are Rad52 and Ku. The former protein initiates repair by homologous recombination; the latter, acting as the heterodimeric DNA-binding component of DNA-protein kinase (PK), is important for repair by nonhomologous end joining. Both proteins bind directly to broken DNA termini as well as single-stranded or hairpin DNA (15, 25, 55).
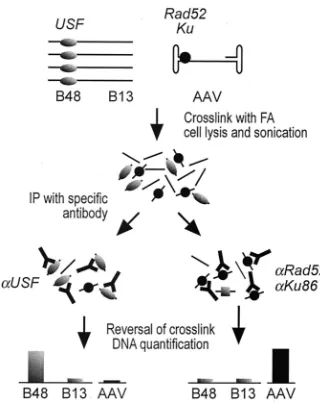
To assess the possible in vivo interaction between Rad52 and/or Ku86 (the larger subunit of the Ku heterodimer) with the AAV genome, we developed a modified ChIP (38). This technique is based on the cross-linking of protein-DNA and protein-protein complexes within the cell by formaldehyde treatment, followed by chromatin sonication, immunoprecipi-tation with specific antibodies, and precise quantification of the immunoprecipitated DNA segments by quantitative PCR (34), as outlined in Fig. 4. Quantification of the immunoprecipitated DNA is carried out by using a competitive procedure that exploits the properties of the competitor DNA fragments shown in Fig. 1C. Should a protein bind to a DNA segment, this segment will be specifically immunoprecipitated with an antibody specific for this protein and will turn out to be en-riched over background in the total immunoprecipitated DNA. Analysis of the protein-DNA interaction at selected DNA regions (Fig. 1C) was performed in wild-type MRC5 cells, untreated or preincubated for 16 h with 1 mM HU, and in AT cells 48 h after infection with AAV-GFP. In each experiment, the technique was validated by immunoprecipitation with an antibody specific for transcription factor USF; our previous results demonstrated the presence of a USF target site in the B48 genomic region (15, 23) and the actual in vivo binding of the factor to this sequence (1, 2). Indeed, immunoprecipitation with the anti-USF antibody resulted in the enrichment (⬇ 35-fold) for the B48 DNA segment with respect to the⬇ 5-kb-distant B13 region in the same locus (Fig. 5B). This enrichment was found both in wild-type cells either treated or not treated with HU and in AT cells.
Figure 5A reports a representative ChIP experiment per-formed on chromatin from untreated 293 cells using antibodies against USF, Ku, and Rad52. DNA fragments recovered from the immunoprecipitated chromatin were mixed with scalar competitor amounts and PCR amplified to detect enrichment for AAV and B13 DNA. The table on the right side of Fig. 5A shows the results of quantification, indicating, in this experi-FIG. 2. AAV transduction of cells defective in DNA damage
checkpoint and repair. (A) Cell lines impaired in different DNA repair pathways were transduced with AAV-GFP without or after treatment with HU and analyzed for GFP fluorescence. wt, wild-type normal MRC5 fibroblasts; XP3 and XP12, xeroderma pigmentosusm XP3BRSV and XP12ROSV cells; CS1, Cockaine’s syndrome CS1AN 5392 cells; HCT116, colorectal carcinoma cells defective in mismatch repair. Shown are means and standard deviations of at least three experiments. (B) Fibroblast cell lines from three patients with ataxia telangectasia were transduced with AAV-GFP. wt, wild-type normal MRC5 fibroblasts; AT1, AT1 BR; AT3, AT3 BR; AT5, AT5BIVA.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.612.86.264.79.318.2]ment, specific 4-fold and 7.5-fold enrichments for AAV DNA using anti-Ku and anti-Rad52 antibodies, respectively. The USF antibody as well as antibodies against glutathione S-trans-ferase (GST) or a rabbit preimmune serum (controls in Fig. 5C) always failed to immunoprecipitate the AAV CMV DNA segment. By contrast, after immunoprecipitation with poly-clonal antibodies against human Rad52 and Ku86, a notable enrichment for this region was consistently observed, both in wild-type and in AT cells (Fig. 5C and 5D, respectively), indi-cating that both proteins interact with the AAV genome inside the cells.
Treatment of wild-type cells with 1 mM HU still permitted detection of protein-bound AAV DNA for both factors. How-ever, binding was decreased with anti-Ku and further increased with anti-Rad52 antibodies, suggesting that HU-induced per-missivity to AAV transduction correlates with a change in the ratio between the number of cellular Ku and AAV-Rad52 complexes. For the ChIP assay shown in Fig. 5D, no HU data were included because this drug does not significantly enhance rAAV transduction in AT cells (Fig. 2B). However, enrichment for Ku and, even greater, for Rad52 is clearly
observed also in AT cells, suggesting that DSB repair proteins are active in these cells as they are in normal cells.
AAV transduction of cells defective in Ku and Rad52.To
assess the functional significance of the molecular interactions between AAV DNA and the Ku and Rad52 proteins, we tested AAV transduction in cells genetically defective for either of these factors. V15B is a Chinese hamster cell line which is deficient for the Ku86 protein and exhibits high sensitivity to ionizing radiation compared to the original parental cell line V79B (28). We complemented the Ku86 defect in V15B by stable transfection of hamster Ku86 cDNA. Among different transduced clones, one (named K8) was selected, exhibiting ␥-ray sensitivity similar to wild-type V79B cells (data not shown).
[image:5.612.54.551.85.437.2]Analysis of GFP-positive cells 2 days after exposure of V15B and K8 cells to AAV-GFP vector showed reproducibly in-creased transduction (⬇5-fold) in mutant V15B compared to reconstituted K8 cells and parental V79B cells (Fig. 6A). Per-missivity to rAAV of both wild-type and mutant cell lines was quite low in untreated cells (0.23 and 0.16% average positive cells for parental V79B and reconstituted wild-type K8 cells, FIG. 3. Sensitivity to AAV transduction of wild-type and ATM fibroblasts after treatment with genotoxic agents. Normal MRC5 (wild type [wt]) and ATM-deficient AT5BIVA (AT5) fibroblasts were transduced with AAV-GFP without treatment (panel A) or after treatment with UV (panel B, upper part), mitomycin C (panel B, lower part), or HU (panel C). Fluorescent cells were visualized and photographed 48 h after transduction. Panel A shows transmitted light (upper part) and fluorescence (lower part) images of the same fields.
on November 8, 2019 by guest
http://jvi.asm.org/
respectively, and 0.85% for mutant V15B cells), but it was induced⬇20-fold by incubation with HU. After HU treatment, the percentage of GFP-positive cells was again⬇5 times higher in Ku-defective than in wild-type cells (Fig. 6A).
To assess the role of Rad52 in AAV vector transduction, we established Rad52⫺/⫺fibroblast cell cultures starting from skin
explants of Rad52⫺/⫺homozygous newborn mice, which were
obtained by crossing Rad52⫹/⫺heterozygotes (42) followed by
F1 progeny screening. In both primary and spontaneously
transformed fibroblast cultures, efficiency of AAV-GFP trans-duction was reproducibly lower than in Rad52⫹/⫹control
pri-mary fibroblasts. In both cases, preincubation of these cells with 5 mM HU increased by 3- to 5-fold the overall number of GFP-positive cells, but the relative permissivity for transduc-tion did not vary (Fig. 6B).
Altogether, these results indicate that both Ku and Rad52 contribute to determine permissivity for rAAV transduction by exerting opposite functions, the former protein being an inhib-itor while the latter is an activator.
DISCUSSION
Postentry events that lead to efficient transduction of AAV vectors are poorly understood. These events include processing of AAV particles in endosomes, lysosomal escape, nuclear import of the AAV genome (6, 19, 27), and single-stranded DNA genome conversion to long-lasting double-stranded DNA molecules (20, 21). In this study we sought to investigate the molecular mechanisms that take part in this last step of rAAV processing.
After infection, in the absence of viral helper functions and AAV Rep proteins, the incoming viral genomes rely solely on cellular proteins for the conversion of their single-stranded
genomes to molecular species suitable for long-term transgene expression. Since agents that alter DNA integrity and metab-olism are able to increase AAV-mediated gene transduction by more than 10-fold, it appears likely that the activation of cel-lular stress response or DNA repair pathways allows vectors to overcome the existing barriers. The survey of cell lines defec-tive for major DNA repair pathways excludes a major involve-ment in AAV DNA processing of DNA repair mechanisms such as nucleotide excision repair, including transcription-cou-pled repair and global genome repair, and mismatch repair. By contrast, we found that cells defective in the ATM protein display considerably higher transduction efficiency, in agree-ment with reported results (45).
The reason for this enhanced permissivity is not clear; how-ever, the metabolic state of these cells is highly likely to be the major determinant of increased AAV transduction, in partic-ular their propensity to accumulate unrepaired DSB damage to genomic DNA. The ATM protein is indeed an important care-taker of genome integrity, and it is central to the activation of the cellular response to DNA DSBs (35). AT cells are charac-terized by hypersensitivity to ionizing radiation together with multiple cell cycle checkpoint abnormalities, hyperrecombina-tion, and excess of apoptosis (10, 35).
In eukaryotic cells, DSBs are repaired through two alterna-tive and competing pathways, nonhomologous end joining and homologous recombination (55). In both cases, these pathways require the interaction of damaged DNA with DNA-binding proteins that recognize the lesion. These proteins are the Ku heterodimer, composed of 70- and 86-kDa subunits, for non-homologous end joining and Rad52 for non-homologous recombi-nation. Both of these factors interact in a sequence-indepen-dent manner to free DNA ends, hairpin loops, single-strand nicks, or gaps (11, 25, 55). The single-stranded AAV genome and its terminal hairpins are likely to represent suitable sub-strates for such proteins inside the cells.
By in vivo quantitative chromatin cross-linking experiments, we indeed found that both Rad52 and Ku physically associate with AAV genomes inside the cells. We also found that stim-ulation of AAV transduction by HU, a drug causing stalling of replication forks, which are unstable and prone to breakage and restoration by recombinational repair (39), modulates binding of AAV DNA to the two proteins by increasing inter-action with Rad52 and decreasing that with Ku86. These re-sults are consistent with a model in which the single-stranded AAV genome is competitively bound by the two proteins once it has entered the cell. While Ku inhibits further maturation of AAV DNA, the fraction interacting with Rad52 is processed through a homologous recombination pathway for double-stranded DNA conversion, leading to a substrate suitable for gene expression. The observation that, even in Rad 52⫺/⫺cells,
[image:6.612.91.251.76.278.2]treatment with HU enhances rAAV transduction efficiency is in line with the poor phenotype of these cells that are not completely defective for homologous recombination (42). In fact, many members of the Rad52 epistasis group appear to have redundant functions in higher eukaryotes. This model is also consistent with the finding that cells defective for Ku86 and impaired in nonhomologous end joining are more permis-sive to AAV transduction than their wild-type, matched coun-terpart. This behavior is similar to what was reported by Tauer FIG. 4. Flow chart of ChIP of proteins binding to AAV DNA. Flow
chart for ChIP. Cells transduced with AAV were treated with formal-dehyde (FA) at 48 h postinfection; chromatin was sonicated down to DNA segments of⬍1 kb and immunoprecipitated (IP) with antibodies specific for AAV-binding proteins and controls. Antibodies against the USF transcription factor (known to bind the B48 region) enrich for this segment; antibodies against DNA-end-binding proteins (Rad52 and Ku86) are investigated for binding to AAV DNA.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 5. ChIP of proteins binding to AAV DNA. (A) Quantification by competitive PCR of DNA recovered from ChIP using control anti-USF antibodies as well as rabbit polyclonal antibodies against Ku86 and Rad52. Immunoprecipitated DNA was mixed with the amount of competitor molecules indicated on top of each lane and PCR amplified with primers for AAV and B13. The amplification products for AAV, total genomic DNA B13 region (G), and competitor (Comp.) are indicated. Lane M, molecular size markers. The table on the right side of the panel shows the results of quantification, indicated as number of DNA molecules immunoprecipitated for each antibody. (B) As a control for ChIP efficiency, immunoprecipitation of the B48 and B13 genomic segments in the lamin B2 locus was investigated. The former region contains a binding site for transcription factor USF (15). Experiments were performed in wild-type (wt) MRC5 fibroblasts either treated or not treated with HU and in AT5 cells. Control antibodies included at least three different antibodies against unrelated proteins. Shown are average values and standard deviations of B48/B13 ratios in at least three different experiments. (C and D) ChIP results using anti-Ku86 and anti-Rad52 antibodies are shown for normal MRC5 fibroblasts (wt), either treated or untreated with HU (C), and for AT5 cells (D). Control antibodies included an antibody against GST, a rabbit preimmune serum, and other antibodies against unrelated proteins (data for controls are pooled). The results are expressed as fold enrichment for AAV DNA with respect to background immunoprecipitation of control genomic DNA (B13 region). Shown are average values and standard deviations in at least three independent experiments.
FIG. 6. AAV transduction in cells impaired in nonhomologous end joining and homologous recombination. (A) Parental Ku⫺V15B cells, wild-type (wt) V79B, and the Ku⫹ K8 clone were transduced with AAV-GPF either without or after treatment with HU. The number of GFP-positive cells was measured by flow cytometry at 48 h after infection. (B) Cell cultures of primary skin fibroblasts from wild-type mice (⫹/⫹) and Rad52 knockout mice (⫺/⫺) were transduced with AAV-GFP either without or after treatment with HU (left side). Immortal fibroblast cell lines were also obtained from both wild-type and Rad⫺/⫺fibroblasts by spontaneous transformation in cell culture and tested for permissivity to AAV-GFP transduction (right side). In both cases, the percentages of GFP-positive cells were measured by flow cytometry at 48 h after infection with AAV-GFP. In both panels, values are means and standard deviations from at least three independent transduction experiments.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.612.138.473.522.666.2]and colleagues for the autonomous parvovirus minute virus of mice in X-ray-sensitive, Ku-defective cell lines (54).
Components of the nonhomologous end-joining machinery have also been shown to play an essential role in chromosome telomere length maintenance by preventing end-to-end fusion of chromosomes. Several experiments indicate that Ku physi-cally associates with telomeric DNA and that this function is conserved from yeasts to humans (9, 24, 29). The exact mech-anism of such interactions is not yet clear, but it is intriguing to imagine that the cell would treat the genome termini of par-voviruses as specialized DSBs. In this case the components of the nonhomologous joining repair would prevent the end-to-end recombination and/or successful single-stranded to double-stranded DNA conversion of the viral vector. Addi-tional support for the involvement of DSB repair pathways in rAAV processing also comes from a recent report by T. Flotte and coworkers, who showed that in vivo transduction of skel-etal muscle of SCID (DNA-PK defective) mice results in al-tered processing of rAAV DNA compared to normal muscle (50).
In contrast to Ku, interaction of AAV DNA with Rad52 can be envisaged as a facilitator of single-stranded DNA process-ing, since in Rad52-defective cells AAV transduction is poorer than in wild-type cells. These functional data and the evidence that the Rad52 protein physically binds to AAV DNA suggest the involvement of the homologous recombination pathway in AAV DNA postentry processing. In transduced tissues in vivo, rAAV genomes mostly persist as episomal DNA molecules, with the formation of high-molecular-weight concatemers sug-gesting the occurrence of frequent intra- and intermolecular recombination events (17, 60). The Rad52 protein might par-ticipate in concatemer formation, since the protein is known to bind DNA ends, protecting them from exonuclease degrada-tion and facilitating repair of gaps by homologous recombina-tion (55).
The involvement of homologous recombination in the pro-cessing of AAV genomes is also suggested by a series of recent findings indicating that annealing of complementary single-stranded rAAV genomes with positive or negative polarity and subsequent intermolecular linking could represent a major mechanism for the formation of double-stranded, high-molec-ular-weight concatameric rAAV species (18, 37). Finally, ad-ditional support for the involvement of homologous recombi-nation in AAV DNA processing is given by the observation that vectors based on AAV can recombine with homologous chromosomal human target sequences at rates close to 1%, which is the highest targeting frequency obtained in normal human cells (30, 44).
In conclusion, these data provide a molecular basis for the understanding of rAAV genome processing in vivo. In partic-ular, our observation that proteins active in DSB repair phys-ically interact with rAAV genomes in vivo provides a link to the longstanding evidence that genotoxic agents boost AAV infection and indicates that interaction of DSB repair proteins with AAV DNA inside the cells modulates transduction effi-ciency. The observation that both Rad52- and Ku-defective cell lines still vigorously respond to HU treatment by increasing permissivity for AAV transduction clearly indicates that these are not the only proteins involved in AAV DNA replication or
solely determine its efficacy. The identification of other cellular partners for this process still represents a challenging task.
ACKNOWLEDGMENTS
This work was supported by a grant from Telethon Italy to M.G. We thank J. Kleinschmidt for the pDG plasmid, A. Pastink for the Rad52⫹/⫺ mice, M. Stefanini for the excision repair-defective cell lines, S. West for the His-tagged human Rad52 protein, N. Muzyczka for the pFU-5 plasmid, and B. Boziglav and M. E. Lopez for excellent technical assistance. We are grateful to F. d’Adda di Fagagna for suggestions and helpful discussions.
REFERENCES
1.Abdurashidova, G., M. Deganuto, R. Klima, S. Riva, G. Biamonti, M. Gi-acca, and A. Falaschi.2000. Start sites of bidirectional DNA synthesis at the human lamin B2 origin. Science287:2023–2026.
2.Abdurashidova, G., S. Riva, G. Biamonti, M. Giacca, and A. Falaschi.1998. Cell cycle modulation of protein-DNA interactions at a human replication origin. EMBO J.17:2961–2969.
3.Acland, G. M., G. D. Aguirre, J. Ray, Q. Zhang, T. S. Aleman, A. V. Cide-ciyan, S. E. Pearce-Kelling, V. Anand, Y. Zeng, A. M. Maguire, S. G. Jacob-son, W. W. Hauswirth, and J. Bennett.2001. Gene therapy restores vision in a canine model of childhood blindness. Nat. Genet.28:92–95.
4.Alexander, I. E., D. W. Russell, and A. D. Miller.1994. DNA-damaging agents greatly increase the transduction of nondividing cells by adeno-asso-ciated virus vectors. J. Virol.68:8282–8287.
5.Banin, S., L. Moyal, S. Shieh, Y. Taya, C. W. Anderson, L. Chessa, N. I. Smorodinsky, C. Prives, Y. Reiss, Y. Shiloh, and Y. Ziv.1998. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science
281:1674–1677.
6.Bartlett, J. S., R. Wilcher, and R. J. Samulski.2000. Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J. Virol.74:
2777–2785.
7.Benson, F. E., P. Baumann, and S. C. West.1998. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature391:401–404. 8.Berns, K. I., and R. M. Linden.1995. The cryptic life style of
adeno-associated virus. BioEssays17:237–245.
9.Bianchi, A., and T. de Lange.1999. Ku binds telomeric DNA in vitro. J. Biol. Chem.274:21223–21227.
10. Bishop, A. J., C. Barlow, A. J. Wynshaw-Boris, and R. H. Schiestl.2000. ATM deficiency causes an increased frequency of intrachromosomal homol-ogous recombination in mice. Cancer Res.60:395–399.
11. Blier, P. R., A. J. Griffith, J. Craft, and J. A. Hardin.1993. Binding of Ku protein to DNA. Measurement of affinity for ends and demonstration of binding to nicks. J. Biol. Chem.268:7594–7601.
12. Canman, C. E., D. S. Lim, K. A. Cimprich, Y. Taya, K. Tamai, K. Sakaguchi, E. Appella, M. B. Kastan, and J. D. Siliciano.1998. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science281:1677– 1679.
13. Celi, F. S., M. E. Zenilman, and A. R. Shuldiner.1993. A rapid and versatile method to synthesize internal standards for competitive PCR. Nucleic Acids Res.21:1047.
14. Chao, H., R. Samulski, D. Bellinger, P. Monahan, T. Nichols, and C. Walsh.
1999. Persistent expression of canine factor IX in hemophilia B canines. Gene Ther.6:1695–1704.
15. Csorda´s To´th, E., L. Marusic, A. Ochem, A. Patthy, S. Pongor, M. Giacca, and A. Falaschi.1993. Interactions of USF and Ku antigen with a human DNA region containing a replication origin. Nucleic Acids Res.21:3257– 3263.
16. Diviacco, S., P. Norio, L. Zentilin, S. Menzo, M. Clementi, A. Falaschi, and M. Giacca.1992. A novel procedure for quantitative polymerase chain re-action by coamplification of competitive templates. Gene122:3013–3020. 17. Duan, D., P. Sharma, J. Yang, Y. Yue, L. Dudus, Y. Zhang, K. J. Fisher, and
J. F. Engelhardt.1998. Circular intermediates of recombinant adeno-asso-ciated virus have defined structural characteristics responsible for long-term episomal persistence in muscle tissue. J. Virol.72:8568–8577.
18. Duan, D., Y. Yue, Z. Yan, and J. F. Engelhardt.2000. A new dual-vector approach to enhance recombinant adeno-associated virus-mediated gene expression through intermolecular cis activation. Nat. Med.6:595–598. 19. Duan, D., Y. Yue, Z. Yan, J. Yang, and J. F. Engelhardt.2000. Endosomal
processing limits gene transfer to polarized airway epithelia by adeno-asso-ciated virus. J. Clin. Investig.105:1573–1587.
20. Ferrari, F. K., T. Samulski, T. Shenk, and R. J. Samulski.1996. Second-strand synthesis is a rate-limiting step for efficient transduction by recombi-nant adeno-associated virus vectors. J. Virol.70:3227–3234.
21. Fisher, K. J., K. Jooss, J. Alston, Y. Yang, S. E. Haecker, K. High, R. Pathak, S. E. Raper, and J. M. Wilson.1997. Recombinant adeno-associated virus for muscle directed gene therapy. Nat. Med.3:306–312.
22. Giacca, M., C. Pelizon, and A. Falaschi.1997. Mapping replication origins by
on November 8, 2019 by guest
http://jvi.asm.org/
quantifying relative abundance of nascent DNA strands using competitive polymerase chain reaction. Methods13:301–312.
23. Giacca, M., L. Zentilin, P. Norio, S. Diviacco, D. Dimitrova, G. Contreas, G. Biamonti, G. Perini, F. Weighardt, S. Riva, and A. Falaschi.1994. Fine mapping of a replication origin of human DNA. Proc. Natl. Acad. Sci. USA
91:7119–7123.
24. Gravel, S., M. Larrivee, P. Labrecque, and R. J. Wellinger.1998. Yeast Ku as a regulator of chromosomal DNA end structure. Science280:741–744. 25. Griffith, A. J., P. R. Blier, T. Mimori, and J. A. Hardin.1992. Ku
polypep-tides synthesized in vitro assemble into complexes which recognize ends of double-strandeded DNA. J. Biol. Chem.267:331–338.
26. Grimm, D., A. Kern, K. Rittner, and J. A. Kleinschmidt.1998. Novel tools for production and purification of recombinant adenoassociated virus vec-tors. Hum. Gene Ther.9:2745–2760.
27. Hansen, J., K. Qing, and A. Srivastava.2001. Adeno-associated virus type 2-mediated gene transfer: altered endocytic processing enhances transduc-tion efficiency in murine fibroblasts. J. Virol.75:4080–4090.
28. Helbig, R., M. Z. Zdzienicka, and G. Speit.1995. The effect of defective DNA double-stranded break repair on mutations and chromosome aberra-tions in the Chinese hamster cell mutant XR-V15B. Radiat. Res.143:151– 157.
29. Hsu, H. L., D. Gilley, E. H. Blackburn, and D. J. Chen.1999. Ku is associated with the telomere in mammals. Proc. Natl. Acad. Sci. USA96:12454–12458. 30. Inoue, N., R. K. Hirata, and D. W. Russell.1999. High-fidelity correction of mutations at multiple chromosomal positions by adeno-associated virus vec-tors. J. Virol.73:7376–7380.
31. Kay, M. A., C. S. Manno, M. V. Ragni, P. J. Larson, L. B. Couto, A. McClelland, B. Glader, A. J. Chew, S. J. Tai, R. W. Herzog, V. Arruda, F. Johnson, C. Scallan, E. Skarsgard, A. W. Flake, and K. A. High.2000. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat. Genet.24:257–261.
32. Kessler, P. D., G. M. Podsakoff, X. Chen, S. A. McQuiston, P. C. Colosi, L. A. Matelis, G. J. Kurtzman, and B. J. Byrne.1996. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc. Natl. Acad. Sci. USA93:14082–14087.
33. Kotin, R. M., M. Siniscalco, R. J. Samulski, X. D. Zhu, L. Hunter, C. A. Laughlin, S. McLaughlin, N. Muzyczka, M. Rocchi, and K. I. Berns.1990. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA87:2211–2215.
34. Marzio, G., M. Tyagi, M. I. Gutierrez, and M. Giacca.1998. HIV-1 Tat transactivator recruits p300 and CBP histone acetyl transferases to the viral promoter. Proc. Natl. Acad. Sci. USA95:13519–13524.
35. Meyn, M. S.1995. Ataxia telangiectasia and cellular responses to DNA damage. Cancer Res.55:5991–6001.
36. Nakai, H., Y. Iwaki, M. A. Kay, and L. B. Couto.1999. Isolation of recom-binant adeno-associated virus vector-cellular DNA junctions from mouse liver. J. Virol.73:5438–5447.
37. Nakai, H., T. A. Storm, and M. A. Kay.2000. Increasing the size of rAAV-mediated expression cassettes in vivo by intermolecular joining of two com-plementary vectors. Nat. Biotechnol.18:527–532.
38. Orlando, V., H. Strutt, and R. Paro.1997. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods11:205–214.
39. Paulovich, A. G., D. P. Toczyski, and L. H. Hartwell.1997. When checkpoints fail. Cell88:315–321.
40. Qing, K., B. Khuntirat, C. Mah, D. M. Kube, X. S. Wang, S. Ponnazhagan, S. Zhou, V. J. Dwarki, M. C. Yoder, and A. Srivastava.1998. Adeno-asso-ciated virus type 2-mediated gene transfer: correlation of tyrosine phosphor-ylation of the cellular single-strandeded D sequence-binding protein with transgene expression in human cells in vitro and murine tissues in vivo. J. Virol.72:1593–1599.
41. Qing, K., C. Mah, J. Hansen, S. Zhou, V. Dwarki, and A. Srivastava.1999. Human fibroblast growth factor receptor 1 is a coreceptor for infection by adeno-associated virus 2. Nat. Med.5:71–77.
42. Rijkers, T., J. Van Den Ouweland, B. Morolli, A. G. Rolink, W. M. Baarends, P. P. Van Sloun, P. H. Lohman, and A. Pastink.1998. Targeted inactivation of mouse RAD52 reduces homologous recombination but not resistance to ionizing radiation. Mol. Cell. Biol.18:6423–6429.
43. Russell, D. W., I. E. Alexander, and A. D. Miller.1995. DNA synthesis and topoisomerase inhibitors increase transduction by adeno-associated virus vectors. Proc. Natl. Acad. Sci. USA92:5719–5723.
44. Russell, D. W., and R. K. Hirata.1998. Human gene targeting by viral vectors. Nat. Genet.18:325–330.
45. Sanlioglu, S., P. Benson, and J. F. Engelhardt.2000. Loss of ATM function enhances recombinant adeno-associated virus transduction and integration through pathways similar to UV irradiation. Virology268:68–78. 46. Sanlioglu, S., D. Duan, and J. F. Engelhardt.1999. Two independent
mo-lecular pathways for recombinant adeno-associated virus genome conversion occur after UV-C and E4orf6 augmentation of transduction. Hum. Gene Ther.10:591–602.
47. Sanlioglu, S., and J. F. Engelhardt.1999. Cellular redox state alters recom-binant adeno-associated virus transduction through tyrosine phosphatase pathways. Gene Ther.6:1427–1437.
48. Snyder, R. O., S. K. Spratt, C. Lagarde, D. Bohl, B. Kaspar, B. Sloan, L. K. Cohen, and O. Danos.1997. Efficient and stable adeno-associated virus-mediated transduction in the skeletal muscle of adult immunocompetent mice. Hum. Gene Ther.8:1891–1900.
49. Snyder, R. O., X. Xiao, and J. Samulski.1996. Production of recombinant adeno-associated viral vectors, p. 12.1.1–12.2.23.InN. Dracopoli, J. Haines, B. Krof, D. Moir, C. Seidman, and J. S. Seidman (ed.), Current protocols in human genetics. John Wiley & Sons, New York, N.Y.
50. Song, S., P. J. Laipis, K. I. Berns, and T. R. Flotte.2001. Effect of DNA-dependent protein kinase on the molecular fate of the rAAV2 genome in skeletal muscle Proc. Natl. Acad. Sci. USA98:4084–4088.
51. Summerford, C., J. S. Bartlett, and R. J. Samulski.1999. AlphaVbeta5 integrin: a coreceptor for adeno-associated virus type 2 infection. Nat. Med.
5:78–82.
52. Summerford, C., and R. J. Samulski.1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol.72:1438–1445.
53. Tafuro, S., L. Zentilin, A. Falaschi, and M. Giacca.1996. Rapid retrovirus titration using competitive polymerase chain reaction. Gene Ther.3:679– 684.
54. Tauer, T. J., M. H. Schneiderman, J. K. Vishwanatha, and S. L. Rhode.1996. DNA double-stranded break repair functions defend against parvovirus in-fection. J. Virol.70:6446–6449.
55. Van Dyck, E., A. Z. Stasiak, A. Stasiak, and S. C. West.1999. Binding of double-stranded breaks in DNA by human Rad52 protein. Nature398:728– 731.
56. Vincent-Lacaze, N., R. O. Snyder, R. Gluzman, D. Bohl, C. Lagarde, and O. Danos. 1999. Structure of adeno-associated virus vector DNA following transduction of the skeletal muscle. J. Virol.73:1949–1955.
57. Xiao, W., S. C. Berta, M. M. Lu, A. D. Moscioni, J. Tazelaar, and J. M. Wilson.1998. Adeno-associated virus as a vector for liver-directed gene therapy. J. Virol.72:10222–10226.
58. Xiao, X.1996. Efficient long-term gene transfer into muscle tissue of immu-nocompetent mice by adeno-associated virus vector. J. Virol.70:8098–8108. 59. Xiao, X., J. Li, T. J. McCown, and R. J. Samulski.1997. Gene transfer by adeno-associated virus vectors into the central nervous system. Exp. Neurol.
144:113–124.
60. Yang, J., W. Zhou, Y. Zhang, T. Zidon, T. Ritchie, and J. F. Engelhardt.
1999. Concatamerization of adeno-associated virus circular genomes occurs through intermolecular recombination. J. Virol.73:9468–9477.
61. Zolotukhin, S., M. Potter, W. W. Hauswirth, J. Guy, and N. Muzyczka.1996. A “humanized” green fluorescent protein cDNA adapted for high-level expression in mammalian cells. J. Virol.70:4646–4654.