Copyright © 2004, American Society for Microbiology. All Rights Reserved.
The N- and C-Terminal Domains of the NS1 Protein of Influenza B
Virus Can Independently Inhibit IRF-3 and Beta Interferon
Promoter Activation
Nicola R. Donelan,
1,2Bianca Dauber,
3Xiuyan Wang,
1,2† Christopher F. Basler,
1Thorsten Wolff,
3and Adolfo García-Sastre
1*
Department of Microbiology1and Microbiology Graduate School Training Program,2Mount Sinai
School of Medicine, New York, New York, and Robert Koch-Institut, Berlin, Germany3
Received 30 March 2004/Accepted 10 June 2004
The NS1 proteins of influenza A and B viruses (A/NS1 and B/NS1 proteins) have only⬃20% amino acid sequence identity. Nevertheless, these proteins show several functional similarities, such as their ability to bind to the same RNA targets and to inhibit the activation of protein kinase R in vitro. A critical function of the A/NS1 protein is the inhibition of synthesis of alpha/beta interferon (IFN-␣/) during viral infection. Recently, it was also found that the B/NS1 protein inhibits IFN-␣/synthesis in virus-infected cells. We have now found that the expression of the B/NS1 protein complements the growth of an influenza A virus with A/NS1 deleted. Expression of the full-length B/NS1 protein (281 amino acids), as well as either its N-terminal RNA-binding domain (amino acids 1 to 93) or C-terminal domain (amino acids 94 to 281), in the absence of any other influenza B virus proteins resulted in the inhibition of IRF-3 nuclear translocation and IFN-promoter ac-tivation. A mutational analysis of the truncated B/NS1(1-93) protein showed that RNA-binding activity cor-related with IFN-promoter inhibition. In addition, a recombinant influenza B virus with NS1 deleted induces higher levels of IRF-3 activation, as determined by its nuclear translocation, and of IFN-␣/synthesis than wild-type influenza B virus. Our results support the hypothesis that the NS1 protein of influenza B virus plays an important role in antagonizing the IRF-3- and IFN-induced antiviral host responses to virus infection.
Influenza A and B viruses are globally distributed pathogens that cause yearly epidemics of respiratory disease in humans. The recommended trivalent influenza vaccine includes anti-gens from recently circulating strains of influenza A and B viruses. The vaccines, together with antiviral therapies such as rimantadine and neuraminidase inhibitors, offer some options for the prophylaxis and treatment of influenza. Despite the availability of these prophylactic and therapeutic measures, influenza viruses continue to be a significant cause of death associated with pneumonia and in the United States alone are responsible for thousands of deaths every winter (29). There remain many unanswered questions as to the factors respon-sible for virulence, especially for influenza B viruses. Under-standing the molecular mechanisms that contribute to viral virulence is likely to help in the design of novel therapeutic countermeasures.
One viral protein that is a candidate virulence factor of influenza B virus is the nonstructural protein 1 (B/NS1). The NS1 protein of influenza A virus (A/NS1) has been shown to have interferon antagonist properties that give the virus an advantage in its fight against the antiviral host response to infection. This effect is mediated at least in part by the ability of the A/NS1 protein to inhibit activation of IRF-3, a tran-scription factor required for the induction of beta interferon (IFN-) synthesis (13, 23, 25, 34, 35) Recent data (6, 28) have
provided evidence that the B/NS1 protein also functions as an interferon antagonist, required for inhibition of IFN synthesis and for efficient viral growth. It was also reported that both the A/NS1 and B/NS1 proteins could block the activation of the IFN-inducible protein protein kinase R (PKR) in vitro (30). Additional studies showed that the B/NS1 protein, and not the A/NS1 protein, blocks the conjugation of ISG15 to its target proteins both in vitro and in virus-infected cells (37). ISG15 is a ubiquitin-like protein whose expression is transcriptionally induced by IFN-␣/. The B/NS1 protein appears to bind to and inhibit the activity of ISG15, although the biological signifi-cance of this is still unknown (36). A second functional distinc-tion between the A/NS1 and B/NS1 proteins is that the B/NS1 protein seems to lack a carboxy-terminal effector domain sim-ilar to the one found in the A/NS1 protein. As a result, the B/NS1 protein does not inhibit cellular mRNA polyadenyla-tion, splicing, and nuclear export (30).
Studies have shown that the first 93 amino-terminal amino acids of the B/NS1 protein can bind in vitro to the same RNA targets as the first 73 amino-terminal amino acids of the A/NS1 protein. Also, the first 93 amino acids of the B/NS1 protein form dimers, similar to the first 73 amino acids of the A/NS1 protein (30). The crystal structure of the B/NS1 protein has not yet been solved, but computer modeling based on the crystal structure of the N-terminal domain of the A/NS1 protein has allowed alignment of the N-terminal RNA-binding domains of the two proteins, which are thought to possess similar␣-helical structures (36). We performed a mutational analysis of the RNA-binding domain of B/NS1 to determine the amino acid residues required for RNA binding and inhibition of IFN syn-thesis. We also tested independently the abilities of the
N-* Corresponding author. Mailing address: Department of Microbi-ology, Box 1124, Mount Sinai School of Medicine, 1 Gustave L. Levy Pl., New York, NY 10029. Phone: (212) 241-7769. Fax: (212) 534-1684. E-mail: adolfo.garcia-sastre@mssm.edu.
† Present address: Section of Immunobiology, Yale University School of Medicine, New Haven, CT 06520.
11574
on November 8, 2019 by guest
http://jvi.asm.org/
terminal and C-terminal domains of B/NS1 to inhibit IFN-
promoter and IRF-3 activation. Finally, we show that in the con-text of influenza B virus infection, elimination of the B/NS1 gene results in enhanced IRF-3 nuclear translocation (activation), and this correlates with higher levels of IFN-␣/induction.
MATERIALS AND METHODS
Cells and viruses.293T, MDCK, Vero, and A549 cells were maintained in Dulbecco modified Eagle medium with 10% fetal bovine serum. Sendai virus, strain Cantell, was propagated at 37°C in 10-day-old embryonated chicken eggs. The construction and growth of Newcastle disease virus-green fluorescent pro-tein (NDV-GFP) was previously described (21). Recombinant wild-type influ-enza B/Lee/40 (rec WT B/Lee) virus and B/Lee/40 virus with NS1 deleted (del NS1 B/Lee virus) were generated by reverse genetics using the transfection of eight plasmids as previously described. The del NS1 B/Lee virus is isogenic to the rec WT B/Lee virus except for the NS gene, in which the NS1-specific sequences were deleted (6). An influenza A virus that lacked expression of the NS1 protein (del NS1/A virus) was generated as previously described (10). Recombinant WT B/Lee virus was grown in 10-day-old embryonated chicken eggs. Del NS1/A and del NS1 B/Lee viruses were grown in 7-day-old embryonated chicken eggs.
Plasmids and GST fusion protein expression.The A/NS1 expression plasmid pCAGGS-PR8 NS1 SAM was previously described (27). Mammalian expres-sion plasmids for B/NS1 (full-length and truncated mutants) were constructed by subcloning the B/NS1 cDNAs from influenza B/Yamagata/1/73 virus into the pCAGGS vector (18) between the EcoRI and XhoI sites. The plasmid pEGFP-C1-hIRF3 expresses human IRF-3 fused to GFP and was constructed by PCR amplification of human IRF-3 and insertion of the hIRF-3 open reading frame (ORF) in frame with the GFP ORF in pEGFP-C1 (Clontech). Glutathione
S-transferase (GST) expression plasmids for the B/NS1 proteins were
con-structed as follows. The plasmid pGEX-B/NS1 encodes a protein consisting of GST fused to the NS1 protein of influenza B/Yamagata/1/73 virus (281 amino acids). The wild-type NS1 cDNA was subcloned into the pGEX-5X-1 vector between the XhoI and EcoRI restriction sites. The pGEX-B/NS1(1-93) plasmid was constructed by in-frame insertion of the GST ORF in front of the coding region of the first 93 amino acids of the B/NS1 protein. Similarly, pGEX-B/ NS1(94-81) was constructed by fusing the GST ORF to a cDNA encoding amino acids 94 to 281 of the B/NS1 protein. Using site-directed mutagenesis, we re-placed, in groups of two or three, each of the codons corresponding to the 16 basic amino acid residues within the N-terminal RNA-binding domain with those encoding alanines. A total of seven alanine replacement mutant proteins in the context of a full-length B/NS1 protein were generated: GST-B/NS1 33/38, 47/50, 52/53/54, 58/60/64, 70/71, 77/78, and 83/86. These fusion proteins were expressed inEscherichia coliBL21 and purified using glutathione-Sepharose beads (Phar-macia) as recommended by the manufacturer. B/NS1 mutant cDNAs were also cloned into pCAGGS vectors for expression in mammalian cells.
EMSA.Electrophoretic mobility shift assays (EMSAs) for B/NS1 binding to double-stranded RNA (dsRNA) were done as previously described (31) with
slight modifications. Briefly,32P-radiolabeled short dsRNA was generated by
annealing complementary SP6 (76 nucleotides) and T7 (80 nucleotides) tran-scripts from the pGEM-11zf vector linearized with SfiI and HindIII, respectively. Gel shift experiments to investigate RNA-binding activity were then performed.
Briefly, 0.4M GST fusion proteins was incubated on ice for 30 min in 20l of
binding buffer (43 mM Tris-HCl, pH 8, 50 mM KCl, 8% glycerol, 2.5 mM
dithiothreitol, 50g ofE. colitRNA/ml, 0.5 U of RNasin/l) containing
radio-labeled dsRNA (10,000 cpm; 1 nM). The protein-RNA complexes were resolved from free RNA by running them on nondenaturing 6% polyacrylamide gels at 4°C for 3 h at 150 V in 0.045 M Tris-borate–0.001 M EDTA running buffer. The gels were dried, and the RNA-containing bands were visualized by autoradiog-raphy.
Del NS1/A virus complementation assay.The del NS1/A virus complementa-tion assay was performed as previously described (1). Briefly, MDCK cells
were transfected with 4.0g of pCAGGS-empty plasmid, pCAGGS-A/NS1 or
pCAGGS-B/NS1 using Lipofectamine 2000 (GIBCO/BRL). At 20 h posttrans-fection, the cells were infected with del NS1/A virus at a multiplicity of infection (MOI) of 0.001. At 8, 24, 36, and 48 h postinfection, the titer of virus in the supernatant was measured by plaque assay on fresh MDCK cells.
Reporter gene assays in 293T cells.For the experiments investigating IFN-
promoter activation, 293T cells were transfected with a mouse IFN-
promoter-driven chloramphenicol acetyltransferase (CAT) reporter plasmid, pIFN-CAT (33). In addition, an internal-control plasmid, pGL2-control (Promega), encod-ing the firefly luciferase protein, and the appropriate pCAGGS-B/NS1
expres-sion plasmid were transfected into the cells. The CAT reporter (0.5g) and 0.5
g of the luciferase plasmid were transfected, along with 4.0g of the
pCAGGS-B/NS1 (wild-type or mutant) plasmid, into 106293T cells using a calcium
phos-phate mammalian transfection kit (Stratagene). Twenty-four hours
posttrans-fection, the cells were infected with Sendai virus at an MOI of⬃10; 24 h
postinfection, the cells were harvested and assayed for CAT expression as pre-viously described (22). Luciferase protein expression was assayed using the Pro-mega luciferase assay system according to the manufacturer’s directions. Levels of CAT activity were normalized among samples according to the luciferase values.
Quantification of IFN-␣/by bioassay.The levels of IFN-␣/secreted by virus-infected cells were determined as previously described (20). A549 cells were either mock infected or infected at an MOI of 2 with WT B/Lee or del NS1 B/Lee virus. Following infection, the cells were incubated with minimal essential medium containing 0.3% bovine serum albumin, and 24 h postinfection, the supernatants were harvested. Viruses present in the supernatant were UV inac-tivated by placing samples on ice 6 in below an 8-W UV lamp for 15 min with constant stirring. Vero cells were seeded in 24-well plates the day before and incubated with the UV-inactivated supernatants for 24 h. The preincubated Vero
cells were then infected with NDV-GFP (MOI⫽2), and 24 h postinfection, the
cells expressing GFP were visualized by fluorescence microscopy. A decrease in
GFP expression was indicative of an antiviral state induced by IFN-␣/present
in A549 cell supernatants.
Quantification of human IFN-by ELISA.A549 cells (106) were either mock
infected or infected at an MOI of 1 with WT B/Lee or del NS1 B/Lee virus. At 18 h postinfection, 1 ml of supernatant was removed and tested for levels of
human IFN-by enzyme-linked immunosorbent assay (ELISA) (human IFN-
ELISA kit; FUJIREBIO Inc., Tokyo, Japan).
Immunofluorescence.Cells were fixed and permeabilized for 20 min at room temperature with 2.5% formaldehyde–0.1% Triton X-100. The fixed cells were washed extensively with phosphate-buffered saline (PBS) and then blocked with 3% milk in PBS–0.1% Triton X-100 for 3 h at room temperature. To visualize B/NS1 protein expression, the cells were then incubated with a rabbit polyclonal antibody directed against the influenza B virus NS1 protein (19) (dilution, 1:200) for 30 min. After being washed two or three times with PBS–0.1% Triton X-100, the cells were incubated for 30 min with Texas red-conjugated donkey anti-rabbit
immunoglobulin G (H⫹L) secondary antibodies (dilution, 1:200; Jackson
Im-munoResearch Laboratories); all antibodies were diluted in PBS–0.1% Triton X-100. The cells were visualized using an Olympus IX-70 fluorescence micro-scope fitted with a Sony Catseye digital camera. For visualizing influenza B virus-infected cells, an NP-specific monoclonal antibody, BM3149 (1:300; DPC Biermann, Bad Nauheim, Germany) was used, followed by a secondary
Alexa-Fluor 594-conjugated goat anti-mouse immunoglobulin G (H⫹L) (1:750;
Mo-lecular Probes). The cells were viewed under a Nikon Axiophot 3000 microscope, and images were recorded by a SPOT RT digital camera.
IRF-3 translocation assay.Vero cells (⬃105) were transfected with 0.5g of
pEGFP-C1-hIRF3 and 1.0 g of pCAGGS-B/NS1, pCAGGS-B/NS1(1-93),
pCAGGS-B/NS1(94-281), or the empty vector (pCAGGS) using Lipofectamine 2000 as instructed by the manufacturer. Transfected cells were incubated at 37°C
for 24 h and then infected at an MOI of⬃10 with Sendai Cantell virus. Infections
were performed in PBS for 1 h at 37°C. After 1 h, medium (Dulbecco modified Eagle medium with 10% fetal bovine serum) was added, and the mixture was incubated at 37°C for 12 h. IRF-3 localization was then visualized by fluorescence microscopy in cells that were fixed and immunostained using a monoclonal antibody against the B/NS1 protein. In experiments to determine IRF-3 trans-location in influenza B virus-infected cells, MDCK cells were transfected with pEGFP-C1-hIRF3 and either mock infected or infected at an MOI of 1 with recombinant WT B/Lee or del NS1 B/Lee virus. The cells were stained for the viral NP protein at 8 h postinfection and analyzed by immunofluorescence microscopy. The percentage of cells with nuclear hIRF-3/GFP was calculated
based on counting⬃100 cells in several different fields at⫻40 magnification
(only cells expressing the B/NS1 proteins or viral NP protein were counted).
RESULTS
Expression of the B/NS1 protein complements the growth of del NS1/A virus.It has been shown that growth of a del NS1/A virus is impaired in IFN-competent systems, such as 10-day-old embryonated eggs and MDCK cells (10). We have developed a complementation assay in which we express the A/NS1 protein intrans by transfecting MDCK cells with an NS1 protein
on November 8, 2019 by guest
http://jvi.asm.org/
pression plasmid (pCAGGS-A/NS1), and as a result, these cells support del NS1/A virus replication (1). In order to de-termine whether expression of the B/NS1 protein complements the growth of del NS1/A virus, we transfected MDCK cells with pCAGGS-B/NS1 plasmid and 20 h later infected them with del NS1/A virus at a low MOI (0.001). Expression of the B/NS1 protein was able to complement the growth of the del NS1/A virus to levels slightly lower than those seen when the A/NS1 protein was expressed intrans. Figure 1 shows that transfection of expression plasmids for the A/NS1 or B/NS1 protein allowed the del NS1/A virus to grow to titers that were 3 to 4 log units higher than in cells transfected with the empty vector. These results indicate that the NS1 protein of influenza B virus can functionally replace the NS1 protein of influenza A virus in this assay. These observations are also in agreement with the pre-viously demonstrated ability of the B/NS1 protein to prevent the activation of the IFN-promoter by del NS1/A virus (6).
Expression of the B/NS1 protein inhibits IFN-promoter activation and IRF-3 nuclear translocation in the absence of any other influenza B virus proteins. The A/NS1 protein is known to inhibit IFN- promoter activation in Sendai virus-infected cells (33). We next investigated whether the B/NS1 protein behaves in a similar fashion. In order to measure
IFN-promoter activation, we transfected 293T cells with a CAT reporter plasmid under the control of a mouse IFN- pro-moter, together with a B/NS1 expression plasmid, pCAGGS-B/NS1. Twenty-four hours posttransfection, we infected the cells with Sendai virus, a known potent inducer of the IFN-
promoter. Plasmid-mediated expression of the B/NS1 protein effectively inhibits Sendai virus-induced IFN-promoter acti-vation, as seen in Fig. 2A, column 3. Stimulation of the IFN-
promoter is mediated by the activation of several transcription factors, including IRF-3. When activated, IRF-3 becomes hy-perphosphorylated and translocates to the nucleus, where it participates in the transcriptional activation of the IFN-gene (8, 26). Next, we tested the ability of the B/NS1 protein to inhibit Sendai virus-induced hIRF-3/GFP nuclear transloca-tion. Consistent with the results from the IFN-promoter ac-tivation studies, the B/NS1 protein prevented hIRF-3/GFP translocation to the nucleus (i.e., activation), as seen in Fig. 2C (B/NS1 WT⫹Sendai panels).
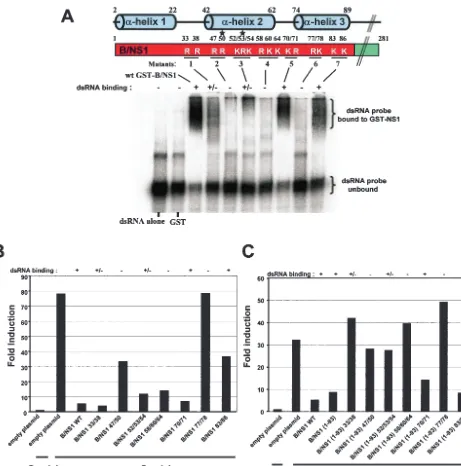
Multiple basic amino acids within the N-terminal domain of the B/NS1 protein contribute to dsRNA binding.The structure of the N-terminal amino acids 1 to 73 that compose the RNA-binding and dimerization domain of the NS1 protein of influ-enza A virus has been described (4, 14). Based on the three-dimensional crystal structure, it was proposed that two basic amino acids, R38 and K41, interact directly with RNA mole-cules to mediate binding. When these basic amino residues were mutated to alanines, the resulting protein was defective in RNA binding (31). Recent biophysical evidence confirms the roles that these basic amino acids play in RNA binding (5). The crystal structure of the B/NS1 protein has not yet been solved. Nevertheless, it is known that N-terminal amino acids 1 to 93 comprise the RNA-binding domain of this protein (30). More-over, homology modeling has predicted that the RNA-binding domain of the B/NS1 protein has a dimeric sixfold ␣-helical structure similar to the A/NS1 protein (31). We have analyzed the contributions to RNA binding of the basic amino acids within the N-terminal domain of the B/NS1 protein. A total of seven mutant B/NS1 proteins were generated in which either two or three basic amino acids were replaced with alanines (Fig. 3A). Each of these mutant proteins was expressed in bacteria as a GST fusion protein and tested for binding to dsRNA by EMSA in vitro. Essentially all of the alanine mu-tants showed impaired dsRNA binding except for B/NS1 70/71 and B/NS1 83/86, and therefore, many of the basic amino acids that were important for dsRNA binding were located within the second␣-helix.
The ability of the B-NS1 protein to inhibit Sendai virus-induced IFN-promoter activation is not dependent on bind-ing to dsRNA.We next went on to test the ability of each of the full-length mutant B/NS1 proteins to inhibit Sendai virus-in-duced IFN-promoter activation by using a transfection-based reporter assay (Fig. 3B). Among all of the mutants, only B/NS1 77/78 was completely inactive in its ability to inhibit IFN-
promoter activation. Interestingly, many of the mutant B/NS1 proteins that were attenuated for binding to dsRNA, like B/ NS1 33/38 and B/NS1 58/60/64, were still able to inhibit Sendai virus-induced promoter activation to levels similar to those of wild-type B/NS1 protein. This was unlike the behavior of the NS1 protein of influenza A virus, which was previously found to be impaired in blocking IFN-promoter activation by mu-tations that adversely affected RNA binding (33).
[image:3.603.60.264.68.196.2]Both the N-terminal and C-terminal domains of the B/NS1 protein inhibit IFN-promoter activation and IRF-3 nuclear translocation.Since an RNA-binding-defective protein such as B/NS1 58/60/64 was still a good inhibitor of IFN- promoter activation, we hypothesized that the C-terminal domain might be involved in this inhibitory activity. Therefore, we tested separately the abilities of the N- and C-terminal portions of the B/NS1 protein to inhibit Sendai virus-induced IFN-promoter activation. The results (Fig. 2A) show that both the N-terminal RNA-binding domain and the C-terminal domain of the B/NS1 protein are independently able to inhibit IFN- pro-moter activation. Next, we tested the abilities of the full-length and N- and C-terminally truncated B/NS1 proteins to inhibit Sendai virus-induced hIRF-3/GFP activation (Fig. 2C and Ta-ble 1). Consistent with the results for the IFN- promoter activation, both the N- and C-terminal domains of the B/NS1 protein were able to inhibit hIRF-3/GFP translocation to
FIG. 1. Expression of the NS1 protein of influenza B virus comple-ments the growth of del NS1/A virus in MDCK cells. MDCK cells were transfected with empty plasmid, pCAGGS-A/NS1, or pCAGGS-B/ NS1. Twenty-four hours posttransfection, the cells were infected with del NS1 A/PR/8 virus at an MOI of 0.001. The virus titer in the super-nantant was measured at different times postinfection.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 2. Functional characterization of full-length B/NS1, B/NS1(1-93), and B/NS1(94-281) proteins. (A) IFN-promoter activation is inhibited by expression of full-length B/NS1, as well as by the expression of either the N- or C-terminal region of the B/NS1 protein. Briefly, 0.5g of the IFN-promoter-driven CAT reporter and 0.5g of the luciferase plasmid were transfected, along with 4.0g of empty plasmid or the pCAGGS-B/NS1, pCAGGS B/NS1(1-93), or pCAGGS B/NS1(94-281) plasmid into 106293T cells. Where indicated (⫹Sendai), 24 h posttransfection, the
cells were infected with Sendai virus at a high MOI (⬃10). Twenty-four hours postinfection, cells were harvested and assayed for CAT and luciferase expression. Theyaxis represents the induction of CAT activity after normalization using the luciferase values. (B) The N-terminal amino acids 1 to 93 possess the RNA-binding activity of the full-length protein. The N- and C-terminal portions of the B/NS1 protein were expressed as GST-B/NS1(1-93) and GST-B/NS1(94-281). Binding of the GST-B/NS1 proteins to dsRNA was tested in vitro. Briefly, an EMSA was performed by incubating 0.4M (each) GST-NS1 fusion protein with32P-radiolabeled short dsRNA. The protein-RNA complexes were then resolved from
free RNA by running them on 6% nondenaturing polyacrylamide gels at 4°C for 3 h at 150 V in 0.045 M Tris-borate–0.001 M EDTA running buffer. (C) hIRF-3/GFP nuclear translocation is inhibited by the expression of full-length B/NS1, as well as by the expression of either the N- or C-terminal region of the B/NS1 protein. Vero cells were cotransfected with pEGFP-C1-hIRF3 and empty plasmid, pCAGGS-B/NS1, pCAGGS-B/NS1(1-93), or pCAGGS-B/NS1(94-281). Where indicated, 24 h posttransfection, the cells were infected with Sendai virus at an MOI of⬃10. Twelve hours postinfection, the cellular localization of the hIRF-3/GFP protein was monitored by fluorescence microscopy. Expression of the B/NS1 wild-type and mutant proteins was shown by immunofluorescence (on the right) by using an anti-B/NS1 specific polyclonal antibody (␣-B/NS1).
on November 8, 2019 by guest
http://jvi.asm.org/
the nucleus (i.e., activation). However, only the N-terminal domain of the B/NS1 protein is able to efficiently bind to dsRNA, similarly to the full-length B/NS1 protein, as shown in Fig. 2B.
[image:5.603.63.524.63.529.2]In the context of the truncated B/NS1(1-93) protein, binding to dsRNA is necessary for the inhibition of Sendai virus-induced IFN-promoter activation.We next investigated the impacts of alanine mutations of the basic amino acids on the
FIG. 3. Role of the RNA-binding activity of the B/NS1 protein in the inhibition of IFN-promoter activation. (A) Basic amino acids within the RNA-binding N-terminal domain of the B/NS1 protein that are required for binding to dsRNA in vitro. A schematic diagram of the full-length B/NS1 protein with the basic amino acid residues within the RNA-binding domain is shown. Predicted␣-helical regions are shown, and the stars indicate basic amino acid residues previously shown to be involved in RNA binding (36). Basic amino acids within the RNA-binding domain of B/NS1 were mutated to alanines, and binding of GST-B/NS1 wild-type and mutant proteins to dsRNA was tested in vitro. Briefly, EMSA was performed by incubating 0.4M (each) GST-NS1 fusion protein with32P-radiolabeled short dsRNAs. The protein-RNA complexes were then resolved from free
RNA by running them on 6% nondenaturing polyacrylamide gels at 4°C for 3 h at 150 V in 0.045 M Tris-borate–0.001 M EDTA running buffer. (B) The ability of the wild-type and mutant full-length B-NS1 proteins to inhibit Sendai virus-induced IFN-promoter activation is not dependent on RNA binding. Empty plasmid (4.0g) or the pCAGGS-B/NS1 (wild type or mutant) was cotransfected with 0.5g of a CAT reporter construct under the control of the IFN-promoter, together with 0.5g of a control luciferase plasmid into 106293T cells. Where indicated (⫹Sendai), 24 h
posttransfection, the cells were infected with Sendai virus at a high MOI (⬃10). Twenty-four hours postinfection, cells were harvested and assayed for CAT and luciferase expression. Induction of the reporter gene activity was measured as previously described for panel A.⫹, strong dsRNA binding activity;⫹/⫺, weak dsRNA binding activity;⫺, no dsRNA activity. (C) In the context of the B-NS1(1-93) protein, binding to dsRNA is necessary for the inhibition of Sendai virus-induced IFN-promoter activation. B/NS1(1-93) wild-type and mutant proteins were tested for the ability to inhibit IFN-promoter activation using the same reporter assay described for panel A.
on November 8, 2019 by guest
http://jvi.asm.org/
inhibition of IFN-promoter activation in the context of the truncated B/NS1(1-93) protein. As shown in Fig. 3C, there is a direct correlation between the ability of the B/NS1(1-93) pro-tein to bind to dsRNA and its ability to inhibit IFN-promoter activation. Only the B/NS1(1-93) 70/71 and B/NS1(1-93) 83/86 mutant proteins, which are not impaired in binding to dsRNA, inhibit IFN-promoter activation at levels similar to the B/NS1 (1-93) protein.
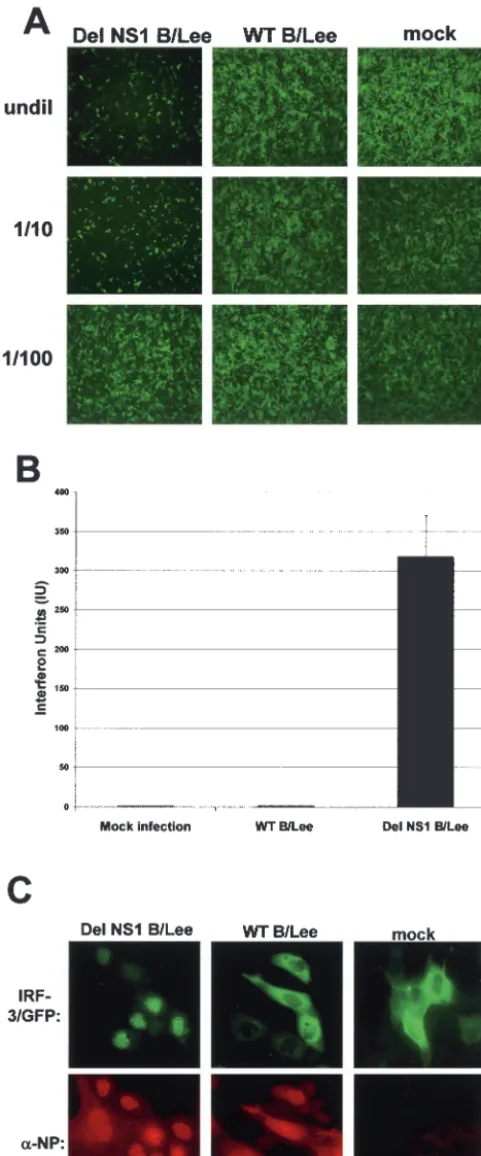
Del NS1/B virus infection of A549 cells induces significantly higher levels of IFN-␣/ production and of IRF-3 nuclear translocation than does wild-type influenza B virus infection.
Thus far, we have shown that the B/NS1 protein acts as an IFN antagonist based on its ability to inhibit IRF-3 and IFN-
promoter activation when expressed by plasmid transfection. A recombinant influenza B virus that does not express the NS1 protein (del NS1 B/Lee) was generated by reverse genetics, and this provided us with an invaluable tool for studying the function of the B/NS1 protein in the context of a viral infection (6). We compared IFN-␣/induction and hIRF-3/GFP nuclear translocation in cells infected with WT B/Lee or del NS1 B/Lee virus. Wild-type influenza B virus infection does not induce high levels of IFN-␣/production, as determined by using both a bioassay and an ELISA kit for human IFN-. (Fig. 4A and B). However, when cells are infected with del NS1 B/Lee virus, there is a significant induction of IFN-␣/production (Fig. 4A, left, and B, third column). Next, we monitored the activation of IRF-3 in cells that were infected with either WT B/Lee or del NS1 B/Lee virus using the hIRF-3/GFP-based translocation assay. Figure 4C and Table 1 show that at 8 h postinfection in del NS1 B/Lee virus-infected cells, the hIRF-3/GFP protein translocates to the nucleus, while the majority of cells infected with WT B/Lee virus have nonactivated, cytoplasmic hIRF-3/ GFP.
DISCUSSION
In this report, we have shown that the B/NS1 protein func-tions as an IFN antagonist. The B/NS1 protein prevents the nuclear translocation of IRF-3 and IFN-promoter induction in virus-infected cells. These properties are similar to those of the A/NS1 protein that acts at the level of inhibition of IFN-␣/
 synthesis by virtue of its ability to inhibit virus-induced IRF-3, NF-B, and c-Jun/ATF2 activation (16, 27, 33). The fact that these two proteins show very little sequence identity yet have such similar functions in the viral life cycle emphasizes
the importance of IFN antagonist proteins in determining the outcome of virus-host interactions. The A/NS1 and B/NS1 proteins share the ability to bind to dsRNA via an N-terminal RNA-binding and dimerization domain, and the ability of the A/NS1 protein to bind to dsRNA is required for optimal IFN antagonist activity (7). In this study, we investigated the role of the dsRNA-binding activity of the B/NS1 protein in its IFN antagonist function.
We have shown that the N- and C-terminal domains of the NS1/B protein can independently inhibit IFN-promoter ac-tivation and hIRF-3/GFP nuclear translocation. There is a correlation between the IFN antagonist activity of the N-ter-minal domain of the B/NS1 protein and its ability to bind dsRNA, suggesting that the sequestration of dsRNA during viral infection may be the mechanism by which the N-terminal RNA-binding domain of B/NS1 functions as an IFN antago-nist. As expected, binding to dsRNA is greatly impaired by the deletion of the B/NS1(1-93) N-terminal domain. Therefore, we propose that the mechanism for IFN antagonist activity for the C-terminal region of the B/NS1 protein is independent of RNA binding. An RNA-binding-independent IFN antagonist function was also demonstrated for the A/NS1 protein, in which a specific mutant A/NS1 protein was found to be defec-tive for binding to dsRNA but still maintained partial IFN antagonist activity (7). Likewise, there seems to be both RNA-binding-dependent (N-terminal) and -independent (C-termi-nal) mechanisms for mediating the IFN antagonist function of the B/NS1 protein. We suggest a mechanism whereby both the N- and C-terminal domains of the B/NS1 protein cooperate to produce a strong IFN antagonist. However, from our studies, we are unable to determine at this point what the relative contribution of each domain of the B/NS1 protein is to the IFN antagonist activity.
[image:6.603.42.544.82.175.2]Mutational analysis of the basic amino acid residues within the RNA-binding and dimerization domain of the B/NS1 pro-tein revealed that binding to dsRNA was adversely affected by many of these mutations. Most of the RNA-binding-defective mutants resulted from mutations of basic amino acids present in the predicted second ␣-helix, as shown in Fig. 3A. The resulting loss of dsRNA binding of the mutant B/NS1 proteins could be accounted for by several different possibilities. The alanine mutations could result in a loss of electrostatic inter-actions between the B/NS1 protein and dsRNA. It has been reported that the basic amino acids R38 and K41 within the
TABLE 1. Quantification of nuclear accumulation of hIRF-3/GFP
Cell line Transfected plasmid(s)a Virus infectionb % Nuclear hIRF-3/GFPc
Vero pEGFP-C1-hIRF-3⫹pCAGGS-empty Mock 8
Vero pEGFP-C1-hIRF-3⫹pCAGGS-empty Sendai virus 97
Vero pEGFP-C1-hIRF-3⫹pCAGGS-B/NS1 Sendai virus 3d
Vero pEGFP-C1-hIRF-3⫹pCAGGS-B/NS1(1–93) Sendai virus 6d
Vero pEGFP-C1-hIRF-3⫹pCAGGS-B/NS1(94–281) Sendai virus 6d
MDCK pEGFP-C1-hIRF-3 Mock 4
MDCK pEGFP-C1-hIRF-3 WT B/Lee 4e
MDCK pEGFP-C1-hIRF-3 Del NS1 B/Lee 82e
aCells were transfected with the indicated plasmids as described in Materials and Methods.
bTwenty-four hours posttransfection, cells were either mock infected or infected with Sendai (MOI⫽10), WT B/Lee (MOI⫽1), or del NS1 B/Lee (MOI⫽1) virus.
cThe percentage of cells with nuclear hIRF-3/GFP was calculated based on counting⬃100 cells in several different fields at⫻40 magnification.
dOnly cells expressing the corresponding B/NS1 proteins as determined by immunofluorescence were taken into consideration.
eOnly cells expressing the viral NP protein as determined by immunofluorescence were taken into consideration.
on November 8, 2019 by guest
http://jvi.asm.org/
RNA-binding domain of the A/NS1 protein directly interact with the dsRNA target. Mutation of these basic amino acids to alanine residues results in an A/NS1 R38AK41A protein that is defective in binding to dsRNA, apparently due to the loss of electrostatic interactions between the dsRNA and the A/NS1 protein (31). Homology modeling of the RNA-binding do-mains of the A/NS1 and B/NS1 proteins revealed that the analogous basic amino acids within the B/NS1 protein are residues R50 and R53, and likewise, mutation of these residues to alanine results in a B/NS1 protein that is defective in binding to dsRNA (36). Although in our studies we did not construct a mutant B/NS1 50/53 protein, both the B/NS1 47/50 and B/NS1 52/53/54 proteins are greatly impaired in their ability to bind to dsRNA.
Another plausible explanation for the loss of dsRNA bind-ing may be that the alanine mutations disrupt dimer formation of the B/NS1 protein. Like the A/NS1 protein, the dimerization of the RNA-binding domain of the B/NS1 protein is required for RNA binding (31). Further analyses will have to be carried out in order to determine the role that the basic amino acids within the RNA-binding domain play in B/NS1 protein dimer formation. Finally, it is also possible that mutation of the basic amino acid residues within the RNA-binding domain of the B/NS1 protein have a significant effect on the overall three-dimensional structure of the protein. With respect to this pos-sibility, we consider the mutant B/NS1 77/78, which unlike the other RNA-binding-defective mutants lost all ability to inhibit IFN-promoter activation. This complete loss of activity of both the N- and C-terminal domains of the B/NS1 protein may be due to a major structural rearrangement resulting in a non-functional protein.
[image:7.603.44.285.68.646.2]The finding that the C-terminal region (amino acids 94 to 281) of the B/NS1 protein can independently inhibit Sendai virus-induced IFN- promoter activation and IRF-3 nuclear translocation was quite intriguing. This is quite distinct from the proposed role of the C-terminal effector domain of the A/NS1 protein that is involved in stabilization and/or facilita-tion of A/NS1 dimer formafacilita-tion, thereby promoting the RNA-binding function of the A/NS1 amino terminus (32). The C-terminal effector domain of the A/NS1 protein has also been implicated in the inhibition of host mRNA splicing (12, 15), polyadenylation (3, 17), and nuclear export (2, 9, 24), all func-tions that are not shared with the C-terminal domain of the B/NS1 protein. Binding to dsRNA is greatly impaired by the deletion of the B/NS1(1-93) N-terminal domain. Therefore, we
FIG. 4. Characterization of an influenza B virus that does not ex-press NS1 protein, del NS1 B/Lee virus. (A) Del NS1 B/Lee virus infection of A549 cells induces significantly higher levels of IFN-␣/ production than wild-type influenza B virus infection. Vero cells were pretreated for 24 h with UV-inactivated supernatant (at the dilutions shown on the left; undil, undiluted) from A549 cells either mock infected or infected with the del NS1 B/Lee or WT B/Lee virus at an MOI of 2. The pretreated Vero cells were then infected with
NDV-GFP, and 24 h postinfection, the cells expressing GFP were monitored by fluorescence microscopy. The presence of IFN-␣/in the supernatant is evidenced by a decrease in the number and intensity of cells expressing GFP. (B) Quantification of IFN-production in virus-infected cells by ELISA. A549 cells were either mock virus-infected or infected with the del NS1 B/Lee or WT B/Lee virus at an MOI of 1. Eighteen hours postinfection, the supernatant was removed and tested by ELISA for levels of IFN-. The error bar indicates standard devi-ation. (C) The NS1/B protein prevents virus-induced nuclear accumu-lation of hIRF-3/GFP in the context of influenza B virus infection. MDCK cells were transfected with pEGFP-C1-hIRF3 and either mock infected or infected with recombinant WT B/Lee or del NS1 B/Lee virus at an MOI of 1. The cells were stained for the viral NP protein at 8 h postinfection and analyzed by immunofluorescence microscopy.
on November 8, 2019 by guest
http://jvi.asm.org/
propose that the mechanism for IFN antagonist activity for the C-terminal region of the B/NS1 protein is independent of RNA binding and may involve direct or indirect interactions with cellular proteins, such as a kinase involved in IRF-3 acti-vation or another factor involved in the transactiacti-vation of the IFN-promoter. Further analysis of the activation status of IRF-3 in the presence or absence of B/NS1 by determining its phosphorylation, dimerization, CBP/p300 association, and DNA binding activities will most likely shed some light on the mo-lecular mechanism by which B/NS1 inhibits IRF-3 activation.
It has recently been demonstrated that the NS1 protein of influenza B virus can suppress activation of the IFN- pro-moter and the induction of IFN-synthesis (6). We have now shown that B/NS1-mediated inhibition of IFN-synthesis cor-relates with the inhibition of IRF-3 nuclear translocation. This was demonstrated in two different assays. First, expression of the B/NS1 protein by plasmid transfection B/NS1 protein in-hibits Sendai virus-induced nuclear translocation of hIRF-3/ GFP. Second, infection with wild-type influenza B virus did not result in IRF-3 activation at 8 h postinfection, while infection with a recombinant influenza B virus lacking the expression of the B/NS1 protein caused hIRF-3/GFP nuclear translocation (i.e., activation). These results are in contrast to a previous report by Kim et al., in which it was shown that infection with WT B/Lee/40 virus induced IRF-3 activation (11). In that study, a higher MOI of B/Lee/40 virus was used for infection and the activation status of IRF-3 was determined at 4 h postinfection. These conditions may inherently favor IRF-3 activation, since the high multiplicity of infection used could result in an abundance of viral dsRNA and other viral com-ponents that may induce the activation of IRF-3. Also at early time points postinfection, such as 4 h, the level of NS1 protein expression might be very low and not sufficient to effectively inhibit IRF-3 activation.
It was demonstrated in previous studies that influenza A and B viruses that expressed altered NS1 proteins could be used as live attenuated viral vaccines in mice (28). In these studies, influenza B viruses containing naturally occurring C-terminal truncations of the B/NS1 protein were found to be attenuated and immunogenic in mice. This observation is supported by our findings in this study that the C-terminal domain of the B/NS1 protein possesses IFN antagonist activity. Therefore, we predict that deletion of this region would result in a virus that is more attenuated than the wild-type virus. Viruses containing mutations within the B/NS1 N-terminal domain that adversely affect binding to dsRNA may also prove to be promising live attenuated vaccine candidates.
Although our studies clearly indicate that the B/NS1 protein is required to inhibit IFN-␣/production during viral infec-tion, at this time we cannot exclude the possibility that addi-tional functions are encoded by the B/NS1 gene. In fact, it has been shown that the B/NS1 protein blocks the conjugation of ISG15 to its target proteins both in vitro and in virus-infected cells (37). ISG15 is a ubiquitin-like protein that is induced to high levels of expression by IFN-␣/. Interestingly, there ap-pear to be additional functions of the B/NS1 protein that are not related to its ability to antagonize the IFN-induced host antiviral response, since del NS1/B/Lee virus is significantly impaired in its ability to replicate even in IFN-deficient sys-tems, such as Vero cells and 6-day-old eggs (6). This
observa-tion suggests that the B/NS1 protein is somehow important for the propagation of the virus independent of its ability to coun-teract the effects of IFN-␣/. Future studies of virus replication and pathogenicity using influenza B virus NS1 mutants in both IFN-competent and IFN-deficient hosts will be needed to fur-ther investigate the functional role of the B/NS1 protein.
ACKNOWLEDGMENTS
We acknowledge members of the A.G.-S. and Peter Palese labora-tories for critical discussions, especially Luis Martinez-Sobrido and Man-Seong Park for their invaluable assistance. We gratefully ac-knowledge Richard Cadagan for excellent technical assistance.
This work was supported by NIH grants to C.F.B and A.G.-S. B.D. received support from the FAZIT Foundation. C.F.B. is an Ellison Medical Foundation New Scholar in Infectious Diseases.
REFERENCES
1. Basler, C. F., X. Wang, E. Muhlberger, V. Volchkov, J. Paragas, H. D. Klenk, A. García-Sastre, and P. Palese.2000. The Ebola virus VP35 protein
func-tions as a type I IFN antagonist. Proc. Natl. Acad. Sci. USA97:12289–12294.
2. Chen, Z., and R. M. Krug.2000. Selective nuclear export of viral mRNAs in
influenza-virus-infected cells. Trends Microbiol.8:376–383.
3. Chen, Z., Y. Li, and R. M. Krug.1999. Influenza A virus NS1 protein targets
poly(A)-binding protein II of the cellular 3⬘-end processing machinery.
EMBO J.18:2273–2283.
4. Chien, C. Y., R. Tejero, Y. Huang, D. E. Zimmerman, C. B. Rios, R. M. Krug, and G. T. Montelione.1997. A novel RNA-binding motif in influenza A virus
non-structural protein 1. Nat. Struct. Biol.4:891–895.
5. Chien, C. Y., Y. Xu, R. Xiao, J. M. Aramini, P. V. Sahasrabudhe, R. M. Krug, and G. T. Montelione.2004. Biophysical characterization of the complex between double-stranded RNA and the N-terminal domain of the NS1 protein from influenza A virus: evidence for a novel RNA-binding mode.
Biochemistry43:1950–1962.
6. Dauber, B., G. Heins, and T. Wolff.2004. The influenza B virus nonstructural NS1 protein is essential for efficient viral growth and antagonizes beta
in-terferon induction. J. Virol.78:1865–1872.
7. Donelan, N. R., C. F. Basler, and A. García-Sastre.2003. A recombinant influenza A virus expressing an RNA-binding-defective NS1 protein induces
high levels of beta interferon and is attenuated in mice. J. Virol.77:13257–
13266.
8. Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz, D. T. Golenbock, A. J. Coyle, S. M. Liao, and T. Maniatis.2003. IKKεand TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol.
4:491–496.
9. Fortes, P., A. Beloso, and J. Ortín.1994. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks mRNA nucleocytoplasmic transport. EMBO
J.13:704–712.
10. García-Sastre, A., A. Egorov, D. Matassov, S. Brandt, D. E. Levy, J. E. Durbin, P. Palese, and T. Muster.1998. Influenza A virus lacking the NS1
gene replicates in interferon-deficient systems. Virology252:324–330.
11. Kim, M. J., A. G. Latham, and R. M. Krug.2002. Human influenza viruses activate an interferon-independent transcription of cellular antiviral genes:
outcome with influenza A virus is unique. Proc. Natl. Acad. Sci. USA99:
10096–10101.
12. Li, Y., Z. Y. Chen, W. Wang, C. C. Baker, and R. M. Krug.2001. The
3⬘-end-processing factor CPSF is required for the splicing of single-intron
pre-mRNAs in vivo. RNA7:920–931.
13. Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott.1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translo-cation, transactivation potential, and proteasome-mediated degradation.
Mol. Cell. Biol.18:2986–2996.
14. Liu, J., P. A. Lynch, C. Y. Chien, G. T. Montelione, R. M. Krug, and H. M. Berman.1997. Crystal structure of the unique RNA-binding domain of the
influenza virus NS1 protein. Nat. Struct. Biol.4:896–899.
15. Lu, Y., X. Y. Qian, and R. M. Krug.1994. The influenza virus NS1 protein:
a novel inhibitor of pre-mRNA splicing. Genes Dev.8:1817–1828.
16. Ludwig, S., X. Wang, C. Ehrhardt, H. Zheng, N. Donelan, O. Planz, S. Pleschka, A. García-Sastre, G. Heins, and T. Wolff.2002. The influenza A virus NS1 protein inhibits activation of Jun N-terminal kinase and AP-1
transcription factors. J. Virol.76:11166–11171.
17. Nemeroff, M. E., S. M. Barabino, Y. Li, W. Keller, and R. M. Krug.1998. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of
CPSF and inhibits 3⬘end formation of cellular pre-mRNAs. Mol. Cell1:991–
1000.
18. Niwa, H., K. Yamamura, and J. Miyazaki.1991. Efficient selection for
high-expression transfectants with a novel eukaryotic vector. Gene108:193–199.
19. Norton, G. P., T. Tanaka, K. Tobita, S. Nakada, D. A. Buonaugurio, D.
on November 8, 2019 by guest
http://jvi.asm.org/
Greenspan, M. Krystal, and P. Palese.1987. Infectious influenza A and B virus variants with long carboxyl terminal deletions in the NS1 polypeptides.
Virology156:204–213.
20. Park, M. S., A. García-Sastre, J. F. Cros, C. F. Basler, and P. Palese.2003. Newcastle disease virus V protein is a determinant of host range restriction.
J. Virol.77:9522–9532.
21. Park, M. S., M. L. Shaw, J. Munoz-Jordan, J. F. Cros, T. Nakaya, N. Bouvier, P. Palese, A. García-Sastre, and C. F. Basler.2003. Newcastle disease virus (NDV)-based assay demonstrates interferon-antagonist activity for the NDV V protein and the Nipah virus V, W, and C proteins. J. Virol.
77:1501–1511.
22. Percy, N., W. S. Barclay, A. García-Sastre, and P. Palese.1994. Expression
of a foreign protein by influenza A virus. J. Virol.68:4486–4492.
23. Pitha, P. M., W. C. Au, W. Lowther, Y. T. Juang, S. L. Schafer, L. Burysek, J. Hiscott, and P. A. Moore.1998. Role of the interferon regulatory factors (IRFs) in virus-mediated signaling and regulation of cell growth. Biochimie
80:651–658.
24. Qian, X. Y., F. Alonso-Caplen, and R. M. Krug.1994. Two functional do-mains of the influenza virus NS1 protein are required for regulation of
nuclear export of mRNA. J. Virol.68:2433–2441.
25. Sato, M., N. Tanaka, N. Hata, E. Oda, and T. Taniguchi.1998. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of
the IFN-gene. FEBS Lett.425:112–116.
26. Sharma, S., B. R. tenOever, N. Grandvaux, G. P. Zhou, R. Lin, and J. Hiscott.2003. Triggering the interferon antiviral response through an
IKK-related pathway. Science300:1148–1151.
27. Talon, J., C. M. Horvath, R. Polley, C. F. Basler, T. Muster, P. Palese, and A. García-Sastre.2000. Activation of interferon regulatory factor 3 is
inhib-ited by the influenza A virus NS1 protein. J. Virol.74:7989–7996.
28. Talon, J., M. Salvatore, R. E. O’Neill, Y. Nakaya, H. Zheng, T. Muster, A. García-Sastre, and P. Palese.2000. Influenza A and B viruses expressing
altered NS1 proteins: a vaccine approach. Proc. Natl. Acad. Sci. USA97:
4309–4314.
29. Thompson, W. W., D. K. Shay, E. Weintraub, L. Brammer, N. Cox, L. J. Anderson, and K. Fukuda.2003. Mortality associated with influenza and
respiratory syncytial virus in the United States. JAMA289:179–186.
30. Wang, W., and R. M. Krug.1996. The RNA-binding and effector domains of the viral NS1 protein are conserved to different extents among influenza A
and B viruses. Virology223:41–50.
31. Wang, W., K. Riedel, P. Lynch, C. Y. Chien, G. T. Montelione, and R. M. Krug.1999. RNA binding by the novel helical domain of the influenza virus NS1 protein requires its dimer structure and a small number of specific basic
amino acids. RNA5:195–205.
32. Wang, X., C. F. Basler, B. R. Williams, R. H. Silverman, P. Palese, and A. García-Sastre.2002. Functional replacement of the carboxy-terminal two-thirds of the influenza A virus NS1 protein with short heterologous
dimer-ization domains. J. Virol.76:12951–12962.
33. Wang, X., M. Li, H. Zheng, T. Muster, P. Palese, A. A. Beg, and A. García-Sastre.2000. Influenza A virus NS1 protein prevents activation of NF-B
and induction of alpha/beta interferon. J. Virol.74:11566–11573.
34. Wathelet, M. G., C. H. Lin, B. S. Parekh, L. V. Ronco, P. M. Howley, and T. Maniatis.1998. Virus infection induces the assembly of coordinately acti-vated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell
1:507–518.
35. Yoneyama, M., W. Suhara, Y. Fukuhara, M. Fukuda, E. Nishida, and T. Fujita.1998. Direct triggering of the type I interferon system by virus infec-tion: activation of a transcription factor complex containing IRF-3 and CBP/
p300. EMBO J.17:1087–1095.
36. Yuan, W., J. M. Aramini, G. T. Montelione, and R. M. Krug.2002. Structural basis for ubiquitin-like ISG 15 protein binding to the NS1 protein of influ-enza B virus: a protein-protein interaction function that is not shared by the corresponding N-terminal domain of the NS1 protein of influenza A virus.
Virology304:291–301.
37. Yuan, W., and R. M. Krug.2001. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein.
EMBO J.20:362–371.