0022-538X/83/060726-11$02.00/0
Copyright©1983, AmericanSocietyforMicrobiology
Long Terminal Repeat Enhancement of
v-mosTransforming
Activity: Identification of Essential Regions
T. G. WOOD,* M. L. McGEADY, D. G. BLAIR,ANDG. F. VANDE WOUDE Laboratory of Molecular Oncology, National Cancer Institute, Bethesda, Maryland20205
Received 29 November 1982/Accepted 9 March 1983
The transforming efficiency of recombinant DNA clones containing the
Mo-loney sarcomavirusv-mos sequence wasenhanced by introducing the Moloney sarcomaviruslong terminalrepeat(LTR) in either the 5' or3' position relativeto
v-mos. We analyzed the polyadenylated RNA expressed in cells transformed by
these recombinant DNA clones and examined the structural integrity of integrated copies of the DNA. In each case, we demonstrated the presence of v-mos
containing RNA transcripts in the polyadenylated RNA and showed that these RNAtranscripts are consistent with the structure of the transfected DNA. The analysis of DNA from these transformed cells showed that the relative positions of thev-mosand LTRsequenceswithin thetransfected DNAwereconservedin
the integrated DNA copies. These results demonstrate that a single LTR can
successfully enhance the transforming activity ofv-mos from eithera5' or a3'
relativeposition. The results from the transfection analysis of recombinant clones containing only portions of the LTR introduced 3' tov-mosdemonstrate that the
essentialregion of the LTR responsible for the enhancement of transformation isa
region within the unique 3' sequences of the LTR containing the 73-base-pair tandem repeat sequence.
The essential components of the Moloney murine sarcoma virus (MSV) proviral genome
responsible
for cell transformation are theac-quired v-mos sequence and the proviral long terminal repeat (LTR) (7, 18). The retroviral
LTRhas been shownto containtranscriptional control elements that function to ensure viral
RNAtranscription of the provirus (9, 15, 29, 30, 33). The results from transfection assays using
subgenomic
MSVproviral
DNA clones haveshown that the LTR enhances the
transforming
activity ofv-moswithequivalent efficiencyfrom
eithera5' or a3'
position
relativeto v-mos(7).
Although these results suggest that asingleLTR can enhance thetransforming activity
ofv-mos, these assays do not provide evidence that will exclude thepossibility
that tandemintegrations
or rearrangements ofthe transfectedrecombi-nant DNA are responsible forthe enhancement
ofthe transforming
activity.
In this report, wepresent the analysis of polyadenylated RNA
expressed in cells transformed by the transfec-tion ofrecombinant DNA clones containing
v-mosandasingleLTRand examine thestructure oftheintegrated formof the transfected DNA in these cells. Furthermore, to determine the
es-sential region of the LTR responsible for en-hancement, weconstructedaseriesof
recombi-nant DNAclonescontaining only portions ofthe
LTR introduced 3' to v-mos and tested these recombinant DNAs intransfection assays.
MATERIALS ANDMETHODS
DNA transfections and developmentof transformed cell lines. Maps of the restriction endonuclease sites present in recombinant DNAclones ofHT1 and ml strains of MSV proviral DNA have been published
elsewhere (19, 34). Descriptions of the subgenomic
proviral recombinant DNAclones used in this study have been reported elsewhere (7) or are described below. DNA transfection of NIH3T3 cells was per-formed by modifications of established procedures (11) as previously described (7). Morphologically transformed cellswereselected from individual fociby single-cell cloning either inagarorby serial dilutionin microwell tissue culture dishes. Thecellswere main-tained inDulbeccomodified minimal essential medium (GIBCO Laboratories) supplemented with 10% (vol/ vol)calfserumand antibiotics.
DNA analysis. The cells were lysed with abuffer containing 0.6% (wt/vol) sodium dodecyl sulfate, 10 mM EDTA,10 mM Tris-hydrochloride (pH 7.5),and 100,ugof pancreaticRNase Aper mlwhich had been incubatedat100°C for 5min.Thelysatewasincubated
at37°C for1 h.ProteinaseK(BoehringerMannheim) wasaddedto afinal concentration of250 ,u.g/ml, and the incubation was continued for 2 h at 37°C. The mixturewasthen extractedoncewithanequal volume
726
on November 10, 2019 by guest
http://jvi.asm.org/
ofphenol saturated with 1 MTris-hydrochloride (pH 8.0), twice with phenol-CHC13 (1:1), and once with CHCl3. DNA was precipitated with 2 volumes of ethanol anddissolved insterileH20.
Restriction endonuclease (New England Biolabs) digests were performed under the conditions recom-mendedby the manufacturer. DNA samples (12to15 ,ug)wereapplied to0.75% (wt/vol) agarose gels con-taining 0.5 ,ug ofethidium bromide per ml, and DNA
fragments were separated by electrophoresis as re-ported by McDonell et al. (20). HindIII digests of lambda DNAwereincluded in eachanalysisfor esti-mating DNA fragmentsize. The transfer of DNAto nitrocellulose membranes(Schleicher& Schuell Co.) was performed as described by Southern (31). The blots were dried for 2 hat80°Candpretreated witha solutioncontaining50%(vol/vol)formamide (Fluka),
5x SSC(1xSSC is0.15 M NaCl plus 0.015 M sodium
citrate), 0.2% (wt/vol) Ficoll (Pharmacia Fine Chemi-cals),0.2%(wt/vol)polyvinylpyrrolidone,50 mM sodi-umphosphate (pH6.5),1% (wt/vol) glycine,0.1% (wt/
vol) sodium dodecyl sulfate, and 250 ,ug of sheared salmon sperm DNA(SigmaChemicalCo.) per mlfor 16 h at 43°C. After hybridization, the blots were washed three times in 2x SSC at room temperature and three times in 0.1x SSC for 15 minat50°C.The blots were then air driedovernightat room tempera-ture.Autoradiography wasperformedwith Kodak SB-5film andanintensifying screen at -70°C.
RNA analysis. The cells (0.3 to 0.5 ml of packed cells) werelysed byhomogenizationinbuffer contain-ing 25 mMTris-hydrochloride (pH 7.5), 25 mM NaCl, 5mMMgCl2,1 mgofheparin (Sigma) per ml, and 2% Triton X-100(Bio-Rad Laboratories).After
centrifuga-tion of thelysateat15,000 x gfor 10 min at 4°C, the
supernatant wasmixed with an equal volume of the abovebuffercontaining200 mMMgCl2.Thismixture wasincubated for 2 h at0°Candthenlayered over a solution of 1 M sucrose, 25 mM Tris-hydrochloride
(pH 7.5), 25 mM NaCl, and 100 mM MgCl2.
Mg2+-complexed polysomes werepelletedby centrifugation at 15,000 x g for 20 min at 4°C. The pellet was suspended in 200
RlI
of 100 mM EDTAcontaining1mg ofproteinase K per ml and then mixed with 5 ml ofa buffercontaining25 mM Trisacetate(pH7.5),600 mM sodium acetate, 2mMEDTA, and0.5% sodium dode-cyl sulfate. The mixture washomogenizedand incu-bated at 45°C for 3 min. Polyadenylated RNA was selectedonoligodeoxythymidylate-cellulose(P-LBio-chemicals, Inc.) as previously described (2). RNA
samples were stored under ethanol at -20°C. RNA was collected by centrifugation at 12,000 x g for5 min, and the RNApellets weredried undervacuum. RNAsamples weredenatured in 15 mMmethyl mer-curyhydroxide (Alfa)for 10 minatroomtemperature and separated by electrophoresis on 1.2% (wt/vol) agarosegels containing5 mMmethyl mercury hydrox-ide (1, 3). The transfer ofRNA to diazobenzyloxy-methyl paper (Schleicher & Schuell) and the subse-quent treatment of the blots for hybridization and
autoradiography wereperformed asdescribedby Al-wineet al.(1).
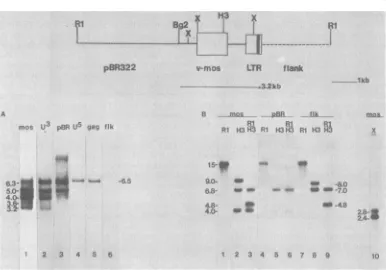
DNA probes. The restriction endonuclease sites usedinisolating specificDNAfragmentsare shownin Fig. 1. p-mos, aplasmid containingtheentire c-mos sequenceand 1.9kilobases(kb) of normalmouseDNA 5'to c-mosclonedintopBRSc7 (see the legend to Fig.
5) at the Sacl and HindIll sties, was used as the source of the mos-specific DNAfragment. pmlsp, a plasmid containing one copy of the ml MSV proviral LTRplus the 5' and 3' cellular sequencesflankingthe ml MSV
pmos
mos
pmlsp
s I
I3
S ~~~~~~1~ 113
AHj3
P2
U3
R
3mlflank
P2 Pe
IL
pml5
989
PL J2
FIG. 1. DNA fragments used to prepare nick-trans-lated DNAprobes. Shown are restriction endonucle-asesites present inthree cloned DNA inserts (p-mos, pmlsp,pmlS) that have been cloned into pBR322 (7, 24). Theserestriction sites were used in isolating DNA fragments (see the text) that represent specific DNA sequences used in the preparation ofnick-translated probes. The size of the DNA fragments was estimated by comparison with the electrophoretic mobility of known DNA size markers in either agarose (20) or polyacrylamide gels (17). p-mos, a plasmid that con-tains theendogenous c-mos sequence (24), was used in isolating a 930-bp AvaI-HindIII mos-specific DNA fragment. pmlsp is a plasmid containing one copy of the ml MSV proviral LTR plus the 5' and 3' cellular sequencesflankingtheml MSVprovirus (19). A 215-bp DNAfragmentthat represents the U3region of the LTR was isolated from PvuII-SacI digests of pmlsp DNA. A 70-bpHhaI-Hinfl DNAfragment containing allof theRsequenceand aHinfl-AluIDNAfragment that contains the entireunique 5' regionof the LTR and 34bp of 3' cellular flanking sequence were also isolated from this plasmid. A700-bp DNA fragment representing the 3' cellular flanking sequences was isolated fromPvuII-PstI digests of pmlsp. pmi5is a
plasmidcontaining the 5' LTR and 2.0 kb of adjacent virus-specific gag sequences cloned from ml MSV proviral DNA (7). A 1.1-kbPvuI-PvuIIDNAfragment representing part of the gagcoding region of ml MSV wasisolated from thisplasmid. Restriction enzymes: A,AluI;Avl,AvaI;Bg2,BgII;H,Hinfl;H3,HindIII;
Hh,HhaI;P1,PvuI; P2,PvuII;Ps,PstI;RI,EcoRI; S, Sacl.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.488.247.441.141.373.2]Ri SB B S H3 H3 B RI
f lank LTP aag v-mos pBR322
45 kb
1 kb
t1 pBR U15 qa, I
(-r
tF11
il.I:
RI.
b~i-, e.H1 -1 pBR u-)ia; t7
ti-3
U _TO-1
U Rn)v
F:i
Ri S .f
13-- a
i;8
4'-4
-..AT 3 4 5 6
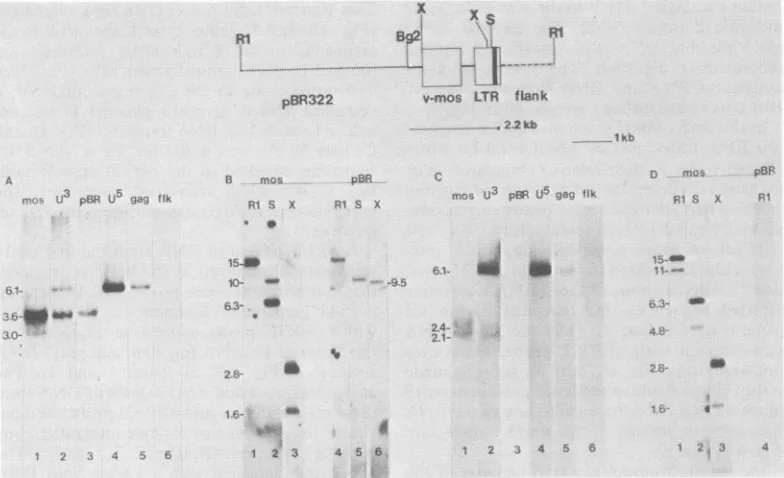
FIG. 2. Analysis of RNA and DNA from pHT25 transfectants. A schematicdiagramof thestructureof the 12.2-kb pHT25plasmid DNA is shown, with various proviral, vector, and cellular flankingDNA sequences labeled. Restriction endonuclease sites utilized inanalyzingthestructureof theintegratedDNAareindicated.
pHT25 DNA was linearized by EcoRI digestion before transfection. The results from the analysis oftwo independentpHT25transfectantsareshown.(A andC)Results fromhybridizationanalysisofpolyadenylated
RNA(2.5,g/lane)isolated from thesetransfectants. Theprobesusedinanalyzingthe RNAblotsareindicatedat
the topof each lane, and theirpreparationis described in thetext.Theresultsfromhybridization analysiswitha mosprobeof restriction endonucleasedigestsof cellular DNAs isolated from nontransformed NIH3T3 cells(B,
lanes 4 and5)and thetwopHT25transfectants(B,lanes 1to4,andD)areshown. Therestriction endonucleases used in the variousdigestsareindicatedatthe top of each lane. Restriction enzymes:B,BamHI; H3,Hindlll;S, Sacl;RI,EcoRI.
provirus (19),wasusedas a sourceof DNAfragments
representing the unique 3' region (U3), the repeat sequence (R), and the unique 5' region (US) of the LTR,aswellasthat ofaDNAfragment representing
the 3' flanking cellular sequences. pmlS, a plasmid containing the 5' end of ml MSV proviral DNA,
including the LTR and 2.0 kb ofvirus-specific gag sequences(7),wasusedtoisolateagag-specificDNA
fragment and the US DNA fragment (HinfI-PvuI),
whichwasusedas aprobe in thehybridization analy-sis ofthe pml3 transfectants. A5.6-kbEcoRI DNA fragment that represents the mink cellularDNAtarget site ofHT1MSVprovirusintegration(34)wasusedas aprobe for theHT1 MSV cellularflankingsequences (datanotshown).
For restriction endonuclease digestion, plasmid DNA fragments were purified by electrophoresis on 5% (wt/vol) polyacrylamide gels (bis:acrylamide,1:25)
(17). The nick translation ofpurified DNAfragments
(1 x 108to2x108cpm/,ug)wasperformedasreported by Rigbyetal.(28),andallhybridizations ofRNA and DNAblots contained5 x 106to10 x 106cpmof the respective DNA probe. EachDNAprobewastested for sequence specificity before use in hybridization
assays.
RESULTS
Analysis ofcells transformedby DNA contain-ing an LTR 5'to v-mos. The resultsfrom DNA transfection assays using cloned recombinant
DNAscontainingv-mosand asingle LTR have previously been reported (7, 18). One of these clones, pHT25, was derived from HT1 MSV proviral DNA and contains the 5' LTR and
adjacent proviral sequences through v-mos
clonedinto pBR322(Fig. 2).
The analysis of RNA and DNA from two
independent pHT25
transfectants
is shown inFig. 2. If the LTRprovides transcriptional
con-trol elements that insure the expression of
v-mos, thentranscriptionshould beinitiatedatthe repeat(R)sequence within the 5' LTR, and the
resulting RNA transcripts would contain U5,
gag, and v-mos sequences. In onepHT25
trans-fectant, a single 10-kb RNA was detectedwith mos, US, and gag probes (Fig. 2A, lanes 1, 3,
and 4). The second pHT25 transfectant
con-tainedtwoRNA
transcripts
(8.9 and7.0kb)thatJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.488.51.447.70.310.2]hybridized with mos, gag, andU5 probes (Fig. 2C, lanes 1, 3, and 4). The predicted primary transcript for an RNA initiated at the R se-quencewithin the LTRandcontaining allofthe viral sequences encodedinpHT25is4.5 kb,but all of the mos-containing RNA transcripts ob-served in these cellswereinexcess of this size. Some of this additional information can be
at-tributed tothe exression of pBR322 vector se-quencessinceaprobetothisDNAdetectedthe same RNA transcriptsas the mos, gag, and U5
probes(Fig.2AandC, lane2).However, the
10-kb RNAtranscript (Fig. 2A) would still require additional sequencesderivedfrom hostor
carri-er DNA even if we assume that a complete
pBR322 vector sequence was expressed. A
proberepresenting the U3 region of theLTRdid
nothybridizeto anyof the mos-containingRNA
transcripts(Fig. 2A and C, lane 5). This
demon-strates that the discrete RNA transcripts ob-servedarenotderived from tandem integrations of the transfected DNA and suggests that the termination andpolyadenylation signals utilized by these RNA transcripts are acquired from either vector, host, or carrier DNA sequences. AU3 probe did detecta1.3-and a1.0-kbRNA
transcript (Fig. 2Aand C,lane 5)in the pHT25 transfectants analyzed; however, these
tran-scripts
did nothybridize
with mos-specificprobes.
Todeterminethenumberofintegrated copies of pHT25 DNA and to assess the structural integrity of the
integrated
DNA sequencesin thetwopHT25transfectants,weanalyzed the cellu-lar DNA by digestion with restriction endonu-cleasesand
by
SoUthern
analysis.
EcoRI-digest-ed DNA from ndntransformed NIH3T3 cells containeda15-kbDNA
fragment
whichhybrid-ized with a mos
probe
(Fig. 2B,
lane5).
ThisDNA
fragment
corresponds
to theendogenous
EcoRI c-mos DNA
fragment
found in normalmouse cellular DNA
(14, 24).
The mosprobe
detected the 15-kb c-mos DNA
fragment
and anew10-kb DNA
fragment (Fig. 2B,
lane1).
The sizeof the latter EcoRI DNAfragment
suggeststhat EcoRI sites at the ends of the transfected pHT25 DNA have not been conserved in the integrated copy and thata
portion
of the trans-fected DNA was deleted. The extent of this deletion could be estimated from Sacl andBamHI
DNA digests (Fig. 2B, lanes 2 and4).
Sacl digests contained a 6.3-kb c-mos DNA
fragment and a 5.4-kb DNA fragment derived
from the transfected pHT25 DNA (Fig. 2B, lanes 2 and4). Asingle Saclsite was present in the LTR, whereas no SacI sites existed in the
adjacentviral orvectorsequences of the
trans-fectedpHT25DNA
(Fig.
2,schema).The 5.4-kbmos-containing DNA fragment is, therefore, generatedfrom a secondSacl siteacquiredfrom
either host or carrier DNA sequences, and its
size implies that aminimum of 3 kb of pBR322 vector sequences has been deleted from the integrated DNA. pHT25 DNA also contained a
7.0-kb BamHI fragment (Fig. 2). This DNA
fragment was not conserved in the integrated
DNA,indicating that potentially all but 340 base
pairs (bp) of the pBR322 sequences have been deletedfrom the integratedcopy. A comparison
of the abundanceof the endogenousc-mos DNA
fragmentrelative to that of the new
mos-contain-ing DNA fragments observed in this pHT25
transfectant suggests that a single copy of the
pHT25 DNA is present in these transformed cells(Fig. 2B,
lanes
1 through 4),which
would imply thatthe10-kb mos-containingRNA (Fig. 2A) detected in these cells results fromtran-scription from this singlecopy of pHT25 DNA
(Fig. 2B).
The 8.9-and 7.0-kbmos-containing RNA
tran-scripts(Fig. 2C) expressed in the second pHT25 transfectant alsoappear tobe transcribedfroma
single integratedcopyofpHT25 DNA(Fig. 2D,
lanes 1 through 4). EcoRI digests of cellular DNAfrom this transfectant contained the 15-kb
c-mos DNA fragment and a new 13-kb DNA
fragment that hybridized with the mos probe
(Fig. 2D, lane1).Probes representing pBR322or
the cellular flanking sequences presentin plas-mid pHT25DNAalso annealwitha13-kbEcoRI
DNAfragment (datanotshown). The mos probe
hybridized to a 7.0-kb BamHI DNA fragment (Fig. 2D,lane4)andan8.8-kb DNA fragment in EcoRI-SacI digests (Fig. 2D, lane 3). Both of theseDNAfragments correspondtothe
predict-ed sizeof theirrespective mos-containing DNA
fragments in pHT25 plasmid DNA. These
re-sultsimplythatessentiallyallofthetransfected pHT25 DNA has been conserved in this
trans-fectant. Sacldigestion of DNA from these cells produceda23-kbmos-containingDNAfragment (Fig. 2D, lane
2),
demonstratingthat the pHT25 DNA sequences are integrated in the cellular DNA. The results from the analysis of DNAfrom both of these pHT25 transfectants show
that a single LTR introduced 5' to the v-mos sequence caneffect the expression of
mos-con-tainingRNAtranscripts.
Analysis of cellstransformedby DNA contain-ing an LTR 3' to v-mos. pHT21 is a recombinant DNA plasmid derived from HT1 MSV
proviral
DNA containing a single LTR 3' to the v-mossequence andcloned intopBR322(Fig. 3). Five mos-specific RNA transcripts ranging in size from 6.3 to 3.2 kb weredetected in thepHT21 transfectant(Fig. 3A,lane1). AnidenticalRNA pattern was obtained by
hybridizations
with aU3probe (Fig. 3A, lane2),whereasnoneofthe
mos-containingRNAtranscriptsannealedtothe U5 probe (Fig. 3A, lane 4). This result shows
on November 10, 2019 by guest
http://jvi.asm.org/
730 AL.
V
pBR322
A
mos LJ3 pBR U5 gag flk
6.3-li
3.0
4.0
.;X
HF3X
xi I x
~~IRI
v-mos LTR flank
8 .mos ...pBR . i
RI RI FI
Rl H3H13 Rl 131H3 RI Hf3 H3
15-9 .., _
9.o- - 0 -8.0
6.8- * Am - - 0-70
_ -4.8
4,8- S
4.0- -o
2 3 4 5 6 1 2 3 4 5 6 7 8 9
FIG. 3. Analysis of RNA and DNA from apHT21 transfectant. A schematicdiagram of the structureof pHT21 plasmid DNA is shown. The 9.9-kb plasmid DNAwaslinearizedby EcoRIdigestion before transfection. (A) Results from hybridization analysis of polyadenylated RNA (5 jig/lane) isolated from the pHT21 transfectant. (B) Results from hybridization analysis of DNA from these transformed cells digested with either EcoRI, HindIII, EcoRI-HindIII, orXbaI restriction endonucleases. The probes used in thehybridization assays are indicatedatthetopof the lanes andaredescribed in thetext.
that the enhancement of the transforming effi-ciency of v-mos by a 3' LTR does not result from the tandem integration of the transfected pHT21 DNA. The mos-containing RNA
tran-scripts expressed in the pHT21 transfectants
presumably terminate in the 3' LTR, utilizing
the LTRpolyadenylation signals. The promoter signals andtranscription initiation sites for these RNAtranscriptsarenotderived from the
trans-fected LTR andpresumablyareeitherpresent in
the Moloney leukemia virus sequences preced-ingv-mosor areacquired fromvector, host, or
carrier DNA sequences. The size of the
mos-containing RNA transcripts expressed in this
transfectant limit the use ofpossible promoter
signals encoded in the proviral DNA to the expression ofonly the 3.2-kb transcript.Two of
the five mos-containing RNA transcripts ex-pressed in the pHT21 transfectant contained
sequences that annealed to the pBR322 probe (Fig. 3A, lane 3).
The U5 probe annealed to a 6.5-kb RNA
transcript in the pHT21 transfectant (Fig. 3A, lane 4). However, the expression of this RNA transcript is apparently not directly related to
theexpression of pHT21-encoded sequences. A
probe made to the cellular flanking sequence downstream from the 3' pHT21 LTR did not
hybridizetothistranscript (Fig. 3A, lane 6), but a probe specific for the gag region of MSV, a region notpresent inpHT21 plasmid DNA, did anneal to the 6.5-kb RNA transcript (Fig. 3A, lane 5). These results are consistent with this transcript representing the expression ofan en-dogenous murine viral RNA. A similar RNA
transcript is sometimesobserved in polyadenyl-ated RNA from normal NIH3T3 cells, but the
level of theexpressionof thisRNA isincreased in this pHT21 transfectant and in several other MSV-transformed cells that have been exam-ined(T. Wood, unpublished data). However,all MSV transfectants do not express this RNA
transcript, asis shown forthetwopHT25
trans-fectantsanalyzed inFig. 2.
To determine the number ofintegrated copies of pHT21 DNA and to assess the structural
integrity oftheintegrated DNAsequences
pres-ent in thesetransformed cells, we analyzedthe
cellularDNA from thepHT21transfectant(Fig. 3B). Hybridization with a mos probe detected
onlya15-kbDNAfragmentinEcoRIdigests of cellularDNAfrom thepHT21 transfectant (Fig.
J.VIROL.
...ktb
mos x
2.8-i
2.4-10
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.488.55.441.68.338.2]X XS
[image:6.488.50.442.76.315.2]Rl Bg2 .i RI
...
pBR322 v-mos LTR flank
... 2.2kb
1kb
B mos pBR
Ri S X RIS X C
mos U3 pBR U5 gae filk
o mob pBP
HiS X Ri
I
6.1- _W
a
24-
2.1-1.6- *
!,23. 456
1 2 3 4 5 6 1 2 3 4 5 6
FIG. 4. Analysisof RNAandDNAfrompml3 transfectants. Aschematic diagram of thestructureof pml3 plasmid DNA is shown.Theresultsfromthehybridization analysis of polyadenylatedRNAisolatedfromtwo
pml3transfectantsareshown in(A) (4,ug/lane) and (C)(5.5Fg/lane). The results from thehybridization analysis ofcellular DNA isolatedfrom thesetransformed cellsareshown in(B andD).The restriction endonucleases used intheDNAdigestsareindicatedatthetopof eachlane.Restrictionenzymes:Rl,EcoRI;S, SacI;X,XbaI. Theprobes (seethetextusedinanalyzing the RNA andDNAblots)areindicatedatthetopof the lanesin each panel.
3B, lane 1). Aprobe representing pBR322orthe cellular flanking sequence in pHT21 plasmid DNAalso detecteda15-kb DNAfragment(Fig.
3B, lanes 4 and 6). Theseprobes didnot hybrid-ize to the endogenous c-mos 15-kb DNA
frag-ment (datanot shown), indicating that a DNA fragmentrepresentinganintegratedcopy ofthe pHT21 DNA comigrates with the c-mos 15-kb DNA fragment. This interpretation was con-firmedby analysis with other restriction endonu-cleases.HindIII digests contained thepredicted 9.0-kbc-mosDNAfragment andtwoadditional mos-containing DNA fragments of6.8 and 4.0 kb (Fig. 3B, lane 2). The lattertwoDNA
frag-ments were notaffectedby additionaldigestion withEcoRI, whereas the c-mosDNA fragment
was cleaved (Fig. 3B, lane 3). The XbaI site
immediately 5' to v-mos was 2.4 kb from the
single XbaI sitepresentin the3' LTR in pHT21 plasmid DNA (Fig. 3, schema). Hybridization
withamosprobe detectedtwoDNAfragments inXbaI digests of DNA from the pHT21
trans-fectant,a2.8-kbc-mosXbaIDNAfragmentand
a2.4-kb XbaI DNAfragment derived from the
integrated pHT21 DNA (Fig. 3B, lane 10). These results demonstratethattherearetwointegrated copies ofpHT21 DNAinthistransfectant,
nei-therof which has undergone rearrangement of the relative positions of the v-mos and LTR
sequences present inthe transfected DNA. Al-though the size of the EcoRI DNA fragment indicated the loss of EcoRI sites from both pHT21 DNA copiespresentinthistransfectant, pBR322 and cellular flanking sequences were retained in both of the integrated DNAs (Fig. 3B, lanes 4 through 9),offering further evidence that a major rearrangement of the transfected DNA has not occurred. Both the pBR322 and
mos probes detected the same DNA fragments in EcoRI, HindIII, and HindIll-EcoRI digests (Fig. 3B, lanes 1 through 6). However, the relativeintensityofpBR322 hybridizationtothe HindIll DNA fragments suggests that different
amounts ofpBR322 sequences are retained in
each of theintegrated copies. It should be noted thatthesetwointegrated copies of pHT21 DNA
areapparently responsible for theexpression of
fivemos-containing RNA transcripts.
Asecondexample of the enhancement of the transforming activity ofv-mos bya single LTR
introduced in a 3' relativeposition is presented
by the analysis ofpml3-transformedcells (Fig. 4). pml3 is derived from ml MSVproviral DNA and, likepHT21, this recombinant DNA plasmid
A
r75 IlJ F3EPBR U5 gag filk
C'- I
,~01i.t~- *
15_a
95
15-00
1$_
--
28-i ? : ,) 6
b.) >- so 4
2-28- i
IC *
1 2 3 4
63- 10
on November 10, 2019 by guest
http://jvi.asm.org/
containsasingle LTR 3' tothev-mos sequence
and cloned into pBR322. The circular 7.7-kb pml3 plasmid DNA was transfected without endonuclease digestion. The results from the
analysis ofRNA and DNA from two indepen-dent pml3 transfectants areshowninFig. 4.
In the first example, the mos probe detected
two RNA transcripts of3.6 and 3.0 kb which differed vastly in their relative abundance (Fig. 4A, lane 1). Theprolongedexposureof this blot revealedtwoadditional faint bands correspond-ingto 2.4and 2.1 kb (note dots inFig. 4A, lane
1). Each of these mos-containing RNA
tran-scripts also hybridizedtotheU3probe (Fig. 4A, lane 2), whereasnoneof these RNA transcripts encoded sequences that annealed to the U5 probe (Fig. 4A, lane 4). Only the 3.6-kb RNA
was detected with pBR322 probe, even after longexposures (Fig. 4A, lane 3). A probe made
tothe cellularflankingsequence presentinpml3 plasmid DNA didnotannealtoanyof the RNA transcripts expressed in this pml3 transfectant (Fig. 4A, lane 6).
The results from an identical analysis of the
RNA
transcripts
expressed inasecondindepen-dent
pm13
transfectant are shown inFig.
4C. After prolonged exposure (10 days), the mosprobe detectedtwo RNA
transcripts
of 2.4 and2.1 kb in these transformed cells
(Fig. 4C,
lane1). Both of these RNA transcripts contained
sequences that
hybridized
to the U3probe,
whereasneithertranscript annealed with the U5, flank, or pBR322
probes (Fig. 4C,
lanes 2through 4 and 6). These results are consistent with the results from theprevious analysis of the pHT21
transfectant
but, inaddition,
show that the levelof
theexpression
ofmos-containing
RNA
transcripts
can varyconsiderably
in cellstransformed with
subgenomic v-mos-containing
proviral DNAs. Note that the sizes of the
mos-containingRNAsshown in
Fig.
4Careidenticaltothoseof the smaller RNAtranscripts detected only after
prolonged
exposure in theprevious
example
(Fig.
4A).Weestimate the level ofmos RNA in the second examplepf
pml3-trans-formed cells tobe between 1 and 10copies percell (T. Wood, unpublished data). This
implies
that only afew
copies
ofmos-containing
RNAarerequired toinduce celltransformation. Fur-thermore, these results suggest that the en-hancement of the transforming
efficiency
ofv-mosdoes not predispose the
expression
ofthe mosgene totranscribehigh
levels ofmosRNA.However, we cannotexcludethe
possibility
thathighlevelsofmosRNAaretranscribed in
pm13-transformed cells butnotprocessedtothe
cyto-plasm.
A 6.1-kb RNA transcript was detected with
theU5andU3
probes
inbothexamples ofpml3-transformed cells
(Fig.
4AandC,
lanes 2 and4).This transcript did not contain mos sequences
(Fig. 4A and C, lane 1) and appeared to be analogous to the 6.5-kb RNA transcript
ex-pressed in pHT21-transformed cells (Fig. 3A). The probe made to the gag
region
ofMSV, a sequence absent in pml3plasmid
DNA,an-nealedtothe6.1-kbRNAtranscript
(Fig.
4AandC,
lane 5). As was noted for the 6.5-kb RNA transcript detected in the pHT21transfectant,
this 6.1-kb RNA transcript represents the expression of endogenous murine retroviral se-quences.
EcoRI
digestion
of DNA from the first pml3 transfectantproduced
a 15-kb DNA fragmentthat
hybridized
to a mosprobe
(Fig.
4B, lane 1).A15-kb EcoRI DNA
fragment
wasalso
detected withapBR322probe,suggesting comigration
of thec-mos15-kb DNAfragment
andpml3 DNAsequences
(Fig.
4B, cf. lanes 1 and 4). Theanalysis
of SacI and XbaIdigests
of DNA fromthesecells withmosand
pBR322 probes
demon-stratesthepresence ofa
single integrated
copyof pml3DNA
(Fig.
4B, lanes 2, 3, 5, and 6). Themos probe annealed with a 1.6-kb XbaI DNA fragment from this pml3 transfectant
(Fig.
4B, lane3). Thiscorresponds
tothepredicted size of
theXbaI DNA
fragment
inpml3plasmid
DNA (Fig. 4, schema) and indicates that the v-mosand LTR sequences in this integrated pml3
DNA have not been
rearranged.
The mos andpBR322probes both detecteda10-kb SacI DNA fragment, suggesting that thevectorsequenceis oriented in a
5' position
relative tov-mos (Fig. 4B, lane 5). This result is consistent with theexpression of
pBR322 sequences in the 3.6-kbRNA
transcript (Fig.
4A,lanes1 and 3).Themos
probe
detecteda 15-kb c-mos DNAfragment
and a new 11-kb DNA fragment inEcoRI
digests of
cellular DNAfrom
the second pml3 transfectant(Fig.
4D, lane 1). The analysis of SacIdigests
of this DNA with themosprobe also demonstrates asingle integrated
copyof
pml3
DNA(Fig.
4D, lane 2).Asnotedwith theprevious pml3
transfectant (Fig. 4B, lane 3),a1.6-kb XbaI DNA
fragment
was detected withthemosprobe, indicatingthat therelative posi-tions of the v-mos and LTR sequences in the
integrated copy of
pml3
DNA have beencon-served
(Fig.
4D, lane 3). The pBR322probe
didnotanneal to DNAfrom this
pml3
transfectant, suggesting that thevector sequenceshave been deleted(Fig. 4D, lane 4).The results from the analysis of RNA and
DNA isolated from cells transformed by the
transfection of cloned subgenomic proviral
DNA(Fig. 2through 4) haveprovided evidence
that a
single
LTR canefficiently
enhance thetransforming activity ofv-mosfrom eithera 5' or a3'relative
position.
Thetransfection of recom-binantDNAcontaining
anLTR 5'to v-mos(i.e.,
on November 10, 2019 by guest
http://jvi.asm.org/
PIasmdW
DNA DNA
H3
pHT1O
C S
:
R
pHT22 L--!--- J 975
LTR
pHTS3 8 LIIILL1170
pHTX3 ] 1150
K
pHTR5
---I--I
-
<5pmI
mmosl
mF
4500LTR
1kb
FIG. 5. Physical maps and transforming efficiency of recombinant DNA clones containingvarious LTR sequencesintroduced3'to v-mos.Descriptionsof pHT10,pHT21, pHT22,andpmlspplasmidDNAshavebeen previously published (7, 19). The recombinant DNA clones and the essential restriction endonuclease sites used inthe construction of the recombinant DNA clonesareshown. The DNAfragmentsused in these constructions werepurified by electrophoresis in agarose gels (20), and the DNAwasrecoveredby electroelution. The DNA fragments were extractedoncewithphenol-CHCl3(1:1) andprecipitatedwithethanol. The DNAfragmentswere
ligated(10), and thesamplewastransfected into competent LE392(32). Colonieswereselected and testedfor confirmation ofstructurebyrestriction endonucleasedigestion.pHTS3wasconstructed from the HindIII-SacI DNAfragmentofpHT22 byligationto aBgtII-HindIIIDNAfragmentfrompHT10andclonedinto the BamHI-Saclsites ofpBRSc7 (a plasmidvectorwithSacl linkers introducedatthe PvuIIsite inpBR322).pHTX3was
constructed inasimilarmannerwiththeHindIII-XbaI DNAfragmentofpHT22ligatedto aBglII-HindIIIDNA
fragment frompHT10and cloned into theBamHi-XbaI sites in pBRX19 (aplasmidvectorwith XbaI linkers introduced at thePvuIIsite inpBR322). pHTR5wasconstructed withaBgII-KpnIDNAfragmentpurifiedfrom apartialdigestofpHT21 DNA. This DNAfragmentwasligatedto aKpnI-EcoRIDNAfragmentderivedfrom pmlsp andcloned into theBamHI-EcoRI sites ofpBR322. Sequences representing v-mosandthe LTRare indicated. The dashed line represents cellularflankingsequences. The cloned DNAswerelinearizedby digestion
witheither EcoRI(pHTS3, pHTX3), SalI (pHT22, pHTR5),orBamHI(pHT10, pml)before DNAtransfection (see the text). Thespecificactivities represent the number of foci induced per 2.5 x I0 cells perpmolof DNA transfected and are expressedasfocus-forming units (ffu). They werecalculated fromatleastfourreplicate
determinations.
pHT25) requires the
acquisition
ofterminationpolyadenylation
of RNAtranscripts
(9, 15, 29,
and
polyadenylation
signals
for RNA expres-30,
33),
the enhancement of thetransforming
sion;
alternatively,
an LTR 3' to v-mos(i.e.,
activity
ofv-mosdoesnotapparently
result from pHT21, pml3)imposes
therequirement
of ob- thesetranscriptional
elements,
and thisimplies
taining signals
fortheinitiation oftranscription.
that the LTR contains other sequences thatAlthough the LTRdoes encode
transcriptional
influence theexpression
ofv-mos.If this istrue, controlsignals
fordirecting
the initiation and theneliminating
thepolyadenylation signals
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.488.119.385.62.378.2]ET AL.
from an LTR introduced 3' to v-mos should not
influence its transforming efficiency and could serve to identify the regions ofthe LTR
respon-siblefor enhancement.
Determination of the sequences responsible for enhancement by an LTR introduced3' to v-mos. To identify the essential sequences required for enhancementbyan LTR3' tov-mos, wetested thetransformingactivity ofaseriesof
recombi-nantDNAclones constructedtocontain various
sequences derived from the LTR (Fig. 5). As
previously shown, pHT10, aplasmid containing
v-mos and 900 bp of Moloney leukemia virus
sequences 5' to v-mos, butno LTRsequences,
exhibits a very lowtransforming efficiency (<5
focus-forming units per pmol of DNA) (7), whereasaplasmidcontaining the entire proviral DNA ofml MSV (pml) produced 4,500 focus-forming units per pmol ofDNA (Fig. 5). The
insertion ofa single LTR 3' to the v-mos se-quence (pHT22) resulted in a 200-fold stimula-tion of the
transforming
efficiency
observed with pHT10 (Fig. 5). Anequivalent
enhancement of thepHT10transformingefficiencywasobserved when DNA fragments containing only se-quencesderivedfromtheU3 region ofthe LTR wereintroduced 3'to v-mos(Fig.
5, pHTS3 and pHTX3). However, when sequences derived from the R and U5 region of the LTR were present 3' to v-mos (pHTR5), thetransformingefficiency
wasequivalent
to the level observedwith the transfection of pHT10 DNA (Fig. 5). These results demonstrate that theregion ofthe
LTR
responsible
for the enhancement of thetransforming
activity
of v-mos consists ofse-quences present in the U3
region
and thatse-quencesinthe RandU5
region (i.e.,
polyadeny-lation
signals)
do notenhance thetransforming
activity
ofv-mos.DISCUSSION
The results from DNA transfection assays
have shown that thetransforming activity of v-mosis enhancedby introducingasingleLTRin
either
a5' or a3'position
relativeto v-mos (7,19). In this report, we have presented results
from the analysis of RNA and DNA isolated from cells transformed by the transfection of recombinantDNAclonescontaining v-mos and a single LTR. In each of the transfectants, we
demonstratedthe presence of additional copies ofv-moswithinthecellularDNAin contrast to a
single haploid DNA copy observed in normal
NIH3T3cells (14,24). Theanalysis ofrestriction
endonuclease digests of cellular DNA from these transfectants by hybridization with vari-ousprobes specific forsequences present in the
transfectedDNA confirms theobservation that asingleLTRis sufficientfor the enhancement of thetransforming activityof v-mos. Tandem
inte-grationsof the transfected DNA,recreating pro-virus-like DNAstructures, are not necessary for theexpression of v-mos RNAtranscriptsorfor
the induction of cellular transformation. It shouldbe notedthatin theseexperimentswedid
not distinguish between the introduction of transfectedDNAinto the hostchromosome and its insertion into high-molecular-weight carrier
DNA in a "pekelasome" structure (27).
The rearrangement of the v-mos and LTR sequencesis not required fortheexpression of
v-mos RNA transcripts, and the relative
posi-tions of the v-mos and LTR sequences within thetransfectedDNAs areconserved inthe inte-grated DNA copies. The fate ofthe vectorand cellularflankingsequences presentin the
trans-fected plasmid DNAs varied for each of the
transfectants that we examined. In one case
(i.e., pml3), theentire pBR322vector sequence present in the transfected plasmid DNA was
deleted from the integrated DNA copy. Our resultsdemonstrate that theLTRprovides
tran-scriptional control elements that insure the expression ofv-mos RNAtranscripts.
The cells transformed by the transfection of
DNA containing an LTR 5' to v-mos express RNAtranscripts thathybridize withmosandU5
probes, butnotwith probesrepresentingthe U3
region ofthe LTR. The termination and
polya-denylation signals utilized by the v-mos RNA
transcripts in these transfectants are not ob-tainedfrom the transfected LTR. Thesesignals
areacquired from eithervector, host,orcarrier DNA. Although downstream promotion
pro-vides one mechanism for the activation ofan oncogene, the
induction
oftransformation
by the insertion ofan LTRin a3' position relativeto v-mosisinconsistent withapromoter activa-tion model. Blairetal. first observed that these
constructs inducedtransformationasefficiently
as the 5' LTR v-mos constructs (7). We have
shown here that cells transformed by the
trans-fection ofDNA
containing
an LTR 3'to v-mos express RNAtranscripts
thathybridize
withmos and U3 probes. The U5
probe
does not annealto the v-mosRNAtranscriptsexpressedinthesecells,demonstratingthattranscript initi-ation sites present in the transfected LTR are not utilized in the expression of these v-mos RNA transcripts. Again, these signals must be acquired from either vector, host, or carrier DNA sequences and,becauseofthehigh trans-formation frequency in their absence, must be
easily
acquired
during
transfection. Becauseof the size ofthe 3.2-kbmos-containing
RNA tran-scriptin thepHT21 transfectant (Fig. 3)and thesize ofthe 2.1-kb
mos-containing
transcript inpml3 transfectants (Fig. 4), we cannot exclude
thepossibleuseofproviralsequencespreceding
v-mos to provide the initiation sites. By
intro-J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
ducing an LTR 5' to v-mos, we provide
tran-scriptional control elements for the expression ofv-mos-containing RNA transcriptsanalogous
to the promoter insertion model suggested for avian leukosis virus(ALV)-inducedbursal lym-phomas (13, 22, 23, 25, 26). These studies show evidence for an increased expression of the chicken c-myc generesulting from the insertion ofan ALV LTR 5' toc-myc. Payneet al. have shown that in one example of ALV-induced bursal lymphomas, the ALV LTR can also in-creasetheexpression ofc-mycfroma3' position (26).
The results obtained witha numberof simian
virus40(SV40)deletionmutantshavesuggested thatimportant elements controlling the expres-sion ofSV40RNAtranscripts are contained in the72-bp tandem repeatslocatedneartheSV40 origin of replication (5,6, 12). Moreauetal. have shown that the 72-bp tandemrepeat fromSV40 can stimulate T-antigen expression in chimeric plasmids where theSV40 TATA box region has beensubstituted with conalbumin oradenovirus 2 major late promoter sequences (21). Further-more, the level of T-antigen expression is not
alteredby reversing the orientation of the 72-bp tandem repeat (21). The enhancement of gene expression forgenesother than thosepresentin
SV40genomic DNA has also been demonstrat-ed. Capecchi reported that the insertion ofan SV40 DNA fragment containing the 72-bp
tan-demrepeatintoaplasmidcontaining the thymi-dine kinase gene of herpes simplex virus in-creases the transformation frequency ofmouse LMTK- cells (8). The expression of the rabbit
P-globin gene is enhanced in HeLa cells when
this gene is inserted into a recombinant vector
that contains SV40 DNA sequences, including the72-bp repeatelement (4).
MSVproviral DNA has been shown to con-taina73-bp tandemrepeatwithin theU3 region of theLTR(9). Levinsonetal. have demonstrat-ed that the 73-bp repeat from ml MSV can replace the SV40 72-bprepeatas anactivator for theexpression of viralsequenceslocated
down-streamfrom the73-bprepeat(16). We tested the ability of various sequences, derived from the LTRandintroduced 3'tov-mos,toenhancethe transforming activity of v-mos (Fig. 5). The results from this analysis demonstrate that
se-quences within the first 300 bases of the LTR
canyield the samelevel ofenhancement of the
transforming activity ofv-mos ascan acomplete LTR.Sequencespresentinthe R andUS regions ofthe LTR(i.e., polyadenylation signals) donot
stimulate the transforming activity of v-mos.
These results show that the region of the LTR responsible for enhancement consists of
se-quences present in the U3 region, a sequence thatcontains the73-bp tandemrepeat.
ACKNOWLEDGMENTS
We aregrateful to A. Woodworth, M. Oskarsson, and T. Robins forcriticalreading of the manuscript and to K. Barry, L. Shaughnessy, and K. Cannon for assistance inpreparing thismanuscript.
LITERATURE CITED
1. Alwine, J. C., D. J.Kemp, and G.R.Stark. 1977.Method fordetection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc. NatI. Acad. Sci. U.S.A. 74:5350-5354.
2. Aviv,H., and P. Leder. 1972.Purification of biologically active globin messenger RNA by chromatography on oligo-thymidylic acid-cellulose. Proc. Natl. Acad. Sci. U.S.A.69:1408-1412.
3. Bailey, J. M., and N. Davison. 1976.Methyl mercury as a reversible denaturing agent for agarose gel electrophore-sis. Anal. Biochem. 70:75-85.
4. Banerji,J., S.Rusconi,and W.Schaffner. 1981. Expres-sionof aB-globingene isenhancedbyremoteSV40 DNA sequences. Cell27:299-308.
5. Benorst, C., and P. Chambon. 1980. Deletionscovering theputative promoterregion ofearlymRNAsof simian virus 40do notabolishT-antigen expression. Proc. Natl. Acad. Sci. U.S.A. 77:3865-3869.
6. Benorst, C., and P. Chambon. 1981. In vivo sequence requirements of the SV40 early promoterregion.Nature (London) 290:304-310.
7. Blair, D. G., W.L.McClements,M. K.Oskarsson,P.J. Flwchinger, and G. F. Vande Woude. 1980. Biological activity of clonedMoloneysarcomavirusDNA: terminal-lyredundant sequences may enhancetransformation effi-ciency.Proc.Natl. Acad. Sci. U.S.A. 77:3504-3508. 8. Capecchi, M. R. 1980.Highefficiencytransformation by
direct microinjection of DNA into cultured mammalian cells.Cell 22:479-488.
9. Dhar,R.,W. L.McClements,L.W.Enquist,andG.F. Vande Woude. 1980.Nucleotide sequences ofintegrated Moloney sarcomaprovirus longterminalrepeatsand their host and viraljunctions. Proc. Natl. Acad. Sci. U.S.A. 77:3937-3941.
10. Ferretti, L., and V.Sgaramella. 1981. Temperature depen-dence of thejoiningbyT4 DNAligaseof terminiproduced bytype IIrestriction endonucleases.NucleicAcids Res. 9:85-93.
11.Graham, F. L., and A. J. van der Eb. 1973. A new techniquefor the assay ofinfectivityof human adenovir-uses5DNA.Virology52:456-461.
12. Gruss, P.,R.Dhar, and G.Khoury.1981.Simian virus 40 tandem repeated sequences as anelement of theearly promoter. Proc. Natl. Acad.Sci. U.S.A. 78:943-947. 13. Hayward, W. S., B. G. Neel, and S. M. Astrin. 1981.
Activation of a cellular onc geneby promoterinsertionin ALV-induced lymphoid leukosis. Nature (London) 290:475-480.
14.Jones, M., R. A. Bosselman, F. H. van der Hoorn, A. Berns,H.Fan, and I. M. Verma. 1980.Identificationand molecular cloning of Moloney mouse sarcoma virus-specific sequences from uninfected mouse cells. Proc. Natl. Acad.Sci. U.S.A. 77:2651-2655.
15. Ju, G., and M. A. Skalka. 1980. Nucleotide sequence analysis of the long terminal repeat (LTR) of avian retroviruses: structural similarities withtransposable ele-ments.Cell 22:379-386.
16. Levinson,B., G.Khoury, G. Vande Woude, and P. Gruss. 1982.Activation of SV40 genomeby 72-basepair tandem repeats of Moloney sarcoma virus. Nature (London) 295:568-572.
17. Maxam, A.M., andW.Gilbert. 1980. Sequencing end-labeled DNA with base-specific chemical cleavages. MethodsEnzymol.65:499-560.
18. McClements,W.L.,R.Dhar,D.G.Blair,L.Enquist,M.
on November 10, 2019 by guest
http://jvi.asm.org/
WOOD ET AL.
Oskarsson, and G. F. Vande Woude. 1981. The long terminal repeat of Moloney sarcoma provirus. Cold Spring Harbor Symp. Quant. Biol. 45:699-705. 19. McClements, W. L., L. W. Enquist, M. Oskarsson, M.
Sullivan, and G. F. Vande Woude. 1980. Frequent site-specific deletion of coliphage X murine sarcoma virus recombinants and its use in the identification of a retro-virus integrationsite.J. Virol. 35:488-497.
20. McDonell, M. W., M. N. Simon, and F. W.Studier. 1977. Analysis ofrestriction fragments of T7 DNA and determi-nation of molecular weights by electrophoresis in neutral andalkaline gels. J. Mol. Biol. 10:119-146.
21. Moreau, P., R. Hen, B. Wasylyk, R. Everett, M. P. Gaub, and P. Chambon. 1981.TheSV40 72 basepair repeat has a strikingeffect on gene expression both in SV40 and other chimeric recombinants. NucleicAcids Res. 9:6047-6068. 22. Neel, B. G., W. S. Hayward, H. L. Robinson, J. Fang, and
S. M. Astrin. 1981.Avian leukosisvirus-induced tumors have common proviral integration sites and synthesize discrete new RNAs:oncogenesis by promoter insertion. Cell 23:323-334.
23. Noori-Dalou, M. R., R. A. Swift, H.-J. Kung, L. B. Crittenden,and R.L. Witter.1982.Specific integrationof REV proviruses in avian bursal lylmphomas. Nature (London) 294:574-576.
24. Oskarsson, M., W. L. McClements, D. G. Blair, J. V. Maizel, and G. F. VandeWoude. 1980. Properties of a normal mouse cell DNA sequence(sarc) homologous to the src sequence of Moloney sarcoma virus. Science 207:1222-1224.
25. Payne, G. S., J. M. Bishop, and H. E. Varmus. 1982. Multiple arrangements of viral DNA and an activated host oncogene in bursal lymphomas. Nature (London) 295:209-214.
26. Payne, G. S., S.A.Courtneidge, L. B. Crittenden, A. M. Fadly, J.M.Bishop, andH.E.Varmus.1981.Analysis of
avianleukosis virus DNA and RNA in bursal tumors: viral geneexpression is not required for maintenance of the tumorstate. Cell 23:311-322.
27. Perucho, M., D. Hanahan, andM.Wigler.1980.Genetic and physical linkage of exogenous sequences in trans-formed cells. Cell 22:309-317.
28. Rigby, P. W.J.,M.Dieckmann, C. Rhodes, and P. Berg. 1977. Labeling deoxyribonucleic acid to high specific activity in vitro bynick translation with DNA polyamer-aseI. J. Mol. Biol. 113:237-251.
29. Shinnick, T. M., R. A. Lerner, and J. G. Sutcliffe. 1981. Nucleotide sequence of Moloney murine leukemia virus. Nature(London) 293:543-548.
30. Shoemaker, C., S.Goff,E.Gilboa,M.Paskind, S. Mitra, and D. Baltimore. 1980. Structure of a cloned circular Moloney murine leukemia virus DNA molecule contain-inganinverted segment:implications for retrovirus inte-gration. Proc. Natl. Acad.Sci. U.S.A. 77:3932-3936. 31. Southern, E. M. 1975. Detection of specific sequences
among DNAfragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517.
32. Tilghman, S. M., D. C. Tiemeier, F.Polsky, M. H. Edgell, J. G.Seidman,A.Leder, L. W. Enquist, B. Norman, and P. Leder.1977.Cloningspecificsegmentsof the mammali-angenome: bacteriophage lambda containing mouse glo-bin andsurroundinggene sequences. Proc. Natl. Acad. Sci. U.S.A. 74:4406-4410.
33. Van Beveren,C.,F.vanStraaten, J.A.Galleshaw,andI. M.Verma.1981.Nucleotide sequence of the genome of a murine sarcoma virus. Cell 27:97-108.
34. VandeWoude, G.F., M. Oskarsson, W. McClements, L. Enquist, D. Blair, P. Fischinger, J. Maizel, and M. Sulli-van. 1980.Characterization of integrated Moloney sarco-maprovirus and flankingsequences cloned in bacterio-phage lambda. Cold SpringHarborSymp. Quant. Biol. 44:735-746.
J. VIROL.