0022-538X/11/$12.00 doi:10.1128/JVI.00876-11
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
The Histone Acetyltransferase CLOCK Is an Essential Component of
the Herpes Simplex Virus 1 Transcriptome That Includes
TFIID, ICP4, ICP27, and ICP22
䌤
Maria Kalamvoki and Bernard Roizman*
Marjorie B. Kovler Viral Oncology Laboratories, the University of Chicago, 910 East 58th Street, Chicago, Illinois 60637
Received 29 April 2011/Accepted 28 June 2011
Studies published elsewhere have shown that the herpes simplex virus regulatory protein ICP0 interacts with BMAL1, a partner and regulator of circadian histone acetyltransferase CLOCK, that both proteins localize at ND10 bodies and are stabilized by viral proteins, that enzymatically active CLOCK partially complements
⌬ICP0 mutants, and that silencing of CLOCK suppresses the expression of viral genes. Here we report that CLOCK is a component of the transcriptional complex that includes TFIID, ICP4, ICP27, and ICP22. The results suggest that the CLOCK histone acetyltransferase is a component of the viral transcriptional machin-ery throughout the replicative cycle of the virus and that ICP27 and ICP22 initiate their involvement in viral gene expression as components of viral transcriptome.
In this report we present evidence that the CLOCK histone acetyltransferase recruited to the ND10 bodies and ultimately to the replication compartments (17) is an integral component of a complex that includes the infected cell proteins ICP4, ICP27, and ICP22 and at least one component of the TFIID transcription complex required for expression of herpes sim-plex virus 1 (HSV-1) genes. The circumstances that led to these studies were as follows.
On entry of viral DNA into the nuclei of infected cells, cellular proteins, including histones and repressors, bind the DNA and attempt to silence it. Studies done in this and other laboratories have shown that at least two checkpoints deter-mine the outcome of a productive infection (9, 12–14, 20, 21,
27, 37). In the first, the viral tegument protein VP16 (␣TIF)
recruits HCF-1, Oct1, and LSD1 to response elements of ␣
promoters (20, 21, 25, 28, 34). LSD1 demethylates histones and
enables the transcription of ␣ genes (20, 21, 25, 37). The
second checkpoint requires the suppression of cellular
repres-sors by ICP0 in order to enable the expression of and ␥
genes. ICP0 displaces HDAC1 or HDAC2 from the REST/ CoREST complex (12–14) and enables transcription of viral genes through recruitment of transcriptional machinery that would include histone acetyltransferases, RNA POL II, and a variety of transcriptional factors. In the course of a search for a histone acetyltransferase that could be involved in chromatin remodeling and initiation of transcription at these checkpoints, we noted that ICP0 binds BMAL1, a circadian regulator of CLOCK histone acetyltransferase (8, 15, 19). Studies of BMAL1 and CLOCK led to the observation that both proteins localize at ND10 bodies, that CLOCK is stabilized, and that
insertion of a wild-type CLOCK gene in a⌬ICP0 virus
aug-mented the growth of the virus mutant whereas incorporation
of a mutated CLOCK gene devoid of enzymatic activity did not. Finally, depletion of CLOCK by small interfering RNA (siRNA) prior to infection delayed or diminished the
expres-sion of viral␣genes (17).
In a series of elegant studies, DeLuca and associates dem-onstrated that ICP4 recruits to promoters of viral gene tran-scriptional factors that would be necessary for transcription of viral genes (4, 24, 29, 33). The question arose whether CLOCK is a component of the same transcriptional complex. Here we report that CLOCK pulls down TBP, a component of TFIID transcriptional factor in both mock-infected and infected cells and that, in addition, it interacts with ICP4, ICP22, and ICP27.
MATERIALS AND METHODS
Cells and viruses.HSV-1(F), a limited passage isolate, is the prototype used in this lab (10). The source and propagation of HEp-2 cells were reported elsewhere (17).
Purification of the GST-CLOCK fusion protein and GST-CLOCK pulldown assays.The full-length CLOCK was PCR amplified from the KIAA0334 clone, digested with EcoRV, and inserted into the SmaI site of pGEX-4T2, in frame with glutathioneS-transferase (GST). The purification of CLOCK was done as previously described with minor modifications (18). Briefly,Escherichia coli XL1-Blue cells transformed with the pGEX-4T2 or with the pGEX-4T2–CLOCK-expressing plasmids were induced with 0.3 mM IPTG (isopropyl- -D-thiogalac-topyranoside; Denville Scientific) for 3 h after the optical density at 600 nm reached a value of 0.5. The harvested cells were lysed by sonication in phosphate-buffered saline (PBS) supplemented with Triton X-100 and Sarkosyl at a final concentration of 1%. The cell debris was removed by centrifugation, and the GST or GST-CLOCK proteins were adsorbed to the glutathione agarose beads (Sigma). Purified proteins were eluted after extensive rinsing of the beads with PBS–1% Triton X-100. For the GST pulldown assays, 2⫻107HEp-2 cells, mock
infected or infected with the wild-type virus at 10 PFU/cell, were lysed in HEPES–1% Triton X-100 buffer consisting of 50 mM HEPES (pH 7.4), 250 mM NaCl, 10 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride (PMSF), and protease
cocktail inhibitor (Sigma) supplemented with 1% Triton X-100. The lysates were reacted overnight with equal amounts of GST or GST-CLOCK proteins immo-bilized on glutathione beads and were rinsed three times with the HEPES–1% Triton X-100 buffer, and the bound protein complexes were dissolved in loading buffer composed of 4% SDS, 100 mM Tris-Cl (pH 6.8), 20% glycerol, and 0.2% bromophenol blue, supplemented with-mercaptoethanol, and boiled for 5 min. After centrifugation to remove the beads, the eluted proteins were subjected to electrophoretic analysis on 9% polyacrylamide gels.
* Corresponding author. Mailing address: The Marjorie B. Kovler Viral Oncology Laboratories, The University of Chicago, 910 East 58th Street, Chicago, IL 60637. Phone: (773) 702-1898. Fax: (773) 702-1631. E-mail: [email protected].
䌤Published ahead of print on 6 July 2011.
9472
on November 7, 2019 by guest
http://jvi.asm.org/
Immunoprecipitation.Approximately 5⫻106
HEp-2 cells mock infected or exposed to 10 PFU of HSV-1(F) per cell were harvested at the times indicated in Results and resuspended in lysis buffer composed of 50 mM HEPES (pH 7.4), 150 mM NaCl, 1% NP-40, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF, 0.5% sodium deoxycholate, 1 mM NaF, 1 mM Na3VO4, and protease cocktail
inhib-itors (Sigma). The cells were incubated for 30 min on ice, cell debris were removed by centrifugation at 7,000 rpm for 10 min, and the supernatant was supplemented with 2g antibody. The CLOCK (H-276) and LAMP-1 (H-228) rabbit polyclonal antibodies were purchased from Santa Cruz. The BMAL-1 rabbit polyclonal antibody has been described elsewhere (19). After overnight incubation at 4°C, protein A beads were added to the samples and incubation continued for 2 h. Finally, the beads were collected and rinsed four times with the lysis buffer. The immunocomplexes bound on beads were dissolved in loading buffer composed of 4% SDS, 100 mM Tris-Cl (pH 6.8), 20% glycerol, and 0.2% bromophenol blue, supplemented with-mercaptoethanol, and boiled for 5 min. After centrifugation to remove the beads, the eluted proteins were electropho-retically separated in denaturing polyacrylamide gels.
Immunoblots. The electrophoretically separated proteins were electrically transferred to nitrocellulose sheets, blocked with PBS supplemented with 0.02% (vol/vol) Tween 20 (PBST) and 5% nonfat milk, and reacted overnight at 4°C with appropriate primary antibodies diluted in PBST-1% nonfat milk. The an-tibody sources and concentrations were as follows: ICP4 (1:1,000) and ICP27 (1:1,000) mouse monoclonal antibodies, Goodwin Institute for Cancer Research, Plantation, FL; TFIID (TBP20) (sc-56796), TAFIIp250 (sc-735), and TFIIB (sc-81399) (1:500) mouse monoclonal antibodies, Santa Cruz. The CLOCK rab-bit polyclonal antibody (Santa Cruz) was used in a 1:500 dilution. The ICP22 (C terminus) rabbit polyclonal antibody has been previously described (23) and was used in a 1:1,000 dilution. After several rinses with PBST-1% nonfat milk, the membranes were reacted with the appropriate secondary antibody conjugated either to alkaline phosphatase or to horseradish peroxidase. Finally, protein bands were visualized with 5-bromo-4-chloro-3-indolyl phosphate (BCIP)–ni-troblue tetrazolium (NBT) (Denville Scientific) or with ECL Western blotting detection reagents (Amersham Biosciences) according to the manufacturer’s instructions.
RESULTS
CLOCK pulls down the TBP subunit of the TFIID complex.
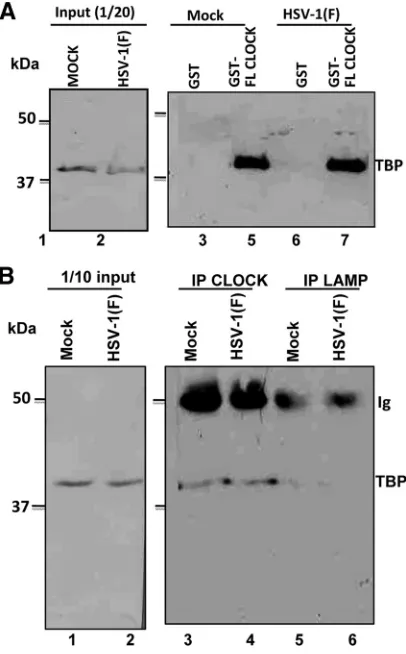
In an earlier study we showed that CLOCK histone acetyl-transferase is recruited to ND10 bodies and plays an essential role in the expression of viral genes (17). A central question posed by this finding is whether CLOCK is a component of the complex of cellular and viral proteins recruited to viral DNA for transcription of viral genes. Earlier studies have shown that key components of the transcription complex TFIID are re-cruited by ICP4 (4, 24, 29, 33). The question arose, therefore, whether CLOCK forms a complex with components of the transcription initiation complex. To resolve this question, ly-sates of mock- and HSV-1(F)-infected cells harvested 8 h after infection were reacted with GST-CLOCK chimeric protein. The proteins bound to the GST-CLOCK protein were sepa-rated on a denaturing polyacrylamide gel and probed with antibody to TBP—a component of TFIID. The results, shown in Fig. 1A, indicate that in contrast to GST, GST-CLOCK chimeric protein pulled down TBP from both infected and uninfected cells.
In the second experiment in this series, lysates of mock-infected and HSV-1(F)-infected cells were reacted with antibody to TBP or to LAMP—an irrelevant antibody. The immune complexes were collected, solubilized, electrophoretically separated in dena-turing gels, and reacted with anti-TBP antibody as described in Materials and Methods. The results (Fig. 1B) show that antibody to CLOCK pulled down TBP from mock-infected and infected cells. In contrast, TBP could not be detected in immune precip-itates brought down by antibody to LAMP.
We conclude that CLOCK forms complexes containing com-ponents of TFIID in both infected and uninfected cells.
GST-CLOCK pulls down TBP but not TAFII250 or TFIIB.
[image:2.585.319.522.70.397.2]Earlier studies have shown that ICP4, a major regulatory pro-tein of HSV-1, interacts with components of the RNA poly-merase II transcription complex. These include TAFII250 and TBP (4, 24, 29, 33). The observation that CLOCK interacts with TBP in both mock-infected and infected cells raised the question whether it also interacts with TAFII250 and TFIIB. To resolve this question, purified GST or GST-CLOCK pro-teins bound to beads were reacted with lysates of HEp-2 cells harvested 8 h after mock infection or infection with wild-type virus. The proteins bound to GST or GST–full-length CLOCK chimeric proteins were solubilized, electrophoretically sepa-rated in a denaturing gel, and probed with antibody to TBP, TFIIB, or TAFII250. The results shown in Fig. 2 indicate that CLOCK pulled down TBP but not TFIIB or TAFII250.
FIG. 1. The TBP subunit of TFIID exists in complex with CLOCK. (A) Lysates from mock- or HSV-1(F)-infected (10 PFU/cell) HEp-2 cells (2⫻107), harvested at 8 h after infection, were mixed with an
equal amount of GST or GST-CLOCK purified proteins immobilized on glutathione beads. The bead-bound complexes were electrophoreti-cally separated on 9% polyacrylamide gels, transferred to nitrocellu-lose sheets, and immunoblotted with the TFIID (TBP) mouse mono-clonal antibody, as described in Materials and Methods. (B) HEp-2 cells (5⫻106) were mock infected or exposed to 10 PFU of
HSV-1(F) per cell. The cells were harvested 8 h after infection and reacted with CLOCK or LAMP-1 rabbit polyclonal antibodies. The electro-phoretically separated immune precipitates were probed with anti-TFIID (TBP) antibody as described above.
on November 7, 2019 by guest
http://jvi.asm.org/
GST-CLOCK pulls down ICP4 and ICP27 from wild-type virus-infected cells. Purified GST or GST-CLOCK proteins immobilized on beads were reacted with lysates of HEp-2 cells harvested 8 h after mock infection or infection with wild-type virus. The electrophoretically separated proteins in denaturing gels were transferred to a nitrocellulose sheet and probed with antibody to ICP4 (Fig. 3A) or ICP27 (Fig. 3B). The results shown in Fig. 3 indicate that GST-CLOCK but not GST pulls down both ICP4 and ICP27.
BMAL1 and CLOCK interact in infected cells, and both proteins pull down ICP22. BMAL1 interacts with and regu-lates the function of CLOCK (8, 15). A central question is whether BMAL1 continues to interact with CLOCK in in-fected cells and whether it is a component of the transcription complex containing viral regulatory proteins. To resolve this question, two series of experiments were done. In the first, lysates of HEp-2 cells harvested 8 h after infection or mock infection were reacted with antibody to BMAL1, CLOCK, or LAMP1. The immune complexes were collected, electropho-retically separated in a denaturing gel, and probed with anti-body to CLOCK. The results (Fig. 4A) showed that both BMAL1 and CLOCK antibodies precipitated CLOCK from lysates of mock-infected or infected cells. The LAMP1
anti-body used in this experiment as a nonspecific control antianti-body failed to precipitate significant amounts of CLOCK.
The objective of the second series of experiments was to determine whether ICP22, a viral protein that regulates late gene expression (5), is also contained in the complexes con-taining CLOCK. In these experiments, lysates of HEp-2 cells harvested 8 h after infection or mock infection were reacted with antibody to BMAL1, CLOCK, or LAMP1. The immune complexes were electrophoretically separated on denaturing gels and reacted with antibody to ICP22. As shown in Fig. 4B, both CLOCK and BMAL1 pulled down ICP22. The control antibody to LAMP1 failed to pull down ICP22.
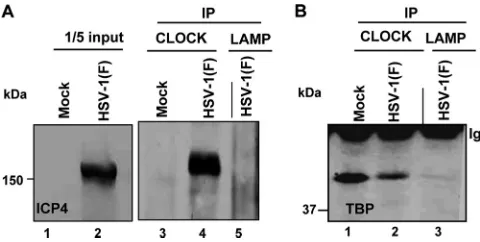
CLOCK interacts with TBP and ICP4 at relatively late times after infection.One of our objectives was to determine whether CLOCK is associated with TBP and ICP4 at late times after infection. We selected the 14-h time after infec-tion with 10 PFU per HEp-2 cell because late viral proteins are made at high rates at that time. Specifically, lysates of mock-infected or infected cells were reacted with antibody to CLOCK. The immune complexes were collected, electro-phoretically separated on denaturing gels, and reacted with either antibody to ICP4 (Fig. 5A) or TBP (Fig. 5B). As shown in Fig. 5, antibody to CLOCK pulled down both TBP
FIG. 2. GST-CLOCK pulls down TFIID (TBP) but not TAFII250 or TFIIB. Purified GST or GST-CLOCK proteins immobilized on beads were reacted with equal amounts of lysates from 2⫻107HEp-2 cells that were mock infected or exposed to 10 PFU of HSV-1(F) per cell.
(A) Ponceau S staining of the bound proteins. (B and C) Immunoblots of the TBP or TFII250 proteins eluted from GST or the GST-CLOCK proteins bound to the glutathione beads. (D) Input of TBP or TFII250 in the lysates reacted with GST or GST-CLOCK proteins. (E and F) Immunoblots of TFIIB pulled down by GST or GST-CLOCK or present in the lysates reacted with the GST or GST-CLOCK proteins. The lysate used for detection of TFIIB was from another experiment but prepared in a manner identical to that used for detection of TBP and TFII250 (A to D). The antibodies against TAFII250, TBP, or TFIIB were used at concentrations indicated in Materials and Methods. (F) Viral or cellular proteins present in 1/10 of the input of the HEp-2 cell lysates used for immune precipitation.
on November 7, 2019 by guest
http://jvi.asm.org/
and ICP4 from lysates of infected cells whereas the control antibody LAMP1 did not.
DISCUSSION
The impetus for the initiation of the studies reported here is the observation that ICP0 interacts with BMAL1 (19), a part-ner and regulator of the circadian histone acetyltransferase CLOCK (8, 15). Both BMAL1 and CLOCK localize at ND10 nuclear bodies in both uninfected and infected cells (17). Si-lencing of CLOCK resulted in a significant delay in the
expres-sion of ␣ genes (17). The role of ICP0 may be 2-fold—to
enhance recruitment of BMAL1 and CLOCK and, along with
other␣proteins, to stabilize these proteins (17). The
signifi-cance of the recruitment of BMAL1 and CLOCK may be deduced from the observation that wild-type, enzymatically active CLOCK complements in part the replication of a mu-tant virus in which ICP0 was replaced with the CLOCK gene. In contrast, enzymatically inactive CLOCK was ineffective (17).
In the initial stages of activation of transcription of␣genes,
the viral transactivator VP16 (␣TIF) recruits OCT1, HCF-1,
[image:4.585.300.540.550.671.2]and LSD1 and numerous other transcriptional factors (9, 12– 14, 20, 21, 25, 27, 28, 34, 37). The function of LSD1 is to demethylate repressive histones, and it would be expected that chromatin remodeling by acetylation of histones would be one of the steps necessary to initiate transcription of viral genes (14, 25). If CLOCK was involved in this process, it would be expected that it would be a component of the complex involved
FIG. 3. GST-CLOCK pulls down ICP4 and ICP27. Purified GST or GST-CLOCK proteins immobilized on beads were reacted with equal amounts of lysates from 2⫻107HEp-2 cells that were mock infected
or exposed to 10 PFU of HSV-1(F) per cell. Immunoblots of the GST or the GST-CLOCK-bound proteins eluted from the glutathione beads with the ICP4 antibody (A) and with the ICP27 antibody (B), as indicated in Materials and Methods. The relevant proteins present in the 1/10 input of the HEp-2 cell lysates used for immune precipitation are shown.
FIG. 4. CLOCK and BMAL1 exist in a complex that also contains ICP22 in wild-type virus-infected cells. HEp-2 cells (5⫻106) either
mock infected or exposed to 10 PFU of HSV-1(F) per cell were harvested at 8 h after infection and lysed. Immunoprecipitations were done with the BMAL1, CLOCK, or control LAMP-1 antibody. Immu-noblots were done with the rabbit polyclonal antibody against CLOCK (A) or with the ICP22 antibody (B), as indicated in Materials and Methods. The proteins present in 1/10 of the lysates used for immu-noprecipitation are shown.
FIG. 5. The ICP4-CLOCK-TFIID (TBP) complex persists even at late hours after infection. HEp-2 cells (5⫻106), either mock infected
or exposed to 10 PFU of HSV-1(F) per cell, were harvested at 14 h after infection. Immunoprecipitations were done with the CLOCK or the control LAMP-1 antibodies. Immunoblots were probed with the anti-ICP4 antibody (A) or the anti-TFIID (TBP) antibody (B).
on November 7, 2019 by guest
http://jvi.asm.org/
in the transcription of viral genes. Earlier studies have shown that the transcriptional complex includes ICP4 and that ICP4 pulls down TBP—a component of TFIID (4, 24, 29, 33). Other transcriptional factors, such as TAFII250, then also associate with the transcriptional complex (4, 24, 29, 33). In the studies reported here, we show that CLOCK interacts with both ICP4 and TBP. We also show that ICP27 and ICP22 are also com-ponents of this complex. Under conditions tested, we were unable to demonstrate an interaction with TFII250 and, as in the case of ICP4, we have not been able to show a direct interaction between CLOCK and TFIIB (4, 24, 29, 33).
The significance of these observations stems from two con-siderations. In an earlier study, we showed that silencing CLOCK with siRNA results in a significant delay in the
ex-pression of␣genes (17). In this study, we show that CLOCK
interacts with ICP4 and that the interaction with TBP and ICP4 is demonstrable as late as 14 h after infection. The results suggest, therefore, that CLOCK is associated with the tran-scriptional complex involved in the expression of viral genes throughout the replication of HSV-1.
The second consideration centers on the key observation that CLOCK pulls down both ICP27 and ICP22. ICP27 plays a
critical role in the accumulation and translation of post-␣
mRNAs. The focus of much of the research on ICP27 is on its role in repression of splicing and transport of viral mRNAs to the cytoplasm. ICP27 has also been reported to bind RNA polymerase II, poly(A) binding protein and the virion host shutoff protein that can degrade mRNA in polyribosomes (6, 7, 11, 16, 30–32, 35, 36). The results reported here suggest the possibility that the interaction of ICP27 with mRNA occurs at the moment of synthesis and may continue throughout the life span of the mRNA.
One of the known functions of ICP22 is to enable efficient accumulation of RNA transcripts of a subset of late genes (5, 26). ICP22 appears to perform two key functions. Thus, ICP22
and the protein kinase UL13 mediate the phosphorylation of
RNA polymerase II. In addition, both proteins play a role in the degradation of cyclins A and B to enable the orphan
partner cdc2 to bind polymerase accessory factor UL42. Cdc2
and its partner UL42 mediate the posttranscriptional activation
and recruitment of topoisomerase II (1–3). It has been sug-gested that the function of the topoisomerase is to enable more
efficient transcription of late (␥2) genes by resolving the tangles
of DNA concatemers accumulating in the course of viral DNA synthesis late in infection (1–3, 5).
Lastly, the recruitment of BMAL1/CLOCK complex to re-model viral chromatin to enable the transcription of viral genes was unexpected. As noted in the introduction, BMAL1 is a regulator of the circadian histone acetyltransferase CLOCK. When activated, BMAL1 activates the transcription of, part-ners with, and translocates CLOCK to the nucleus. Both pro-teins in the complex are phosphorylated, and, in addition, CLOCK also acetylates BMAL1. The half-life of the protein in the nucleus is relatively short (8, 15, 22). As noted earlier, CLOCK is stabilized in infected cells, and in this report we show that it is present in complexes containing TFIID and ICP4 at both relatively early and late stages of infection. In principle, the necessity to remodel viral chromatin does not explain the choice of CLOCK for this function. Viruses do not throw dice. Why did HSV-1 evolve a relationship with CLOCK
rather than the myriad other acetyltransferases available in the cell? Is it because BMAL1/CLOCK complex is subject to reg-ulatory processes that are less complex and it is more readily conscripted by the virus?
ACKNOWLEDGMENT
These studies were aided by a grant from the National Cancer Institute (grant R37 CA78766).
REFERENCES
1.Advani, S. J., R. Brandimarti, R. R. Weichselbaum, and B. Roizman.2000. The disappearance of cyclins A and B and the increase in activity of the G2/M-phase cellular kinase cdc2 in herpes simplex virus 1-infected cells
require expression of the␣22/US1.5 and UL13 viral genes. J. Virol.74:8–15.
2.Advani, S. J., R. R. Weichselbaum, and B. Roizman. 2001. cdc2 cyclin-dependent kinase binds and phosphorylates herpes simplex virus 1 UL42
DNA synthesis processivity factor. J. Virol.75:10326–10333.
3.Advani, S. J., R. R. Weichselbaum, and B. Roizman.2003. Herpes simplex virus 1 activates cdc2 to recruit topoisomerase II␣for post-DNA synthesis expression of late genes. Proc. Natl. Acad. Sci. U. S. A.100:4825–4830. 4.Carrozza, M. J., and N. A. Deluca.1996. Interaction of the viral activator
protein ICP4 with TFIID through TAF250. Mol. Cell. Biol.16:3085–3093. 5.Carter, K. L., and B. Roizman.1996. The promoter and transcriptional unit
of a novel herpes simplex virus 1␣gene are contained in, and encode a protein in frame with, the open reading frame of the␣22 gene. J. Virol. 70:172–178.
6.Dai-Ju, J. Q., L. Li, L. A. Johnson, and R. M. Sandri-Goldin.2006. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it under-goes proteasomal degradation during infection. J. Virol.80:3567–3581. 7.Dobrikova, E., M. Shveygert, R. Walters, and M. Gromeier.2010. Herpes
simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J. Virol.84:270–279. 8.Doi, M., J. Hirayama, and P. Sassone-Corsi. 2006. Circadian regulator
CLOCK is a histone acetyltransferase. Cell125:497–508.
9.Du, T., G. Zhou, S. Khan, H. Gu, and B. Roizman.2010. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc. Natl. Acad. Sci. U. S. A. 107:15904–15909.
10.Ejercito, P. M., E. D. Kieff, and B. Roizman.1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J. Gen. Virol.2:357–364.
11.Fontaine-Rodriguez, E. C., T. J. Taylor, M. Olesky, and D. M. Knipe.2004. Proteomics of herpes simplex virus infected cell protein 27: association with translation initiation factors. Virology330:487–492.
12.Gu, H., Y. Liang, G. Mandel, and B. Roizman.2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, mod-ified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:7571–7576.
13.Gu, H., and B. Roizman.2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A.104:17134– 17139.
14.Gu, H., and B. Roizman.2009. Engagement of the lysine-specific demethyl-ase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 83:4376–4385.
15.Hardin, P. E., and W. Yu.2006. Circadian transcription: passing the HAT to CLOCK. Cell125:424–426.
16.Hardy, W. R., and R. M. Sandri-Goldin.1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J. Virol.68:7790–7799.
17.Kalamvoki, M., and B. Roizman.2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. U. S. A.107:17721–17726. 18.Kalamvoki, M., and B. Roizman.2010. Interwoven roles of cyclin D3 and
cdk4 recruited by ICP0 and ICP4 in the expression of herpes simplex virus genes. J. Virol.84:9709–9717.
19.Kawaguchi, Y., et al.2001. Herpes simplex virus 1 alpha regulatory protein ICP0 functionally interacts with cellular transcription factor BMAL1. Proc. Natl. Acad. Sci. U. S. A.98:1877–1882.
20.Kristie, T. M., Y. Liang, and J. L. Vogel.2010. Control of alpha-herpesvirus IE gene expression by HCF-1 coupled chromatin modification activities. Biochim. Biophys. Acta1799:257–265.
21.Kristie, T. M., and B. Roizman.1987. Host cell proteins bind to the cis-acting site required for virion-mediated induction of herpes simplex virus 1␣genes. Proc. Natl. Acad. Sci. U. S. A.84:71–75.
22.Kwon, I., et al.2006. BMAL1 shuttling controls transactivation and degra-dation of the CLOCK/BMAL1 heterodimer. Mol. Cell. Biol.26:7318–7330. 23.Leopardi, R., P. L. Ward, W. O. Ogle, and B. Roizman.1997. Association of
on November 7, 2019 by guest
http://jvi.asm.org/
herpes simplex virus regulatory protein ICP22 with transcriptional com-plexes containing EAP, ICP4, RNA polymerase II, and viral DNA requires posttranslational modification by the UL13 protein kinase. J. Virol.71:1133–
1139.
24.Lester, J. T., and N. A. DeLuca.2011. Herpes simplex virus 1 ICP4 forms complexes with TFIID and mediator in virus-infected cells. J. Virol.85:5733– 5744.
25.Liang, Y., J. L. Vogel, A. Narayanan, H. Peng, and T. M. Kristie.2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med.15:1312–1317. 26.Roizman, B., and D. M. Knipe. 2007. Herpes simplex viruses and their
replication, p. 2501–2601.InD. M. Knipe et al. (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
27.Roizman, B.2011. The checkpoints of viral gene expression in productive and latent infection: the role of the HDAC/CoREST/LSD1/REST repressor complex. J. Virol.85:7474–7482.
28.Roizman, B., et al.1988. The trans-activation of herpes simplex virus gene expression: comparison of two factors and their cis sites. Biochimie70:1031– 1043.
29.Sampath, P., and N. A. Deluca. 2008. Binding of ICP4, TATA-binding protein, and RNA polymerase II to herpes simplex virus type 1 immediate-early, immediate-early, and late promoters in virus-infected cells. J. Virol.82:2339–2349.
30.Sandri-Goldin, R. M.1994. Properties of an HSV-1 regulatory protein that appears to impair host cell splicing. Infect. Agents Dis.3:59–67. 31.Sandri-Goldin, R. M.2001. Nuclear export of herpes virus RNA. Curr. Top.
Microbiol. Immunol.259:2–23.
32.Sandri-Goldin, R. M.2008. The many roles of the regulatory protein ICP27 during herpes simplex virus infection. Front. Biosci.13:5241–5256. 33.Smith, C. A., P. Bates, R. Rivera-Gonzalez, B. Gu, and N. A. Deluca.1993.
ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol.67:4676–4687.
34.Spector, D., F. Purves, and B. Roizman.1991. Role of␣-transinducing factor (VP16) in the induction of alpha genes within the context of viral genomes. J. Virol.65:3504–3513.
35.Taddeo, B., W. Zhang, and B. Roizman.2010. Role of herpes simplex virus ICP27 in the degradation of mRNA by virion host shutoff RNase. J. Virol. 84:10182–10190.
36.Zhou, C., and D. M. Knipe.2002. Association of herpes simplex virus type 1 ICP8 and ICP27 proteins with cellular RNA polymerase II holoenzyme. J. Virol.76:5893–5904.
37.Zhou, G., D. Te, and B. Roizman.2010. The CoREST/REST repressor is both necessary and inimical for expression of herpes simplex virus genes. mBio2:e00313–e00310.