Initiation Factor 4B Contributes to Viral Replication by Suppressing
IFITM3 Protein Expression
Song Wang,aXiaojuan Chi,bHaitao Wei,aYuhai Chen,aZhilong Chen,bShile Huang,cJi-Long Chena,b
CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences (CAS), Beijing, Chinaa
; College of Animal Science, Fujian Agriculture and Forestry University, Fuzhou, Chinab
; Department of Biochemistry and Molecular Biology, Louisiana State University Health Sciences Center, Shreveport, Louisiana, USAc
ABSTRACT
Although alteration in host cellular translation machinery occurs in virus-infected cells, the role of such alteration and the pre-cise pathogenic processes are not well understood. Influenza A virus (IAV) infection shuts off host cell gene expression at tran-scriptional and translational levels. Here, we found that the protein level of eukaryotic translation initiation factor 4B (eIF4B), an integral component of the translation initiation apparatus, was dramatically reduced in A549 cells as well as in the lung, spleen, and thymus of mice infected with IAV. The decrease in eIF4B level was attributed to lysosomal degradation of eIF4B, which was induced by viral NS1 protein. Silencing eIF4B expression in A549 cells significantly promoted IAV replication, and conversely, overexpression of eIF4B markedly inhibited the viral replication. Importantly, we observed that eIF4B knockdown transgenic mice were more susceptible to IAV infection, exhibiting faster weight loss, shorter survival time, and more-severe organ damage. Furthermore, we demonstrated that eIF4B regulated the expression of interferon-induced transmembrane pro-tein 3 (IFITM3), a critical propro-tein involved in immune defense against a variety of RNA viruses, including influenza virus. Taken together, our findings reveal that eIF4B plays an important role in host defense against IAV infection at least by regulating the expression of IFITM3, which restricts viral entry and thereby blocks early stages of viral production. These data also indicate that influenza virus has evolved a strategy to overcome host innate immunity by downregulating eIF4B protein.
IMPORTANCE
Influenza A virus (IAV) infection stimulates the host innate immune system, in part, by inducing interferons (IFNs). Secreted IFNs activate the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway, leading to elevated tran-scription of a large group of IFN-stimulated genes that have antiviral function. To circumvent the host innate immune response, influenza virus has evolved multiple strategies for suppressing the production of IFNs. Here, we show that IAV infection induces lysosomal degradation of eIF4B protein; and eIF4B inhibits IAV replication by upregulating expression of interferon-induced transmembrane protein 3 (IFITM3), a key protein that protects the host from virus infection. Our finding illustrates a critical role of eIF4B in the host innate immune response and provides novel insights into the complex mechanisms by which influenza virus interacts with its host.
I
nfluenza A virus (IAV), whose genome consists of 8 segments of negative-sense single-stranded RNA, is still a major public health threat, causing a disease that leads to enormous morbidity and economic loss annually in the world (1). Remarkably, as a result of antigenic drift or shift undergone in IAV, several devas-tating epidemics and pandemics have occurred over the past cen-tury. To defend against influenza virus infection, the host mobi-lizes the innate immune system, including induction of antiviral cytokines, as the first line to destroy most of the invaders (2). Among the cytokines induced, the activated interferon (IFN) sys-tem is an important component of innate immunity and has been implicated in the anti-influenza virus defense (3,4).Host cells produce a complex mixture of IFNs upon sensing of pathogen-associated molecular patterns (PAMPs) by pattern rec-ognition receptors (PRRs). Three types of IFNs have been docu-mented: type I includes mainly IFN-␣and IFN-; type II is IFN-␥; and type III consists of three members in humans, IFN lambda 1 (IFN-1), IFN-2, and IFN-3 (also named interleukin-29 [IL-29], IL-28A, and IL-28B, respectively) (5). Secreted IFNs bind to their receptors to activate the Janus kinase (JAK)/signal transduc-ers and activators of transcription (STAT) pathway, resulting in
increased transcription of a large group of IFN-stimulated genes (ISGs) such as ISG15, MxA, 2=-5=oligoadenylate synthetase (OAS), and interferon-induced transmembrane proteins (IFITMs) (6–9). These ISGs play key roles in host immune defense against viral infections (10). On the other hand, to establish a successful infec-tion, viruses have evolved multiple strategies to circumvent the host innate immune response, including suppression of IFN sig-naling (11). However, the precise mechanisms by which influenza virus counteracts the IFN-dependent innate immune signaling are not fully understood.
Received16 January 2014 Accepted7 May 2014
Published ahead of print14 May 2014
Editor:B. Williams
Address correspondence to Ji-Long Chen, [email protected].
Supplemental material for this article may be found athttp://dx.doi.org/10.1128 /JVI.00126-14.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.00126-14
on November 7, 2019 by guest
http://jvi.asm.org/
Recently, alteration in host cellular translation machinery and its role during the viral infection have come into the focus of interest (12–14). It has been demonstrated that many viruses are able to acquire mechanisms by which they selectively translate viral RNAs by using the host translational machinery but inhibit the translation of host mRNAs, thereby suppressing host innate defenses (15). Strikingly, viruses use diverse strategies to subvert host protein synthesis functions. These strategies frequently in-volve the virus-induced modification of eukaryotic translation initiation factors (eIFs) that control translation initiation, a major rate-limiting step of protein synthesis (12). It is well known that during viral infection, the protein kinase R (PKR) can be activated by binding virus-derived double-stranded RNA (dsRNA) (16). Activated PKR limits viral replication through phosphorylation of translation initiation factor 2␣(eIF2␣), a component of the cel-lular translation machinery, which, in turn, leads to the shutdown of cellular and viralde novoprotein synthesis (17). However, vi-ruses have evolved mechanisms to counteract PKR activation. For example, paramyxoviruses have developed a strategy to block PKR-dependent phosphorylation of eIF2␣through viral P and V proteins (16). Influenza virus can activate a cellular protein, p58IPK, to inhibit PKR activity, and herpesviruses encode a pro-tein,␥34.5, which recruits a cellular phosphatase to dephosphor-ylate eIF2␣, overcoming the PKR-mediated translational inhibi-tion (18).
In host cells, eIF4E, eIF4G, and eIF4A constitute a complex named eIF4F that controls the mRNA translation initiation. In-terestingly, it has been shown that eIF4E is phosphorylated upon infection with herpesvirus or poxvirus, leading to increased trans-lation of inhibitor of nuclear factor kappa B (NF-B) alpha (IB␣) (19–21). As a result, the host produces less IFN-, attenuating innate antiviral response and causing higher susceptibility to virus infection (21). eIF4GI can be recruited specifically to the 5= un-translated region (5=UTR) of the viral mRNA through interacting with influenza virus NS1 protein, resulting in the preferential translation of the viral mRNAs (22). Also, eIF4A can be exploited by influenza virus to ensure the production of viral proteins (23). Recently, we have further found that Abelson murine leukemia virus-induced Pim-dependent eIF4B phosphorylation plays a critical role in cellular transformation by this virus (24). eIF4B, a member of the eIF family, is involved in the ribosome recruitment to the 5=ends of mRNAs and is thought to stimulate eIF4F activity through enhancing the helicase activity of eIF4A to unwind the mRNA 5=secondary structure (25). Therefore, eIF4B is a key com-ponent in the regulation of mRNA translation initiation. Previous studies have demonstrated the functional involvement of eIF4B in viral replication. For example, it can interact with the 3=region of the poliovirus internal ribosome entry site (IRES) to regulate viral RNA translation initiation (26). Open reading frame 45 (ORF45) of Kaposi’s sarcoma-associated herpesvirus (KSHV) can increase the phosphorylation of eIF4B to promote protein synthesis during lytic replication (27). Importantly, eIF4B also plays a key role in regulating the IFN-induced expression of ISG15 protein (28). ISG15 possesses potent antiviral activity, such as suppression of influenza virus replication by inhibiting the viral gene expression and conjugating itself to the viral NS1 protein, thereby disrupting the NS1 function (29–31). However, although alterations in cel-lular translation machinery have been observed in virus-infected cells, the precise roles of these alterations remain elusive. In
par-ticular, little is known about whether and how eIF4B affects influ-enza virus infection.
In this study, we examined the expression and role of eIF4B during IAV infection. Surprisingly, we observed that the protein level of eIF4B was dramatically reduced in influenza virus-infected A549 human alveolar epithelial cells and mice tissues. Bothin vitro andin vivoexperiments using eIF4B-silencing or -overexpressing cells as well as transgenic mice demonstrated that eIF4B was in-volved in the regulation of IAV replication. Furthermore, we found that eIF4B was required for efficient expression of IFITM3, a critical antiviral factor. Therefore, our results indicate that eIF4B plays an important role in host innate immune defense against influenza virus infection.
MATERIALS AND METHODS
Ethics statement.The animal protocol used in this study was approved by the Research Ethics Committee of Institute of Microbiology, Chinese Academy of Sciences (permit number PZIMCAS2012001). All mouse ex-perimental procedures were performed in accordance with the Regula-tions for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People’s Republic of China.
Virus and reagents.Influenza virus strains A/WSN/33 (H1N1), A/PR/ 8/34 wild type (WT), and deltaNS1 (delNS1) were generated and propa-gated in specific-pathogen-free (SPF) chicken embryo as previously de-scribed (32). The following antibodies were used in this study: anti-eIF4B, anti-phospho-eIF4B (Ser422), anti-NS1, and anti-ISG15 (Santa Cruz Bio-technology, Santa Cruz, CA); anti-IFITM3, anti-MxA, and anti-PKR (Proteintech, Chicago, IL); anti-p58IPK (Cell signaling Technology, Bev-erly, MA); anti-influenza A virus NP (kindly provided by Wenjun Liu, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China); and other antibodies (obtained as described previously) (32). The protein synthesis inhibitor cycloheximide (CHX) and the lysosome inhibitors NH4Cl and chloroquine were purchased from Sigma-Aldrich (St. Louis, MO). The proteasome inhibitor MG132 was obtained from Merck (Darmstadt, Germany).
DNA construction.cDNA encoding IFITM3 or ISG15 was subcloned into the pFLAG-CMV-5a vector with a Flag tag in the COOH terminus. Flag-eIF4B (WT) and Flag-eIF4B (S406A/S422A) were constructed as previously described (24). The eight reverse genetic plasmids for the res-cue of influenza virus A/PR/8/34 were kindly provided by Ron A. M. Fouchier (National Influenza Center and Department of Virology, Eras-mus MC, Rotterdam, Netherlands). DelNS1 plasmid is a derivative of the PR8 NS segment (lacking nucleotides 57 to 528), which was cloned into the vector pHW2000 between the HindIII and SphI sites.
Cell culture and infection.293T, A549, MDCK cells (American Type Culture Collection, Manassas, VA), and mouse embryonic fibroblasts (MEFs) (24) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal calf serum (FCS) supplemented with pen-icillin (100 U/ml) and streptomycin (100g/ml). For virus infection, cells were infected with A/WSN/33 virus at the indicated multiplicity of infec-tion (MOI). After adsorpinfec-tion for 1 h at 37°C, the cells were washed with phosphate-buffered saline (PBS) and cultured in DMEM containing 2
g/ml trypsin.
Mouse experiments.Female mice (5 to 6 weeks old, 18 to 20 g) were inoculated intranasally with 5⫻104PFU of the A/WSN/33 virus. At the indicated time postinfection (p.i.), the mice were then euthanized and their organs (lung, spleen, thymus, and liver) were removed aseptically for further analysis.
Cell extracts and Western blotting.Cell lysates were prepared, and Western blotting was performed as previously described (33). Briefly, samples were separated on SDS-polyacrylamide gel, transferred onto a nitrocellulose membrane, and probed with antibodies as indicated.
RNA preparation, RT-PCR, and quantitative real-time PCR.Total RNA was extracted from cells or tissues using TRIzol reagent (Invitrogen,
on November 7, 2019 by guest
http://jvi.asm.org/
Carlsbad, CA). cDNA was synthesized using 2g of total RNA and Molo-ney murine leukemia virus (MMLV) reverse transcriptase (RT; Promega, Madison, WI), followed by PCR using rTaq DNA polymerase and quan-titative PCR using SYBR PremixEx TaqII (TaKaRa, Tokyo, Japan) with the following primers: eIF4B forward, 5=-GCACCTATGTCCCCAAACC A-3=, eIF4B reverse, 5=-GAAGACGGCTCCGGTCAATA-3=; NP forward, 5=-TCAAACGTGGGATCAATG-3=, NP reverse, 5=-GTGCAGACCGTGC TAAAA-3=; GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (hu-man) forward, 5=-AGAAGGCTGGGGCTCATTTG-3=, GAPDH (hu-man) reverse, 5=-AGGGGCCATCCACAGTCTTC-3=; GAPDH (mouse) forward, 5=-GCCTCGTCCCGTAGACAAAA-3=, GAPDH (mouse) re-verse, 5=-CCCTTTTGGCTCCACCCTTC-3=. GAPDH was chosen as a reference gene for internal standardization.
Plaque assay and HA assay.Plaque assay and hemagglutinin (HA) assay were performed as previously described (32). Briefly, for plaque assay, MDCK cells infected with serial dilutions of the supernatants of cell cultures were washed with PBS and overlaid with␣-minimal essential medium containing low-melting-point agarose (Promega, Madison, WI) and TPCK (tolylsulfonyl phenylalanyl chloromethyl ketone)-treated trypsin (Sigma-Aldrich, St. Louis, MO). After incubation for 72 h, plaques were stained and counted. For hemagglutinin assay, the supernatants were diluted with PBS and mixed with an equal volume of 0.5% chicken eryth-rocytes. Then, viral titers were counted from the highest dilution factors that produced a positive reading.
Lung virus determination.Lung virus titers were determined 72 h postinfection. Lungs of infected mice were homogenized in 1 ml of ice-cold PBS and frozen at⫺80°C for 14 h. Then, thawed samples were cen-trifuged at 2,000⫻gfor 10 min, and the supernatants were titrated by plaque assay as described above.
shRNA-based knockdown and generation of stable cell lines.Short hairpin RNA (shRNA)-based knockdown cell lines were generated by infection of A549 cells with lentiviruses expressing specific shRNAs in pSIH-H1-GFP vector as described previously (32). The sequences used in the shRNAs targeting specific genes were as follows: human eIF4B shRNA-1, 5=-GACAAGTATCGAGATCGTTAT-3=, and shRNA-2, 5=-GC CAAAGAAACCTGAGGAGAA-3=(24); IAV NS1 shRNA-1, 5=-GCAAAT AGTGGAGCGGATTCT-3=, and shRNA-2, 5=-GGACAATTGTTGGCGA AAT-3=; IFITM3 shRNA, 5=-TCCTCATGACCATTCTGCTCAT-3= (34); ISG15 shRNA, 5=-TGCGACGAACCTCTGAACA-3=(35); and luciferase control shRNA, 5=-CTTACGCTGAGTACTTCGA-3=. A549 cell lines sta-bly expressing eIF4B or empty vector (EV) were generated by infecting the cells with lentiviruses encoding these genes in pNL-GFP vector as previ-ously described (24).
Generation of TG mice.eIF4B knockdown transgenic (TG) mice were generated as previously described (24). Briefly, the shRNA sequences tar-geting eIF4B were cloned into an expression vector under an H1 moter. Then, the plasmid was linearized and microinjected into the pro-nucleus of fertilized zygotes harvested from C57BL/6J mice. Genotyping of transgenic mice was performed by PCR using primers with the follow-ing sequences: 5=-AAATCCTGGTTGCTGTCTCTTTATG-3=and 5=-GG AAGGTCCGCTGGATTGA-3=.
Histopathological analysis.Mouse organs were fixed in 4% parafor-maldehyde and embedded in paraffin. Then, 4-mm-thick sections were prepared and stained with hematoxylin and eosin (H&E). The slides were visualized under an Olympus BH-2 microscope (Tokyo, Japan).
Statistical analysis.Data represent the mean values⫾standard errors [SE]). Statistical analysis was performed by Student’sttest. APlevel of
⬍0.05 was considered to be significant.
RESULTS
IAV infection reduces eIF4B protein levels bothin vitroandin
vivo.To determine whether eIF4B is involved in influenza virus infection and replication, A549 human alveolar epithelial cells were infected with A/WSN/33 virus and harvested at different time points postinfection, followed by Western blotting. We
found that protein and phosphorylation levels of eIF4B were con-sistently reduced after infection with the viruses (Fig. 1A). The reduction of eIF4B protein was also observed in MEFs following the viral infection (Fig. 1B). However, expression of eIF4B was not affected by herpes simplex virus (HSV) infection (see Fig. S1A and B in the supplemental material). Furthermore, after mice were intranasally infected with A/WSN/33 for 3 days, protein levels of eIF4B were markedly reduced in the lung samples (Fig. 1C). Since our previous experiments showed that IAV infection resulted in destruction of spleen and thymus in mice when a lethal dose of the A/WSN/33 was used (36), we investigated the effect of the virus infection on the eIF4B level in these lymphoid organs. Surpris-ingly, protein levels of eIF4B decreased considerably in the spleen and thymus but not in the liver of infected mice (Fig. 1D). To-gether, our data indicate that IAV infection reduced expression of eIF4B protein bothin vitroandin vivo.
It is now well established that influenza virus infection leads to the shutdown of some cellular protein expression by inhibiting transcription or inducing mRNA degradation in host cells (37). To determine whether the reduction of eIF4B protein level is a consequence of a decrease in its mRNA level, A549 cells were infected with the WSN viruses and examined by RT-PCR and quantitative real-time PCR. We observed that the amount of eIF4B mRNA was not significantly decreased after infection of A549 cells with the virus (Fig. 1EandF). Similarly, eIF4B mRNA levels in MEFs infected with the WSN viruses remained un-changed compared with the control (see Fig. S1C in the supple-mental material). Furthermore, influenza virus infection did not obviously alter eIF4B mRNA levels in mouse lung, spleen, and
FIG 1Influenza A virus infection decreases eIF4B protein levels bothin vitro andin vivo. (A) A549 cells were infected with WSN viruses (MOI⫽3) and harvested at the indicated times, followed by Western blotting with the indi-cated antibodies. The results are representative of three independent experi-ments. (B) MEFs were uninfected or infected with WSN viruses for 12 h, followed by Western blotting with the indicated antibodies. (C, D) Mice intra-nasally infected with or without WSN for 3 days were sacrificed, and the lungs (C) and thymus, spleens, and livers (D) were homogenized, followed by West-ern blotting with the indicated antibodies. Shown are representative immuno-blots from three experiments with similar results. (E, F) A549 cells were in-fected with WSN as described for panel A, followed by RT-PCR (E) and quantitative real-time PCR (F) to detect eIF4B mRNA levels. GAPDH served as an internal control.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.301.542.65.259.2]thymus (see Fig. S1D and E in the supplemental material). The results indicate that IAV-induced reduction of the eIF4B protein level is not due to a decrease of the eIF4B mRNA level.
IAV infection induces lysosomal degradation of eIF4B
pro-tein.Since the decrease of eIF4B protein level was not correlated
with its mRNA level, we next estimated the relative half-life of eIF4B protein in A549 cells infected or not infected with the WSN viruses. Cells were treated with cycloheximide (CHX), a protein synthesis inhibitor, at 6 h postinfection and lysed after an incuba-tion for the indicated times, and the rates of eIF4B remaining in the cells were given as percentages of eIF4B protein levels at the zero time point. As shown inFig. 2AandB, the half-life of eIF4B was estimated to be approximately 32 h in uninfected control cells. Infection with the virus dramatically shortened the half-life of eIF4B to⬃16 h, suggesting that IAV infection promotes protein degradation of eIF4B in the cells.
There are two main pathways governing the intracellular pro-tein degradation: one is the proteasome pathway, and the other is the lysosome pathway (38). To define how IAV infection causes protein degradation of eIF4B, proteasome inhibitor MG132 and lysosome inhibitors NH4Cl and chloroquine were employed. We
found that treatment with MG132 clearly increased the amount of ubiquitinated proteins (Fig. 2C) but did not reverse the effect of IAV infection on the eIF4B protein level in A549 cells (Fig. 2D). In contrast, treatment with either NH4Cl or chloroquine
al-most completely prevented IAV-induced protein degradation of eIF4B (Fig. 2E). These data suggest that IAV-induced eIF4B degradation is by the lysosome pathway and not via the protea-some pathway.
NS1 protein of influenza virus is involved in the regulation of
eIF4B protein stability.Next, we investigated how IAV infection
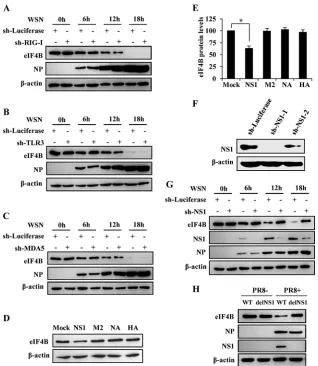
triggers the protein degradation of eIF4B. We speculated that the reduction of eIF4B in IAV-infected cells was a host innate defense against the viral infection. To this end, we tested if the influenza virus-induced degradation of eIF4B was dependent on retinoic acid-inducing gene 1 protein (RIG-I), Toll-like receptor 3 (TLR3), and MDA5, the most important pattern recognition receptors sensing the infection of influenza virus. A549 cell lines expressing specific shRNAs targeting RIG-I, TLR3, MDA5, or luciferase (control) were generated as previously described (5). We found that silencing RIG-I, TLR3, or MDA5 had no obvious effect on the virus-induced reduction of eIF4B protein (Fig. 3A,B, andC; see also Fig. S2A, B, and C in the supplemental material), suggesting that this process is not mediated through RIG-I-, TLR3-, or MDA5-dependent mechanisms.
Next, the influence of viral proteins on the eIF4B protein level was examined by transient expression of WSN viral proteins. In-terestingly, we observed that overexpression of NS1, but not other viral proteins including hemagglutinin (HA), neuraminidase (NA), and matrix protein 2 (M2), significantly decreased the
FIG 2Influenza virus infection induces lysosomal degradation of eIF4B protein. (A) Half-lives of eIF4B in uninfected and infected A549 cells were examined. A549 cells uninfected or infected with WSN viruses (MOI⫽1) were treated with CHX (100g/ml) at 6 h postinfection. At the indicated times after treatment, cells were harvested and cell extracts were prepared for Western blotting to analyze eIF4B protein levels. (B) eIF4B levels shown in panel A were quantitated by densitometry and normalized to-actin levels. In each experiment, the highest level of eIF4B is set to 100. Plotted are the average levels from three independent experiments. The error bars represent the SE. **,P⬍0.01 (Student’sttest). (C) Western blot analysis of extracts of 293T cells transfected with plasmid carrying HA-Ub and treated with dimethyl sulfoxide (DMSO) or MG132 (10M) as indicated. (D) A549 cells infected with WSN viruses (MOI⫽1) were either mock treated or treated with MG132 (10M). At the indicated times, cells were harvested and cell extracts were prepared for Western blotting using the indicated antibodies. This result is representative of three identical experiments. (E) A549 cells infected with WSN viruses (MOI⫽1) were either mock treated or treated with NH4Cl (20 mM) or chloroquine (50M). At the indicated times, cells were analyzed by Western blotting with the indicated antibodies. Shown are
representative data of three independent experiments with similar results.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.139.452.66.328.2]amount of eIF4B protein (Fig. 3DandE). To confirm this finding, we generated cell lines stably expressing specific shRNAs targeting NS1 or luciferase (control) (see Fig. S2D in the supplemental ma-terial) and performed infection assays with WSN viruses. Western blot analysis showed that NS1 was markedly reduced in the gen-erated cell lines compared to the control cells at 6 h, 12 h, and 18 h postinfection (Fig. 3FandG). Remarkably, although the eIF4B protein level was dramatically reduced in the infected control cells as expected, the effect of influenza virus infection on eIF4B pro-tein level was reversed by silencing NS1 expression (Fig. 3G). Sim-ilar results were obtained from experiments using delNS1 A/PR/ 8/34 virus (Fig. 3H). Together, our results suggest that NS1 protein of influenza virus may be responsible for induction of eIF4B degradation during the viral infection.
Altering eIF4B expression has profound effects on influenza
virus replication.Since influenza virus infection induced the
deg-radation of eIF4B protein, we were interested in unveiling the role of eIF4B in influenza virus replication. For this, A549 cell lines stably expressing specific shRNA targeting eIF4B or ectopically overexpressing eIF4B were generated. As shown inFig. 4AandB, Western blot analysis demonstrated the significantly altered ex-pression of eIF4B in these cell lines. The cells were then infected with WSN, and the viral replication was measured by hemagglu-tinin assay and plaque assay. Using hemaggluhemagglu-tinin assay, we ob-served that the viral titers were significantly increased when ex-pression of the endogenous eIF4B in A549 cells was silenced by the specific shRNA (Fig. 4C). This was further verified by plaque as-say. As shown inFig. 4D(see also Fig. S3A in the supplemental material), disruption of eIF4B expression caused a significant in-crease in the viral titers examined at 18 h and 22 h postinfection. In contrast, both hemagglutinin assay and plaque assay revealed that forced overexpression of eIF4B resulted in a significant reduction
FIG 3Influenza virus NS1 is involved in inducing the degradation of eIF4B. (A, B, C) A549 cell lines stably expressing specific shRNAs targeting RIG-I (A), TLR3 (B), MDA5 (C), or luciferase control were infected with WSN viruses (MOI⫽1) and harvested at the indicated times, followed by Western blotting using the indicated antibodies. Shown are representative immunoblots from three experiments with similar results. (D) Western blot analysis of lysate derived from A549 cells transfected with empty vector (Mock) or plasmid expressing NS1, M2, NA, HA protein, of influenza A virus. (E) The eIF4B levels shown in panel D were quantitated by densitometry and normalized to-actin levels. In each experiment, the eIF4B level in A549 cells transfected with empty vector is 100. Plotted are the average levels from three independent experiments. The error bars represent the SE. *,P⬍0.05 (Student’sttest). (F) shRNA-based knockdown of NS1 was analyzed by Western blotting to determine the interference efficiency. shRNA targeting luciferase (sh-Luciferase) served as a control. (G) A549 cells stably expressing specific shRNAs targeting NS1 or luciferase (control) were infected with WSN viruses (MOI⫽1) and analyzed by Western blotting using the indicated antibodies. The results are representative of three independent experiments. (H) A549 cells were infected with PR8 wild-type (WT) or deltaNS1 (delNS1) viruses for 12 h and analyzed by Western blotting using the indicated antibodies. This result is representative of three identical experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.133.451.64.430.2]of the viral titers (Fig. 4EandF; see also Fig. S3B in the supple-mental material).
To further confirm the effect of eIF4B on influenza virus rep-lication, we used MEFs derived from wild-type and eIF4B
knock-down transgenic mice previously generated in the laboratory (24). As shown inFig. 4G, the eIF4B protein level was markedly reduced in the MEFs derived from eIF4B knockdown transgenic mice. Af-ter infection of the MEFs with WSN for 18 h, the viral tiAf-ters in the supernatants of cell cultures were examined by plaque assay. We found that the production of influenza virus was greatly increased in the eIF4B knockdown MEFs, compared with that in wild-type counterparts (Fig. 4H). These findings demonstrate that eIF4B negatively regulates the replication of influenza virusin vitro.
Silencing eIF4B increases the susceptibility of transgenic
mice to IAV infection.To define the functional relevance of eIF4B
in IAV infection, we wished to employ a more physiological model system for determining the role of eIF4B in this process. There-fore, eIF4B knockdown transgenic mice (TG) were used as a model and analyzed after infection with WSN virus, an influenza A virus commonly used to create mouse lethal infections. Inter-estingly, we found that eIF4B knockdown mice displayed a faster body weight loss than wild-type (WT) littermates during the viral infection (Fig. 5A). Furthermore, the survival rate of infected mice was evaluated. We observed that all eIF4B knockdown mice died within 6 days postinfection, whereas approximately 46% of wild-type littermates remained alive on day 6 postinfection, and even about 23% of the wild-type mice survived at the end of the exper-iments (at day 12 postinfection) under our experimental condi-tions (Fig. 5B). Consistent with these observations, the plaque assay showed that the viral titers in lung tissues of infected eIF4B knockdown mice were higher than those of wild-type littermates (Fig. 5C), suggesting that silencing eIF4B expression promotes the production of influenza virusin vivo.
In addition, the pathological changes of mouse organs were examined following the virus infection. Notably, eIF4B knock-down led to more-severe organ damage caused by influenza virus infection, exhibiting a greater degree of lung injury and thymus atrophy (see Fig. S4A and B in the supplemental material). To further investigate the pathological features of these organs, he-matoxylin and eosin (H&E) staining was performed (Fig. 5D). In the lungs of infected wild-type mice, there were edema and infil-tration of inflammatory cells around the bronchiole and vessels, and a small number of inflammatory cells were seen in the alveoli. In contrast, abundant inflammatory cells were present in the al-veoli and diffuse in the peribronchiolar and perivascular regions in the lungs of infected eIF4B knockdown mice. In addition, ex-tensive edema was seen in these lungs, and a large amount of epithelial cell debris was observed in the lumen of many bronchi-oles. These results indicate that eIF4B knockdown makes mice more susceptible to influenza virus infection.
eIF4B regulates IFITM3 protein expression to influence the
replication of influenza virus.Since our experiments presented
above demonstrated that silencing eIF4B expression promoted the production of influenza virusin vitro andin vivo, we next investigated how eIF4B affects this process. Using cDNA microar-ray to profile the cellular transcriptional response to WSN influ-enza virus infection in A549 cells (www.ncbi.nlm.nih.gov/geo/; GenBank accession number GSE32878), we found that MxA, ISG15, and IFITMs, the important ISGs implicated in the antiviral defense, were obviously upregulated (see Fig. S5A in the supple-mental material). As eIF4B is a key component in the regulation of translation initiation of mRNAs with structured 5=UTR and is required for efficient expression of ISG15 protein induced by IFNs (28), we further examined the effect of eIF4B expression on the
FIG 4Alteration of eIF4B levels significantly influences influenza virus replica-tion. (A) Interference efficiency of shRNA-based knockdown of eIF4B was deter-mined by Western blotting. shRNA targeting luciferase (sh-Luciferase) served as a control. (B) eIF4B-overexpressing A549 cells were generated by stably infecting the cells with lentivirus encoding eIF4B, and its expression in the eIF4B-overexpress-ing cells (pNL-eIF4B) and the control cells (Control) was analyzed by Western blotting. (C) A549 cells stably expressing the shRNAs targeting eIF4B or luciferase were infected with WSN viruses (MOI⫽0.1). The supernatants of cell culture were examined for the viral titers by hemagglutinin assay at the indicated time points postinfection (p.i.). (D) eIF4B knockdown A549 cells and control cells were infected with WSN viruses as described for panel C. Viral titers in the supernatants of these cells were examined by plaque assay (18 h p.i.). (E, F) A549 cells overex-pressing eIF4B and control cells were infected with WSN viruses as described for panels C and D. Viral titers in the supernatants of these cells were examined by hemagglutinin assay (E) and plaque assay (F). (G) Shown is an immunoblot of the MEF lysates derived from wild-type (WT) mice and eIF4B knockdown transgenic (TG) mice. WT, TG1, and TG2 were three representative embryos from the same female mouse. (H) MEFs shown in panel G were infected with WSN viruses, and viral titers in the supernatants of these cells were examined by plaque assay as described for panel D. The error bars in panels D, F, and H represent the SE.n⫽3; *,P⬍0.05; **,P⬍0.01 (Student’sttest).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.42.284.70.473.2]protein levels of these ISGs during the influenza virus infection. Interestingly, silencing eIF4B greatly diminished the protein ex-pression of ISG15 and IFITM3, but not MxA, after infection with influenza virus (Fig. 6AandB). However, silencing eIF4B had no effect on the mRNA levels of these ISGs (see Fig. S5B in the sup-plemental material), indicating that eIF4B did not regulate expres-sion of ISG15 and IFITM3 at the transcriptional level. Because PKR signaling plays an important role in influenza virus infection, we also determined the protein expression of PKR and its inhibi-tor p58IPK in eIF4B knockdown cells. Western blotting showed that silencing eIF4B had no significant effects on protein levels of PKR and p58IPK in IAV-infected or -noninfected cells (see Fig. S5C in the supplemental material).
Since expression of ISGs is critically important for host defense against virus infection, we further investigated whether increased replication of influenza virus in eIF4B knockdown cells was due to the decrease of these ISG proteins. To this end, forced expression of ISG15 or IFITM3 in these cells was performed and influenza virus replication was examined. We found that overexpression of ISG15 only slightly decreased the viral replication triggered by eIF4B knockdown, whereas overexpression of IFITM3 almost completely prevented the viral replication induced by eIF4B si-lencing (Fig. 6C). To confirm this finding, we generated A549 cell lines stably expressing shRNA targeting either IFITM3 or ISG15 and tested whether disrupting expression of these ISGs had any effect on the viral production. Western blot analysis demonstrated that the specific shRNAs strongly downregulated the expression of
IFITM3 or ISG15 compared with the control shRNA to luciferase (Fig. 6DandE; see also Fig. S5D and E in the supplemental mate-rial). Indeed, we observed that IFITM3 knockdown significantly increased influenza virus production, whereas ISG15 knockdown only slightly increased the viral titers (Fig. 6F). Previous studies have shown that IFITM3 restricts IAV infection by preventing virus cytosolic entry (34), suggesting that IFITM3 blocks early stages of IAV replication.
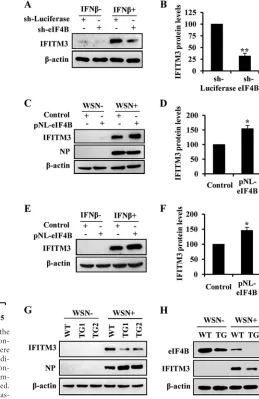
In addition, our experiments showed that disruption of eIF4B expression caused an obvious reduction of IFN--induced IFITM3 and ISG15 protein expression (Fig. 7AandB; see also Fig. S6A in the supplemental material). Conversely, eIF4B overexpres-sion increased IFITM3 protein levels after IAV infection or IFN- treatment (Fig. 7CtoF). Furthermore, mutation of eIF4B (S406A/ S422A) dramatically attenuated the effect of eIF4B on IFITM3 protein expression (see Fig. S6B in the supplemental material). Similarly, eIF4B knockdown also resulted in a decreased expres-sion of IFITM3 in MEFs infected with the influenza virus (Fig. 7G; see also Fig. S6C and D in the supplemental material). To confirm this findingin vivo, eIF4B knockdown mice and wild-type litter-mates were infected with WSN viruses and IFITM3 protein was examined. Consistent with thein vitroobservations, Western blot analysis showed that expression of the IFITM3 protein was also obviously reduced in eIF4B knockdown mice compared with the wild-type control (Fig. 7H; see also Fig. S6E to G in the supple-mental material). Collectively, these results indicate that eIF4B is required for efficient expression of IFITM3 protein induced by
FIG 5eIF4B knockdown transgenic mice are more susceptible to influenza A virus. (A) Shown is the body weight change of wild-type (WT) and eIF4B knockdown mice (TG) mock infected or infected intranasally with WSN. Body weight was measured daily. The results are shown as mean percentage weight changes from three independent experiments. The dashed line indicates 75% of initial body weight. (B) Survival rate of wild-type mice (n⫽14) and eIF4B knockdown mice (n⫽14) infected intranasally with WSN. Mice were monitored for up to 12 days. During that period, mice were sacrificed when showing hind limb paralysis. *,P⬍0.05 (Kaplan-Meier test). (C) Shown is the viral load in the lungs of wild-type and eIF4B knockdown mice infected for 3 days with WSN. Each symbol represents an individual mouse. **,P⬍0.01 (Student’sttest). (D) Representative micrographs (magnification,⫻400) of the wild-type and eIF4B knockdown mouse lungs stained with hematoxylin and eosin (HE). The pathological changes of lungs in TG mice were more serious than in their WT counterparts after infection for 3 days.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.135.454.65.321.2]IFNs during the IAV infection and suggest that eIF4B inhibits influenza viral production, possibly by upregulating IFITM3 pro-tein synthesis.
DISCUSSION
Interaction between influenza virus and host is essential for its pathogenesis. To successfully establish an infection and effectively propagate in host cells, influenza virus has evolved multiple strat-egies to circumvent the host antiviral responses, including shut-ting down expression of certain host genes (37) and causing deg-radation of some host proteins (39). Because expression of IFNs is an early powerful response against influenza virus infection, it is not surprising that this pathogen has coevolved gene products
restricting IFN expression (40) as well as blocking IFN-induced signaling (11). For example, it is well known that NS1 of influenza virus, a multifunctional nonstructural protein, plays a critical role in interfering with the type I IFN response. NS1 prevents virally
FIG 6eIF4B influences influenza virus replication through regulating the expression of IFITM3 protein. (A) eIF4B knockdown A549 cells and the con-trol cells were uninfected or infected with WSN viruses for 12 h. The cells were harvested, and cell extracts were prepared for Western blotting using the indi-cated antibodies. (B) IFITM3, ISG15, and MxA protein levels under the con-ditions of WSN infection described for panel A were quantitated by densitom-etry and normalized to-actin levels. (C) Rescue assay was performed. Control and eIF4B knockdown A549 cells transfected transiently with plas-mids encoding empty vector (Mock), ISG15, or IFITM3 were infected with WSN viruses for 18 h. Next, the viral titers in the supernatants of these cells were examined by plaque assay (upper panel). Flag-tagged ISG15 and IFITM3 protein levels after introduction of the plasmids were analyzed by Western blotting (lower panel). (D, E) shRNA-based knockdown of IFITM3 (D) and ISG15 (E) under the conditions of WSN infection was analyzed by Western blotting using the antibodies as indicated. (F) A549 cells stably expressing the shRNAs targeting IFITM3, ISG15, or luciferase were infected with WSN vi-ruses (MOI⫽0.1). The supernatants of cell culture were examined for the viral titers by plaque assay (18 h p.i.). **,P⬍0.01 (Student’sttest).
FIG 7eIF4B regulates IFITM3 protein expressionin vitroandin vivo. (A) Shown is Western blot analysis of lysates of eIF4B knockdown A549 cells and control cells treated with or without IFN-(500 U/ml) for 12 h. (B) IFITM3 protein levels from panel A after IFN-treatment were quantitated by densi-tometry and normalized to-actin levels. In each experiment, the IFITM3 level in control cells (sh-Luciferase) is 100. (C) Shown is Western blot analysis of lysates of eIF4B overexpressing A549 cells and control cells infected with or without WSN viruses for 12 h. (D) IFITM3 protein levels after WSN infection shown in panel C were quantitated by densitometry and normalized to-actin levels. In each experiment, the IFITM3 level in control cells is 100. (E) Shown is Western blot analysis of lysates of eIF4B overexpressing A549 cells and con-trol cells treated with or without IFN-(500 U/ml) for 12 h. (F) IFITM3 protein levels after IFN-treatment in panel E were quantitated by densitom-etry and normalized to-actin levels. In each experiment, the IFITM3 level in control cells is 100. (G) MEFs derived from wild-type and eIF4B knockdown transgenic mice were infected with or without WSN viruses for 12 h, and cell lysates were analyzed by Western blotting using the indicated antibodies. (H) Wild-type (WT) and eIF4B knockdown transgenic (TG) mice intranasally infected with or without WSN viruses for 3 days were sacrificed, and the spleens were homogenized, followed by Western blotting using the indicated antibodies. *,P⬍0.05; **,P⬍0.01 (Student’sttest).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.46.307.66.391.2] [image:8.585.277.536.68.467.2]induced type I IFN production by suppressing RIG-I signal trans-duction and inhibiting the activation of interferon regulatory fac-tor 3 (IRF-3) (41–43), which is crucial for countering type I IFN-mediated antiviral responses. Importantly, the first line of defense against influenza virus infection relies on the innate immune re-sponse that is mediated by ISGs, the key antiviral components induced by IFNs (44). However, the precise mechanisms by which influenza virus overcomes ISGs remain largely unknown. In this study, we found that influenza virus induced a marked degrada-tion of eIF4B protein, which plays a critical role in protein synthe-sis of some ISGs as indicated by previous studies (28). Further-more, we observed that influenza virus infection caused a drastic decrease of eIF4B protein levels in mouse lungs, spleens, and thy-muses. Subsequently, our experiments demonstrated that the deg-radation of eIF4B protein was mediated by the lysosome-depen-dent mechanism. Importantly, our results revealed that NS1 protein of influenza virus was responsible for induction of eIF4B degradation during the viral infection.
eIF4B is a key part of the protein complex involved in the control of eukaryotic mRNA translation that is preferentially ex-erted at the initiation phase (45). It has been shown that eIF4B is crucial for the specific expression of genes important for develop-ment, cell growth, proliferation, and survival (46). Moreover, a recent study has defined a key regulatory role of eIF4B in IFN-dependent biological effects (28). Here we found that disruption of the eIF4B expression promoted the replication of influenza virus in A549 cells whereas forced expression of eIF4B impeded this process. Of note, ourin vivostudies showed that silencing eIF4B expression led to a greatly increased susceptibility of trans-genic mice to influenza A virus infection. Taken together, these data suggest that eIF4B contributes to the host defense against influenza virus infection likely through regulating the protein ex-pression of some critical ISGs. Thus, influenza virus-induced eIF4B degradation is in favor of the viral replication.
Indeed, we found that eIF4B knockdown obviously reduced the protein expression of ISG15 and IFITM3 after infection with influenza virus, although silencing eIF4B did not affect the mRNA levels of these ISGs. Remarkably, IFN--induced ISG15 and IFITM3 protein expression was reduced by eIF4B knockdown, indicating a requirement for eIF4B in mRNA translation/protein expression of some critical ISGs induced by IFNs. Although it is well recognized that the JAK/STAT pathway plays crucial roles in type I, II, and III IFN-dependent transcription of ISGs (47), the regulation of mRNA translation for these IFN-sensitive genes re-mains elusive. Our finding here represents the first report of a key regulatory role for eIF4B in the ISG protein expression during the influenza virus infection and suggests that influenza virus has evolved a strategy to target eIF4B protein to circumvent the host antiviral responses.
Our study has also addressed whether the change of expression of these ISG proteins in eIF4B-overexpressing or knockdown cells is associated with the altered replication of influenza virus. Inter-estingly, we observed that the effect of eIF4B deficiency on the viral replication could be completely reversed by the forced expression of IFITM3. IFITM3 is a critical antiviral factor that mediates cel-lular innate immunity to multiple pathogenic viruses, including influenza virus (9,48–51). Investigations in both mice and hu-mans have established that IFITM3 is essential for the host defense against influenza virus (52). In addition, IFITM3 plays an impor-tant role in the host adaptive immune response. Its expression is
found in lung resident memory T cells (TRMcells) and specifically
endows TRMcells with prophylactic resistance to future
encoun-ters with influenza virus (53). Here, we, for the first time, show that influenza virus may impair the IFITM3-dependent biological effects through downregulating eIF4B protein expression. To rep-licate, viruses must enter into the host and use the host cell’s sources. Recently, several studies have revealed that IFITMs re-strict viral membrane fusion and entry and thereby block early stages of viral replication (34,54–56). Thus, IAV-induced degra-dation of eIF4B contributes to the viral replication by suppressing IFITM3 protein expression.
In summary, our results provide novel insights into the com-plex mechanisms by which influenza virus interacts with its host during pathogenesis. In addition, our findings establish a key role for eIF4B in the innate immune response that relies on ISGs. It is worth noting that the number of ISGs examined in the present study is limited. There may exist other ISGs that are also regulated by eIF4B and have synergistic effects with IFITM3 on influenza virus infection. A large-scale study of the regulation of ISG mRNA translation/protein expression by eukaryotic translation initiation factors may enhance our understanding of innate immune re-sponse to influenza virus infection. In addition, further studies are needed to address the biological significance of influenza virus-induced eIF4B reduction in spleens and thymusesin vivo.
ACKNOWLEDGMENTS
This work was supported by grants from the National Key Technologies Research and Development Program of China (2013ZX10004-611) and the Natural Science Foundation of China (U1305212), by an Intramural grant of the Chinese Academy of Sciences (KJZD-EW-L01-3), and by the Hundreds of Talents Program of the Chinese Academy of Sciences 2009 – 2014 to J.-L. Chen.
We declare that we have no conflicts of interest.
REFERENCES
1.Lam TT, Wang J, Shen Y, Zhou B, Duan L, Cheung CL, Ma C, Lycett SJ, Leung CY, Chen X, Li L, Hong W, Chai Y, Zhou L, Liang H, Ou Z, Liu Y, Farooqui A, Kelvin DJ, Poon LL, Smith DK, Pybus OG, Leung GM, Shu Y, Webster RG, Webby RJ, Peiris JS, Rambaut A, Zhu H, Guan Y.2013. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature502:241–244.http://dx.doi .org/10.1038/nature12515.
2.Takeuchi O, Akira S.2009. Innate immunity to virus infection. Immunol. Rev.227:75– 86.http://dx.doi.org/10.1111/j.1600-065X.2008.00737.x. 3.Takaoka A, Yanai H.2006. Interferon signalling network in innate
de-fence. Cell. Microbiol.8:907–922.http://dx.doi.org/10.1111/j.1462-5822 .2006.00716.x.
4.Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, Sorgeloos F, Ehl S, Mayer D, Kochs G, Schwemmle M, Gunther S, Drosten C, Michiels T, Staeheli P. 2010. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 84:5670 –5677. http://dx.doi.org/10.1128/JVI .00272-10.
5.Wei H, Wang S, Chen Q, Chen Y, Chi X, Zhang L, Huang S, Gao GF, Chen J-L. 2014. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus Infection. PLoS Pathog.10:e1003845.http://dx.doi.org/10.1371/journal.ppat .1003845.
6.D’Cunha J, Knight E, Haas AL, Truitt RL, Borden EC.1996. Immuno-regulatory properties of ISG15, an interferon-induced cytokine. Proc. Natl. Acad. Sci. U. S. A.93:211–215.http://dx.doi.org/10.1073/pnas.93.1 .211.
7. Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova J-L, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III
on November 7, 2019 by guest
http://jvi.asm.org/
feron signaling. J. Virol. 81:7776 –7785. http://dx.doi.org/10.1128/JVI .00546-06.
8.Rebouillat D, Hovanessian AG.1999. The human 2=, 5=-oligoadenylate synthetase family: interferon-induced proteins with unique enzymatic properties. J. Interferon Cytokine Res.19:295–308.http://dx.doi.org/10 .1089/107999099313992.
9.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ.2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell139:1243– 1254.http://dx.doi.org/10.1016/j.cell.2009.12.017.
10. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM.2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature472:481– 485.http://dx.doi.org/10 .1038/nature09907.
11. Pauli E-K, Schmolke M, Wolff T, Viemann D, Roth J, Bode JG, Ludwig S.2008. Influenza A virus inhibits type I IFN signaling via NF- B-dependent induction of SOCS-3 expression. PLoS Pathog.4:e1000196. http://dx.doi.org/10.1371/journal.ppat.1000196.
12. Walsh D, Mohr I.2011. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol.9:860 – 875.http://dx.doi.org/10.1038 /nrmicro2655.
13. Mohr I, Sonenberg N.2012. Host translation at the nexus of infection and immunity. Cell Host Microbe 12:470 – 483.http://dx.doi.org/10.1016/j .chom.2012.09.006.
14. Walsh D, Mathews MB, Mohr I.2013. Tinkering with translation: pro-tein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol.
5:a012351.http://dx.doi.org/10.1101/cshperspect.a012351.
15. García-Sastre A, Biron CA.2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science312:879 – 882.http://dx.doi.org /10.1126/science.1125676.
16. Gainey MD, Dillon PJ, Clark KM, Manuse MJ, Parks GD. 2008. Paramyxovirus-induced shutoff of host and viral protein synthesis: role of the P and V proteins in limiting PKR activation. J. Virol.82:828 – 839. http://dx.doi.org/10.1128/JVI.02023-07.
17. Williams B.1999. PKR; a sentinel kinase for cellular stress. Oncogene
18:6112– 6120.http://dx.doi.org/10.1038/sj.onc.1203127.
18. Katze MG, He Y, Gale M. 2002. Viruses and interferon: a fight for supremacy. Nat. Rev. Immunol. 2:675– 687.http://dx.doi.org/10.1038 /nri888.
19. Walsh D, Arias C, Perez C, Halladin D, Escandon M, Ueda T, Wa-tanabe-Fukunaga R, Fukunaga R, Mohr I.2008. Eukaryotic translation initiation factor 4F architectural alterations accompany translation initi-ation factor redistribution in poxvirus-infected cells. Mol. Cell. Biol.28:
2648 –2658.http://dx.doi.org/10.1128/MCB.01631-07.
20. Walsh D, Perez C, Notary J, Mohr I.2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovi-rus-infected cells. J. Virol.79:8057– 8064.http://dx.doi.org/10.1128/JVI .79.13.8057-8064.2005.
21. Herdy B, Jaramillo M, Svitkin YV, Rosenfeld AB, Kobayashi M, Walsh D, Alain T, Sean P, Robichaud N, Topisirovic I, Furic L, Dowling RJ, Sylvestre A, Rong L, Colina R, Costa-Mattioli M, Fritz JH, Olivier M, Brown E, Mohr I, Sonenberg N. 2012. Translational control of the activation of transcription factor NF-kappaB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nat. Immu-nol.13:543–550.http://dx.doi.org/10.1038/ni.2291.
22. Aragón T, de la Luna S, Novoa I, Carrasco L, Ortín J, Nieto A.2000. Eukaryotic translation initiation factor 4GI is a cellular target for NS1 protein, a translational activator of influenza virus. Mol. Cell. Biol.20:
6259 – 6268.http://dx.doi.org/10.1128/MCB.20.17.6259-6268.2000. 23. Yángüez E, Castello A, Welnowska E, Carrasco L, Goodfellow I, Nieto
A.2011. Functional impairment of eIF4A and eIF4G factors correlates with inhibition of influenza virus mRNA translation. Virology413:93– 102.http://dx.doi.org/10.1016/j.virol.2011.02.012.
24. Yang J, Wang J, Chen K, Guo G, Xi R, Rothman PB, Whitten D, Zhang L, Huang S, Chen J-L.2013. eIF4B phosphorylation by Pim kinases plays a critical role in cellular transformation by Abl oncogenes. Cancer Res.
73:4898 – 4908.http://dx.doi.org/10.1158/0008-5472.CAN-12-4277. 25. Rogers GW, Richter NJ, Merrick WC.1999. Biochemical and kinetic
characterization of the RNA helicase activity of eukaryotic initiation factor 4A. J. Biol. Chem.274:12236 –12244.http://dx.doi.org/10.1074/jbc.274 .18.12236.
26. Ochs K, Saleh L, Bassili G, Sonntag VH, Zeller A, Niepmann M.2002.
Interaction of translation initiation factor eIF4B with the poliovirus inter-nal ribosome entry site. J. Virol.76:2113–2122.http://dx.doi.org/10.1128 /jvi.76.5.2113-2122.2002.
27. Kuang E, Fu B, Liang Q, Myoung J, Zhu F.2011. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J. Biol. Chem.286:41171– 41182.http://dx.doi.org/10.1074 /jbc.M111.280982.
28. Kroczynska B, Kaur S, Katsoulidis E, Majchrzak-Kita B, Sassano A, Kozma SC, Fish EN, Platanias LC.2009. Interferon-dependent engage-ment of eukaryotic initiation factor 4B via S6 kinase (S6K)-and ribosomal protein S6K-mediated signals. Mol. Cell. Biol.29:2865–2875.http://dx .doi.org/10.1128/MCB.01537-08.
29. Hsiang T-Y, Zhao C, Krug RM.2009. Interferon-induced ISG15 conju-gation inhibits influenza A virus gene expression and replication in human cells. J. Virol.83:5971–5977.http://dx.doi.org/10.1128/JVI.01667-08. 30. Zhao C, Hsiang T-Y, Kuo R-L, Krug RM. 2010. ISG15 conjugation
system targets the viral NS1 protein in influenza A virus–infected cells. Proc. Natl. Acad. Sci. U. S. A.107:2253–2258.http://dx.doi.org/10.1073 /pnas.0909144107.
31. Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, García-Sastre A.2007. IFN-stimulated gene 15 functions as a critical antiviral molecule against influ-enza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. U. S. A.104:1371– 1376.http://dx.doi.org/10.1073/pnas.0607038104.
32. Wang S, Li H, Chen Y, Wei H, Gao GF, Liu H, Huang S, Chen J-L.2012. Transport of influenza virus neuraminidase (NA) to host cell surface is regulated by ARHGAP21 and Cdc42 proteins. J. Biol. Chem.287:9804 – 9816.http://dx.doi.org/10.1074/jbc.M111.312959.
33. Guo G, Qiu X, Wang S, Chen Y, Rothman P, Wang Z, Wang G, Chen J.2010. Oncogenic E17K mutation in the pleckstrin homology domain of AKT1 promotes v-Abl-mediated pre-B-cell transformation and survival of Pim-deficient cells. Oncogene29:3845–3853.http://dx.doi.org/10.1038 /onc.2010.149.
34. Feeley EM, Sims JS, John SP, Chin CR, Pertel T, Chen L-M, Gaiha GD, Ryan BJ, Donis RO, Elledge SJ.2011. IFITM3 inhibits influenza A virus infection by preventing cytosolic entry. PLoS Pathog.7:e1002337.http: //dx.doi.org/10.1371/journal.ppat.1002337.
35. Desai SD, Wood LM, Tsai Y-C, Hsieh T-S, Marks JR, Scott GL, Giovanella BC, Liu LF.2008. ISG15 as a novel tumor biomarker for drug sensitivity. Mol. Cancer Ther. 7:1430 –1439. http://dx.doi.org/10.1158 /1535-7163.MCT-07-2345.
36. Fan K, Jia Y, Wang S, Li H, Wu D, Wang G, Chen J-L.2012. Role of Itk signalling in the interaction between influenza A virus and T-cells. J. Gen. Virol.93:987–997.http://dx.doi.org/10.1099/vir.0.041228-0.
37. Lyles DS.2000. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev.64:709 –724.http://dx.doi.org /10.1128/MMBR.64.4.709-724.2000.
38. Adams J.2004. The proteasome: a suitable antineoplastic target. Nat. Rev. Cancer4:349 –360.http://dx.doi.org/10.1038/nrc1361.
39. Alfonso R, Rodriguez A, Rodriguez P, Lutz T, Nieto A.2013. CHD6, a cellular repressor of influenza virus replication, is degraded in human alveolar epithelial cells and mice lungs during infection. J. Virol.87:4534 – 4544.http://dx.doi.org/10.1128/JVI.00554-12.
40. Haye K, Burmakina S, Moran T, García-Sastre A, Fernandez-Sesma A.
2009. The NS1 protein of a human influenza virus inhibits type I inter-feron production and the induction of antiviral responses in primary hu-man dendritic and respiratory epithelial cells. J. Virol.83:6849 – 6862. http://dx.doi.org/10.1128/JVI.02323-08.
41. Gack MU, Albrecht RA, Urano T, Inn K-S, Huang I, Carnero E, Farzan M, Inoue S, Jung JU, García-Sastre A.2009. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe5:439 – 449.http://dx.doi.org/10.1016/j .chom.2009.04.006.
42. Rajsbaum R, Albrecht RA, Wang MK, Maharaj NP, Versteeg GA, Nistal-Villán E, García-Sastre A, Gack MU.2012. Species-specific inhi-bition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog.8:e1003059.http://dx.doi.org/10.1371/journal .ppat.1003059.
43. Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, García-Sastre A.2000. Activation of interferon regulatory factor 3 is inhibited by
on November 7, 2019 by guest
http://jvi.asm.org/
the influenza A virus NS1 protein. J. Virol.74:7989 –7996.http://dx.doi .org/10.1128/JVI.74.17.7989-7996.2000.
44. Schoggins JW, Macduff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM.2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature505:691– 695.http://dx.doi .org/10.1038/nature12862.
45. Raught B, Peiretti F, Gingras A-C, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW.2004. Phos-phorylation of eucaryotic translation initiation factor 4B Ser422 is modu-lated by S6 kinases. EMBO J.23:1761–1769.http://dx.doi.org/10.1038/sj .emboj.7600193.
46. Shahbazian D, Parsyan A, Petroulakis E, Topisirovic I, Martineau Y, Gibbs BF, Svitkin Y, Sonenberg N.2010. Control of cell survival and proliferation by mammalian eukaryotic initiation factor 4B. Mol. Cell. Biol.30:1478 –1485.http://dx.doi.org/10.1128/MCB.01218-09. 47. Darnell JE, Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and
transcriptional activation in response to IFNs and other extracellular signaling proteins. Science264:1415–1420.http://dx.doi.org/10.1126 /science.8197455.
48. Weidner JM, Jiang D, Pan X-B, Chang J, Block TM, Guo J-T.2010. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol.84:
12646 –12657.http://dx.doi.org/10.1128/JVI.01328-10.
49. Lu J, Pan Q, Rong L, Liu S-L, Liang C.2011. The IFITM proteins inhibit HIV-1 infection. J. Virol.85:2126 –2137.http://dx.doi.org/10.1128/JVI .01531-10.
50. Jiang D, Weidner JM, Qing M, Pan XB, Guo H, Xu C, Zhang X, Birk A, Chang J, Shi PY, Block TM, Guo JT. 2010. Identification of five interferon-induced cellular proteins that inhibit West Nile virus and
den-gue virus infections. J. Virol.84:8332– 8341.http://dx.doi.org/10.1128 /JVI.02199-09.
51. Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, Ahmed AA, Chi X, Dong L, Longobardi LE, Boltz D, Kuhn JH, Elledge SJ, Bavari S, Denison MR, Choe H, Farzan M.2011. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 7:e1001258.http://dx.doi.org/10 .1371/journal.ppat.1001258.
52. Everitt AR, Clare S, Pertel T, John SP, Wash RS, Smith SE, Chin CR, Feeley EM, Sims JS, Adams DJ, Wise HM, Kane L, Goulding D, Digard P, Anttila V, Baillie JK, Walsh TS, Hume DA, Palotie A, Xue Y, Colonna V, Tyler-Smith C, Dunning J, Gordon SB, Smyth RL, Open-shaw PJ, Dougan G, Brass AL, Kellam P.2012. IFITM3 restricts the morbidity and mortality associated with influenza. Nature484:519 –523. http://dx.doi.org/10.1038/nature10921.
53. Wakim LM, Gupta N, Mintern JD, Villadangos JA. 2013. Enhanced survival of lung tissue-resident memory CD8⫹T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol.
14:238 –245.http://dx.doi.org/10.1038/ni.2525.
54. Li K, Markosyan RM, Zheng Y-M, Golfetto O, Bungart B, Li M, Ding S, He Y, Liang C, Lee JC, Gratton E, Cohen FS, Liu S-L.2013. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog.9:e1003124. http://dx.doi.org/10.1371/journal.ppat.1003124.
55. Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang I, Farzan M, Jung JU.2013. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe13:452– 464.http://dx .doi.org/10.1016/j.chom.2013.03.006.
56. Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB.2014. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog.10:
e1004048.http://dx.doi.org/10.1371/journal.ppat.1004048.