Copyright C) 1994, American Society for Microbiology
Modulation of
Cyclin Gene
Expression
by Adenovirus ElA in
a
Cell Line with ElA-Dependent Conditional Proliferation
DIMITRY SPITKOVSKY,' PHILIPP STEINER,2 JIRI LUKAS,3 EMMA LEES,4 MICHELEPAGANO,SALMUT
SCHULZE,'
SILVIAJOSWIG,1 DIDIER PICARD,6MASSIMOTOMMASINO,7MARTIN EILERS,2ANDPIDDERJANSEN-DURRl*
Angewandte Tumorvirologie, DeutschesKrebsforschungszentrum,1 and Zentrumfur MolekulareBiologie,2D-69120 Heidelberg, Germany; Divisionfor CancerBiology, Danish Cancer Society Research Center, DK-2100Copenhagen,
Denmark3;Massachusetts GeneralHospital Cancer Center Boston, Massachusetts
021294;
Mitotix, Cambridge, Massachusetts021395;DepartmentdeBiologieCellulaire, UniversitedeGeneve, CH-1211 Geneve 4,Switzerland6; and Tumor VirusGroup, ImperialCancer ResearchFund, Cambridge CB21QP,
UnitedKingdom7
Received 1October 1993/Accepted 27 December 1993
Toinvestigate howadenovirus ElA controls cell proliferation,wehave fusedElAto the hormone-binding domain of the human estrogen receptor (ER) and introduced the EIA-ER chimeric gene together with an
activatedrasgeneintoprimaryratembryofibroblasts. Cell linesderived from thistransfection proliferate in
an estrogen-dependent manner. Estrogen-dependent activation of ElA-ER led to a rapidinduction of both
cyclin Eand cyclinAgeneexpression. Incontrast,levels ofcyclin Dl were stronglyreduced by activation of ElA-ER. Similar changes in cyclin gene expression were observed when primary human fibroblasts were
infected with wild-type adenovirus and when adenovirus ElA was stably expressed in NIH 3T3 cells. Our
findings suggest that activation ofcyclinA and E,but not Dl, gene expression byElAprecedesandmaybe
responsible for ElA-dependent cell proliferation. In contrast, we found that quantitative disruption of
complexes between the E2F transcription factor and the retinoblastoma protein is not required for
ElA-dependent S-phaseentry.
Recent evidence suggests a correlation between the devel-opment of certain cancers and infection with different DNA tumorviruses (for a review, see reference 42). One character-istic property of these viruses is their ability to stimulate proliferation in quiescent cells; to carryoutthisfunction, they have evolved specific genes, such as the E7 gene of human papillomaviruses (35 and references therein), the large T
antigen of simian virus 40 (reviewed in reference 9), and the ElA gene of adenovirus (see below). The deregulation of cell cycle control achieved by such oncogenes may contributetothe immortalization and transformation of mammalian cells by theseviruses.
The mechanism(s) by which these genes override cell cycle control is largely unknown. Adenovirus ElA protein may interfere with growth control by physically interacting with several cell cycle control proteins (39), including the retino-blastoma protein (pRB), p107, cyclin A, cyclin E, and cdk2
(reviewed in reference 7). The interaction between EIA and pRB (38) has attracted particular attention, as pRB has been shownto negatively regulate cell cycle progression during the G1 phase (10). One intracellular target for pRB is the E2F transcription factor (4, 5). E2F is involved in the control of several genes required for cell proliferation (reviewed in reference 23). In untransformed mammalian cells, E2F is complexedtopRB(2), and the activity of E2F is repressed by pRB in transient cotransfection experiments (11, 12, 41). Binding of ElA to pRB can disrupt the association of pRB
with E2Fin vitro (2). Disruption of pRB-E2F complexes by
*Corresponding author. Mailing address: Angewandte Tumorvi-rologie, Abteilung 620, Deutsches Krebsforschungszentrum, Im Neuenheimer Feld 242, D-69120 Heidelberg, Germany. Phone: 49-6221-424628. Fax: 49-6221-424902.
ElAmight, therefore, contribute to the stimulation of DNA
synthesis bythe viral oncoprotein.
Progression through the mammalian cell cycle is thought to
be regulated by a set of related protein kinases, termed
cyclin-dependent kinases(cdkgenefamily [20, 36]),and their regulatorysubunits,termedcyclins (reviewedin reference32). Several classes of cyclins which differ in the timing of their
expression in the cell cycle have been identified (for reviews,
seereferences6, 22, 26, and34).Evidence thatcyclins regulate
cell cycle progression isprovidedby the observations (i) that injection of antibodies to several cyclins blocks progression through the cell cycle (1, 29), (ii) that overexpression of any
oneof thecyclingenesA,Dl,orE can overcomethe cellcycle blockimposed by theRBgene(13), and (iii) that overexpres-sion ofcyclinE(25)aswellascyclinDl(33)canaccelerate the
G,
phase in mammalian fibroblasts and alter their growth factorrequirements.Since there isample evidence that adenovirus ElA can act as atranscriptionfactor(forarecentreview, see reference 24), it ispossible that cell cycle entry may be correlated to specific changesofcyclin gene expressionmediatedby EIA. However, suchchangesare notdocumented; inparticular, it is not clear fromprevious data which cyclin genes, if any, may be targets forElA.Toaddressthis question, primary rat fibroblasts were transfected with the EJ ras oncogene and a second vector encoding a chimeric protein (ElA-ER), in which the ElA protein was rendered conditionally active by fusing it to the hormone-binding domain of the human estrogen receptor
(ER). Wefound that in cells expressing the chimeric protein ElA-ER (IREE-1 cells), proliferation is dependent on the addition of the appropriate steroid hormone. Expression of both thecyclinAandcyclinEgenesis induced byElAinthis system,while expression of cyclin Dl is downregulated.
EIA-dependent modulation of cyclin gene expression was
con-2206
on November 9, 2019 by guest
http://jvi.asm.org/
CYCLIN GENE REGULATION BY ElA 2207
firmed in control experiments in which ElA was delivered to fibroblasts by an adenovirus infection or, alternatively, by
stable transfection.
The potential to manipulate ElA activity by an external stimulus, as in IREE-1 cells, offers the unique opportunity to perform kinetic experiments on the stability of E2F-pRB complexes when challenged by ElA. We found that indeed ElAinterferes with E2F-pRB complexes inIREE-1 cells when grown in the presence of estrogen. However, complexes of E2F withpRBpersisted through several cell cycles after the addi-tion ofhormone to quiescent cells, indicating that disruption of suchcomplexes occurs with slow kinetics. Taken together, our dataindicate that ElA-dependent S-phase entry is correlated to specific modulation of cyclin gene expression, whereas disruption of complexes of E2F with pRB may not be required for the onset of DNA synthesis.
MATERIALSAND METHODS
Construction of plasmid pElAER. ER sequences from plasmid HE14 (17) were first subcloned into the BamHI site of pSP64 to introduce a SmaI site upstream of the coding sequences. ER sequences excised from this intermediate plas-mid weresubsequentlyligated to adenovirus-2ElA sequences at the SmaI site in plasmid pm975 (31). In this construct expression of a chimeric protein containing the 150 amino-terminal amino acids of ElA (containing CR1 and CR2; see Results) fused to the ER hormone-binding domain is driven by theElApromoter.
Cell lines. To establish cells expressing the ElA-ER con-struct, rat embryo fibroblasts were transfected with 5 ,Lg of pElAER and 5 ,ug of pEJras. Transfections were plated in Dulbecco modified Eagle medium (DMEM) containing
10%
fetal calf serum(FCS), to which estrogen was added at a finalconcentration of 100 nM. In this experiment six foci of
transformed cells appeared, whereas in a control experiment notransformed cells appeared in the absence of estrogen. Cells from one focus oftransformationwere established and gave rise to the cell lineIREE-1. To obtain cells that constitutively expressadenovirus ElA, NIH 3T3 fibroblasts were infected by
a recombinant retrovirus (pMXSVneo-18) carrying the ElA
13S cDNA (a gift of R. Ralston). Subsequently, colonies were selected for growth in soft agar. One of the resulting clones was used toestablish the NIH 3T3/13S cell line. Control cells were
derivedbyinfecting NIH 3T3 cells with the empty vector. Cell cycle analysis. Cells were washed with phosphate-buffered saline (PBS), fixed in 70% ethanol, and stained by
propidium iodide as described previously (8).
Fluorescence-activated cell scanning(FACScan) analysis wasperformed by the cell-fit program, with a Becton-Dickinson FACScan sys-tem. Thymidine incorporation was measured as described previously (8).
Antibodies and Western immunoblotting. Antibodies to human cyclin Dl were obtained by immunizing a rabbit with
purifiedcyclinDl proteins as described previously (1).
Mono-clonal antibodies to cyclin E were produced by standard
procedures after BALB/c mice were immunized with a
gluta-thione-S-transferase-cyclin
Efusionprotein, as described pre-viously (18). Polyclonalantibodies to cyclin A (29) andcdk2 (27), respectively, were prepared as described previously.Monoclonal antibodies recognizing the C terminus of ElA
(M73) were obtained from Dianova (Hamburg, Germany). Antibodies to the N terminus of ElA (M37) were obtained fromEd Harlow.Western blots were performed as described previously (27), with the enhanced chemiluminescence system
(Amersham, Inc.).
Northern (RNA) blotting. Total cellular RNA was extracted by the guanidinium thiocyanate-acid phenol method. Total RNA (10,ug) was electrophoresed on 1% agarose formalde-hyde gels and transferred to nylon membranes. Expression of
ElA
and theElA-ER
construct was analyzed with a 0.3-kb BstXI fragment prepared from anElA
cDNA clone. Expres-sion ofcyclin A was monitored with a 1.6-kb mouse probe (15) or a2.2-kbhuman probe, expression of cyclin E was monitored with a 1.5-kb mouse probe (15) or a 2.5-kb human (16) cDNA probe, and expression of cyclinDl
was monitored with the mouse cyll probe (19). Glyceraldehyde-3-phosphate dehydro-genase (GAPDH) expression was analyzed with a rat cDNA probe (15).Band shift assays. A synthetic oligonucleotide encompass-ing the E2F-binding site of the adenovirus E2 promoter was incubated with extracts from different cell lines, as described previously (28). E2F-associated proteins were analyzed by incubation of the band shift reaction with specific antibodies on ice for 50 min prior to electrophoresis. For detection of cyclin A and cdk2 proteins, a polyclonal antiserum was used after affinity purification (28). Human and rat pRB were detected by monoclonal antibody XZ 55 (Dianova), which has been shownpreviously to recognize native rodent pRB (15).
Infection of fibroblasts with adenovirus. Primary human fibroblasts
(MS107,
provided by E.-M. deVilliers) (28) that were obtained from the oral cavity of a healthy human were infected at about 50% confluency by adenovirus 5 or theElA-deficient mutant
d1312,
as described previously (14). For infection, cells were placed in serum-free medium and incu-bated for 1 h with virus at 10 PFU per cell. After 1 h the medium wasreplaced by DMEM supplemented by 10% FCS. For detection of ElA in infected fibroblasts, immunofluo-rescence was performed on coverslips with infected MS 107 cells. Coverslips were rinsed twice in PBS, and cells were fixed for 6 minatroomtemperature in 3.7% formaldehyde in PBS. Coverslips were dipped in ice-cold acetone for 20 s and then washed in 70% ethanol and PBS. The monoclonal anti-ElA antibody M73, diluted 1:20 in PBS containing 5,ug of bovine serum albumin, was applied for 60min
at 37°C. After three PBSwashes fluorescein-conjugated goat anti-mouse immuno-globulin G was applied. After further incubation for 45 minat 37°C, cells were washed three times with PBS and thencounterstained with DAPI (4',6-diamidino-2-phenylindole) (1
,ug/ml)
inPBS for 15minat37°C. Cells were washed twice with PBS,mounted on glass slides with Mowiol (Calbiochem), and examined with a Leitzfluorescence microscope.RESULTS
Characterization of a cell line expressing
EIA-ER
fusion proteins. Primary rat embryo fibroblasts were transfected with a vector encoding the EJ ras oncogene together with an expression vector encoding a chimeric protein, in which ElAis fused inframe to asteroidreceptor hormone-binding domain. Initial experiments using full-length ElA fused to thehor-mone-binding domain of the glucocorticoid receptor (31) revealedtransformingactivity even in the absence of hormone (data notshown). Since this result may reflect inappropriate spacing between the hormone-bindingdomain and the active domain of ElA (reviewed inreference 30), we constructed a vector coding for a fusionprotein
(ElA-ER)
(Fig. 1A)inwhich theN-terminal 150 amino acids of adenovirus type 2ElAare linked to thehormone-bindingdomain of the human ER. Both conserved regions 1 and 2 of ElA (CR1 and CR2 [21]) are retained in this construct. Two weeks after transfection, foci wereobserved on plates in which cultures were grown in theVOL.68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
A EIAcDI
A(codonI
Ad EIA-5'region
B
-EIA ER
-EIA
200.
150 E o 150
5-E00
2 3 4
time (days)
5 6
time (days)
90.
80.
70.
60.
0 50.
40.
30.
20.
10.
() 4 12 16 24 48 72 96 120
time(hrs)
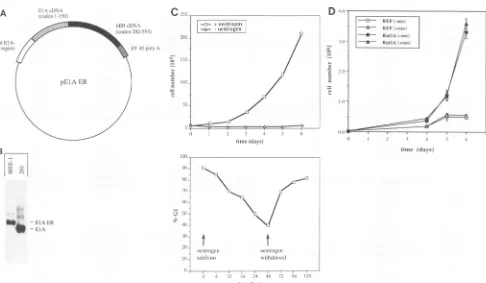
FIG. 1. Characterization ofIREE-1 cells. (A) Structure ofpElAER.pElAERcontains theEIApromoter-enhancer driving expressionofthe
ElA-ERfusion protein, comprising amino acids 1 to 150 of Ad2 EIAfused to amino acids 282 to 595 of the human ER.Furthermore, this
pSP64-derivedvectorcontains the simianvirus 40 BamHI-BclIfragment (nucleotides2533 to2770) comprisingthe simian virus 40poly(A)site.
(B) Expression of the EIA-ERgeneinIREE-1 cells. RNA from 293 cellsandIREE-1cellswasisolated andhybridizedtoaprobederivedfrom
EIAsequences.EIA expressionin293 cellsis indicatedbytheappearanceofa0.8-kbmRNA,whereas inIREE-1 cellsanmRNA withalength
ofabout 1.6 kbwasdetected.(C) Growth propertiesofIREE-1cells.(Upper panel)IREE-1cellswereseeded induplicatein DMEM(10% FCS), containing either100 nM estrogenor noestrogen. Cellswerecountedevery24 h.(Lower panel)Cellswerekeptin hormone-free mediumfor1
week and restimulated by the addition of 100 nMestrogen.At the indicatedtime, samplesof the cellswereanalyzed byFACScan. Theproportion of cells inG,phase isshown.(D) Growth propertiesof control cells. Ratembryofibroblasts(REF)and RatIA cellswereseeded induplicate in DMEM(10% FCS), containing either 100 nMestrogen(+ oes)or noestrogen(- oes).Cellswerecountedevery24 h.
presence ofestrogen but not on plates from which estrogen
was omitted. Cell lines were established from individual foci
and grown in DMEM supplemented with 10% FCS in the
presenceof 00nMestrogen.One of these celllines,IREE-1,
wasused forsubsequent experiments. To monitor expression of the EIA-ERgeneinIREE-1 cells, RNAwasprepared from
IREE-1 cells and hybridizedtoanElA-derived labelledprobe.
The presenceofanmRNAwith alength of 1.6 kb in IREE-1
cells demonstrates expression of the transfected gene (Fig.
IB). Using amonoclonal antibody to the N-terminal part of ElA (M37), we detected a protein of the expected size (75 kDa) inextracts from IREE-1 butnot from control cells (see below) (Fig. 2B).
As shown in Fig. IC (upper panel), IREE-1 cells grow
exponentially in thepresenceofserumandestrogen;therewas
nosign of growth arrestunder these conditions foratleast 12 months(datanotshown). In the absence ofestrogen,the cells do not grow but remain quiescent for several weeks. To analyze the kinetics of ElA-dependent S-phase entry,IREE-1 cellgrowthwasarrested by withdrawal ofestrogenfor 1 week and then restimulated by the addition ofestrogen. At several times after the estrogen addition, samples were taken and
analyzed for their distribution in different phases of the cell cycle by FACScan. The results of this experimentareshown in
Fig. IC (lower panel). Itappearsthat IREE-1 cell growthwas
arrested in
GI
in the absence of estrogen, and cells entered S-phase after readdition of hormone, reaching a peak afterabout 48 h. Withdrawal ofestrogenafter 48 h ledto rearrestof cell growth into G1. Estrogen-dependent S-phase entry of IREE-1 cellswasconfirmed inaparallel experiment measuring
[3H]thymidine incorporation (data not shown). To
demon-stratethatestrogen-dependent proliferation of IREE-1 cells is mediated by the EIA-ER chimeric gene, we established
growth curves for nontransfected primary rat embryo fibro-blasts and foran establishedratfibroblast cell line, RatlA, in theabsence andpresenceofestrogen.For bothcell types,the proliferation ratewasnot significantly altered by the addition of 100 nM estrogen tothe medium (Fig. 1D). The results of theseexperiments indicate that estrogen-dependent prolifera-tion of IREE-1 cells reflects their dependence on an EtA
function forgrowth, rather than activation of the endogenous ERby the steroid. This conclusion is supported byourfinding
thatrat fibroblasts do not express detectable levels of the rat
ER(7a).
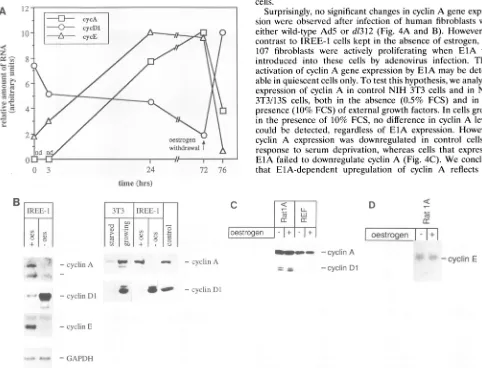
Activation ofcyclin A and cyclin Egeneexpression by ElA.
We determined whether activation of cell proliferation in IREE-1 cells was accompanied by changes in cyclin gene
expression. RNAwas prepared from quiescent IREE-1 cells
and from IREE-1 cells at different times after readdition of estrogenandanalyzed by Northern blotting.
Estrogen-depen-Io +oestroen
-oestrogen
tt
oestrogen
addition withdrawal
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.63.549.70.353.2]CYCLIN GENE REGULATION BY ElA 2209
dl312 Ad5
A
anti ETA
DAPI
)
cn
<t
-OOr-mxco a:: cm 22 2 4m
B
-E1A-ER
IV ]-ElA
dent proliferation was accompanied by a rapid induction of
cyclinA and cyclin E mRNA levels (Fig. 3A). In additional experiments,wedeterminedthatmaximalinduction of cyclinE
mRNAlevelswasreachedat8 hafter theaddition ofestrogen, whileby 4 h half-maximal activation was observed (Fig. 3A)
(datanotshown).Toanalyzewhetherthisinduction is
revers-ible,RNAwasalso preparedfromgrowing IREE-1cells and from cells shifted to estrogen-free medium for 4 h. mRNA levels ofbothcyclinsAand Ewerefoundtobemuchhigherin cells grown in the presence of estrogen than in the cells
withdrawn from estrogen(Fig. 3B). Higherlevels ofcyclinA mRNAwerereflected in similarchangesof theproteinlevels
(Fig. 3B). Rat cyclin E was not detectable by antibodies to humancyclinEavailable tous.
Controlexperiments demonstratedthattheadditionof 100 nMestrogendidnotinducesignificant changesinthe level of
cyclin Ainratembryo fibroblasts and RatlAcells (Fig. 3C).
Similarly, the level of cyclin E mRNA was not affected by
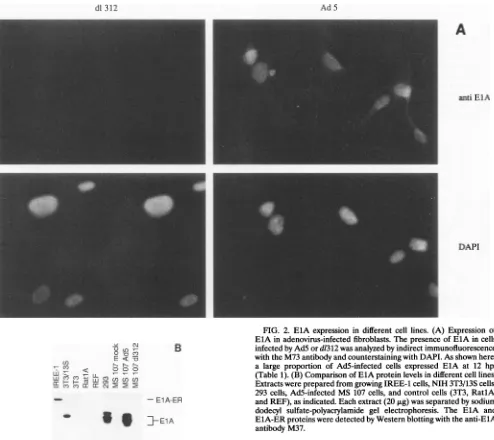
FIG. 2. ElA expression in different cell lines. (A) Expression of ElA in adenovirus-infected fibroblasts. Thepresenceof ElA in cells infected by AdSord1312wasanalyzed by indirect immunofluorescence with the M73 antibodyandcounterstaining with DAPI. As shown here,
a large proportion ofAd5-infected cells expressed ElA at 12 hpi (Table 1). (B)Comparison of ElA protein levels in different cell lines. Extractswereprepared from growing IREE-1 cells, NIH 3T3/13S cells,
293 cells,Ad5-infected MS 107 cells, and control cells (3T3, RatlA,
andREF),asindicated. Eachextract(20,ug)wasseparated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The EIA and ElA-ERproteinsweredetected by Western blotting with the anti-ElA
antibody M37.
estrogeninRatlA cells(Fig. 3D).Takentogether,thesedata indicate that activation of cyclin Aand cyclin Egene
expres-sion, observed in IREE-1 cells after theadditionofestrogen, reflects activation of the ElA-ER fusion protein and is not
[image:4.612.72.566.71.511.2]mediated by activation ofthe ratER.
TABLE 1. ElA-positive nucleiwerecountedat differenttimes of adenovirusinfection,asoutlined in thelegendtoFig. 2A
Time(hpi) ElA-positivenuclei()
0.0~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
O
...6...0
12... 85±5
16... 80±6
24... 84±3
31...3 aResultsareaverages ± standarddeviations fortwoindependentinfection
experiments. VOL.68,1994
on November 9, 2019 by guest
http://jvi.asm.org/
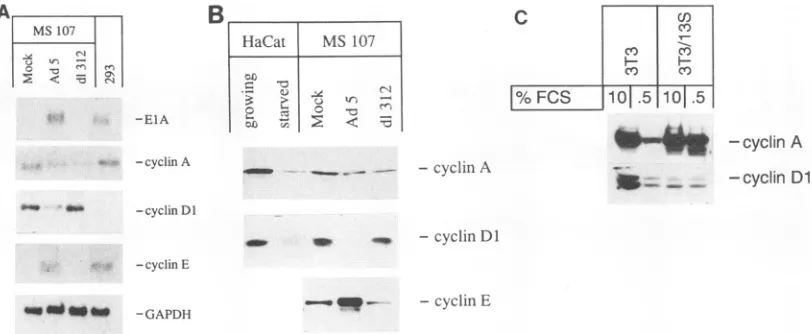
[image:4.612.329.571.625.703.2]Toconfirm thatadenovirus EIAcanupregulate expression
of cyclins E and A, we infected primary human fibroblasts
(isolate MS 107) with adenovirus type 5 (Ad5) and the ElA-deficientmutantd1312.Immunofluorescence showed that expressionof EIAcouldfirst be detectedat 12 hpostinfection
(hpi) in cells infectedwith Ad5,atwhich time about85% of the nucleiwerebrightly stained(Fig. 2A); d1312-infected cells did
notshow anysignal. During theAd5infection, nuclearstaining disappeared at about 30 hpi (Table 1), consistent with the
previous observation that EIA proteins have a short half-life andnegatively regulate theirownsynthesis(3). Control
exper-imentswerealsoperformedwith 293cells,expressingEIAand
A
z
cb. ._
.o-
=: ct-*
CZ*_
&
ef~C
12
0 3 24 72 76
time(hrs)
ElB,and in NIH3T3/13Scells. NIH3T3/13Scclls, derived by infection with a recombinant retrovirus (see Materials and Methods), express the 289-amino-acid (13S) ElA protein.
Expression of EIA in the cell lines used in this study was
demonstratedbyWesternblotting (Fig. 2B).
In Ad5-infected, but not d1312-infected MS 107 cells, we
observed a strong increase in cyclin E mRNA (Fig.4A) and protein (Fig. 4B) at 18 hpi,demonstrating that cyclinE gene
expression is induced in an ElA-dependent fashion. High
levels ofcyclinE werealsoobservedin293 cells(Fig. 4A).As
already suggested by the rapid induction ofcyclin E mRNA upon activation ofElA-ER in IREE-1 cells, these data
indi-cate that EIA acts as a potent inducer of cyclin E gene
expression,independentof theproliferativestatusof thetarget
cells.
Surprisingly, nosignificant changesincyclinA gene
expres-sion were observed after infection of human fibroblasts with either wild-type Ad5 ord1312 (Fig. 4A and B). However, in
contrast to IREE-1 cellskept inthe absence ofestrogen, MS
107 fibroblasts were actively proliferating when ElA was
introduced into these cells by adenovirus infection. Thus,
activation ofcyclinA gene expression byEIA maybe detect-able inquiescent cellsonly.Totestthishypothesis,weanalyzed
expression ofcyclin A in control NIH 3T3 cells and in NIH
3T3/13S cells, both in the absence (0.5% FCS) and in the
presence (10% FCS)of externalgrowthfactors.Incellsgrown
in the presence of 10% FCS, nodifference in cyclin Alevels could be detected, regardless of ElA expression. However, cyclin A expression was downregulated in control cells in
response to serum deprivation, whereas cells that expressed
EIAfailed to downregulatecyclinA (Fig.4C). Weconclude that EIA-dependent upregulation of cyclin A reflects the
B
_- ;: -cyclin A
c -n
*
7i
-cYclin DA
-* 0., cyliD
_0 -cyclinE
3T3
IREE-i
Q .-
o1
> u0
ta 2
rA o +
-cyclinA
C
c --[
LL~~~U15 wL
oestrogen I-
I
+I
-1I+
I_ - -cyclin A D
oestrogen
I
- |+|4*
A - cyclin E -cyclin Dl0
_ -cyclinDl-GAPDH
FIG. 3. Estrogen-dependent changes of cyclin gene expression in IREE-1 cells. (A) Changes ofcyclin mRNAs during estrogen stimulation of IREE-1 cellsby Northern blot.IREE-1 cell growth was arrested by hormone-free medium and restimulated by the addition of estrogen. After 72 hthe medium was replaced by hormone-free medium, and the cells were kept for 4 h in the absence of estrogen. At the time indicated RNA was prepared and hybridized to probes derived from cyclin A, cyclin
Dl,
and cyclin E, as indicated (also see panel B). mRNA levels were quantitated by densitometric scanning. (B) Changes of cyclin gene expression induced by estrogen are reversible. (Left panel) RNA was prepared fromIREE-I cellsgrowinginthe presence of estrogen (+ oes) or from cells shifted to estrogen-free medium for 4 h (-oes). Total cellular RNA was hybridizedtoprobes derived from cyclin A, cyclin DI, cyclin E, and GAPDH, as indicated. (Right panel) Extracts were prepared from IREE-1 cells growing inthe presence of estrogen (+ oes) or from cells shifted to estrogen-free medium for 24 h (-oes).Extracts were also prepared from growing NIH 3T3-cells(10% FCS) or serum-starved NIH 3T3 cells (0.5% FCS). Control lanes contained either cyclin A, in vitro translated from a bacterial expression vector (29), or extracts fromRTI12 cells, derived from a human tumor expressing cyclinDI (18a).CyclinDIappears as a double band in Westernblots performed with extracts from rodent cells, whereas only one band is visible in extracts of human cells (Fig.4B).(C) Expression ofcyclinAandcyclin
Dl
isnot changed by estrogen in rat embryo fibroblasts (REF) andRatlAcells.Rat embryo fibroblasts andRatIAcells were grown inthe absence of oestrogen (-) or treated with 100 nM estrogen for 48 h (+). Levels of cyclinAand cyclinDI were analyzed by Western blotting.(D) Expression of cyclinEis notchanged by estrogen inRatlAcells.RatlA cells were grown as described for panel C; expression of cyclin Ewasanalyzedby Northern blotting._= ,
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.54.536.215.583.2]CYCLIN GENE REGULATION BY ElA 2211
A
MS4~r~ -cycinA
-cyclinDI
-~~cyclinE
qp1~O~i-GAPDH
R
HaCat r
OD tO
S
107CV-0 e
-cyclin A
I:: ENWW
-.9 m 4
C
CO)CO
C'I) C') CO Co)
I%FCS
J101.5
110
.51
lw
-o_7_t
11--tw=
mo-cyclinA -cyclinDl
- cyclin DI
_"_*mw-. -
cyclin
EFIG. 4. ElA-dependent changes of cyclin gene expression in adenovirus-infected human fibroblasts. (A) ElA-dependent changes of cyclin mRNAlevels in infected MS 107 cells.MS107 cells were infected byAdS or d1312 or mock infected. RNA was prepared at 18 hpi and probed with cyclin A-, Dl-, and E- and ElA-specific probes, as indicated. For a control, RNA was also prepared from 293 cells and included as a reference. (B) ElA-dependent changes of cyclin protein levels in infected MS 107 cells. Extracts were prepared from MS 107 cells infected as described for panelA.Therelative amount of cyclin A, Dl, and E was determined by Western blotting. For a control, extracts were also prepared from HaCaT cells, either growing in the presence of 10% FCS or serum starved (0.5% FCS). (C) Modulation of cyclin A and cyclinDl gene expression byserum
starvationinNIH3T3/13S cells and control cells. Extracts were prepared from NIH3T3/13Scells that were either grown in the presence of 10% FCS or kept in the presence of0.5% FCS for 48 h, as indicated. Extracts were also prepared from control NIH 3T3 cells that had been treated inthe same way. Each extract (20pLg)was analyzed byWestern blotting.
proliferativestatusof the targetcells and may, therefore, be an indirect consequence of ElA-induced cell proliferation.
Repression of cyclinDl gene expression by ElA. In contrast
tocyclins A and E, activation of ElA-ER in quiescent IREE-1 cells led to agradual decrease of cyclinDl mRNA(Fig. 3A) andprotein (datanotshown; also see below). Full repression ofcyclin DlmRNArequired exposure to estrogen for at least 72 h(Fig. 3A). Subsequent removal of estrogen for 4 h led to
arapid increaseincyclinDl mRNA and protein (Fig. 3B). In other rodent fibroblasts, e.g., NIH 3T3, cyclin Dl is barely
detectable in serum-starved cells but highly expressed in
growing cells (Fig. 3B) (40). Furthermore, the addition of estrogen did not affect the level ofcyclin Dl in rat embryo fibroblasts andRatlAcells(Fig.3C), indicating that repression of cyclin Dl, as observed in IREE-1 cells, depends on the presence of the ElA-ER protein. These data suggest that
expression ofElAcandownregulate expression of the cyclin Dl gene.
This conclusion was confirmed by the observation that
infection of MS 107 fibroblasts by Ad5 led to a substantial reduction ofcyclin DlmRNA(Fig. 4A)andprotein(Fig. 4B).
Infection withd1312didnotaffectcyclinDl expressionin MS 107 cells, suggesting that repression requires an active ElA protein. To investigatethe possibilitythat expression ofElA might change the cell cycle profile of an infected culture,
FACScan analyses were performed with Ad5-infected and
mock-infectedMS107cells. From theseexperiments itappears thatat16hpiaslight increasein the
G1
fraction resulted from adenovirus infection (52% inGl)
compared with mock-in-fected cells (45% inGl)
(data
notshown).
This observation rulesoutthe formalpossibilitythat thechangesincyclinEand Dl levels whichwe observedaredueto amajoralteration in the cellcycle profileof cellsat16hpi duringanAdS infection.Cyclin Dl expressionwas notdetectable in293cells,further
supporting the
hypothesis
thatthis gene may berepressed by
an adenovirusearlygene(Fig.4A).
In NIH3T3 controlcells,as well as in established human keratinocytes (HaCaT) (Fig.
4B), cyclin Dl is highly expressed when the cells are grown in medium containing 10% FCS but is reduced upon serum starvation.Incontrast tothisobservation, cyclin Dl expression wasstrongly downregulated in proliferating NIH 3T3/13S cells, compared with proliferating control NIH 3T3 cells (Fig. 4C), indicating that ElA-expressing cells fail to upregulate cyclin Dl expression upon cell proliferation. Taken together, our data suggestthat expression of the cyclin Dl gene is repressed
byElA.
ElA-dependent changes ofE2Fmultiprotein complexes. We nextaddressed thequestion of whether complexes of E2F and pRB would be sensitive to ElA-dependent progression of IREE-1cells toS phase. First, we monitored the status ofE2F in rapidly growing IREE cells (with estrogen) and after the removal of estrogen for2 days. Complexes betweenE2Fand pRB were not detected in extracts from cells which were
growingin the presence of estrogen and, hence, containedan
active ElA protein. The removal of estrogen led to the appearance ofa newcomplex which contains the RBprotein (Fig. 5A). The protein composition of the other complexes
seeninFig.5Aremains obscure. However, the results shown in Fig.5Arule out the association of either cyclinAorcdk2with E2Fin any of thecomplexes observed. The antibodiestocyclin
A and cdk2, whichwere used in this experiment, have been shown previously to recognize the relevant antigen in E2F bandshiftexperiments using extractsfrom rat cells (15) (Fig. 6). These results demonstrate that activation of ElA-ER by
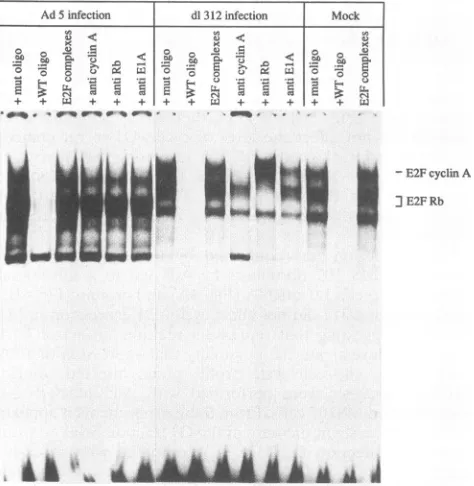
estrogen can indeed disrupt E2F-pRB complexes. Similar changesintheE2Fcompositionwereobservedduring adeno-virus infection of MS 107 fibroblasts(Fig. 6),inagreement with
current hypotheses (23). In a control experiment, we found that ElA-ERprotein iscoprecipitatedwhenpRB is immuno-precipitated from IREE-1 cells (data not shown),
indicating
association of bothproteins.Second, we asked whether dissociation of E2F-pRB
com-plexes would bean earlyeventduring restimulation of
prolif-eration in cells in which growth was arrested by hormoneVOL.68,1994
Ws
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.114.520.73.240.2]IREE-I+ oestrogen IREE-1-oestrogen
x<
<
+-+ WC+- + L + + + + +
3c v~ S EvE >=S
+ +++; + + +
qVf04,<S_rO_t t
LA
B +oestrogen [hrs] . 4 24 48 72 control
Rbantibody | + . + + + + +
E2FRb[
wEM
]E2F Rb
FIG. 5. E2F complexesinIREE-1cells.(A)E2F-RBcomplexeswereinducedby estrogenwithdrawal fromgrowing IREE-1cells.IREE-1cells
weregrowninthepresence ofestrogen.Half ofthe culturewaskeptinthe absence ofestrogenfor 48h,while control cellswerefurther grown
inthepresenceofestrogen.Extractswerepreparedfrom the cells and used for band shiftexperiments. Multiprotein complexesformedonthe
labelled DNAfragmentaredesignated"E2F-complexes";thecompositionof suchcomplexeswasanalyzed bythe addition ofwild-type (WT)or mutant(mut) competitor oligonucleotides (oligo)and antibodiestopRB, p107, cyclin A, cdk2,EIA,cdc2,andp13suc,asindicated (seeMaterials andMethods). (B) Disruption ofE2F-pRB complexes byaddition of estrogentoquiescent IREE-1cells occurredslowly. IREE-1cellgrowthwas
arrestedbyhormone-free mediumand restimulatedbythe addition of estrogen. At the timesindicated, samplesweretaken. Extracts of thesecells
wereanalyzedinband shiftexperimentsin thepresenceorabsence ofpRBantibodies,asindicated.In the controllanes,extractsofIREE-1cells
growninthepresenceofestrogenforseveral weekswereanalyzed.
withdrawal. Cell growth was arrested by keeping the cells in hormone-free medium for 1 week. Estrogen was added, and thecellswere harvested at several times after theaddition of thehormone. Thestatusof E2Fwasanalyzed bythebandshift
technique. Asexpected, E2F-pRB complexesweredetectedin cells in the absence ofestrogen. Surprisingly, suchcomplexes persisted for atleast 3 days after the hormone addition(Fig. 5B), althoughatthat time thecells hadgone throughthe first cellcycles(Fig. 1).Weconcludethatquantitativedisruptionof
E2F-pRB complexes byElA isnotrequiredfor ElA-mediated entry intoS phase.
DISCUSSION
We have analyzed the effects of adenovirus ElA on the
expression ofcyclingenes and thecomposition of E2F
multi-protein complexes in two experimental systems. First, we
constructed a cell line expressing EJ ras together with a
chimeric gene (ElA-ER) encompassing the N-terminal 150 codons of the ElA cDNA fused in frame to the hormone-binding domain of the human ER. Cells that express this
chimericgeneproliferate inahormone-dependentmanner.As
discussed in Results, appropriate controls were included to
ruleouteffects ofestrogenthatarenotrelatedtothepresence
of the ElA-ER protein. Second,we developed aprotocol for
quantitative infectionofgrowing primary human fibroblastsby
adenovirus 5,expressingElA. Conclusionsderived from such
experimentswerecontrolled by additional experiments
involv-ingthe ElA-deficient virusd1312aswell astwo different cell
lines, expressing either ElA and E1B (293 cells) or only the
13Sform of ElA(NIH3T3/13S cells).Fromtheseexperiments
we make the following conclusions.
(i) expression of the cyclin E gene is rapidly induced in
adenovirus-infected growing fibroblasts but not in cells
in-Ad 5 infection dl312 infection
I
Mock0
<U0
~~~~~000
0 .w0
-E2FcyclinA JE2F Rb
I*Mh'
I!!fI
NJ
FIG. 6. ElA-dependent changesinE2Fmultiprotein complexes in
adenovirus-infected MS 107cells. Extractswere prepared from
AdS-infected cells, d1312-infected cells, ormock-infected cells at 16 hpi.
E2Fcomplexeswereanalyzedasdescribed inthe legendtoFig. 5.mut
oligo, mutantoligonucleotide; WToligo,wild-type oligonucleotide. A
I I
m
I ;.,I ll.
L.,&"LA
m
Ai th
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.65.545.75.280.2] [image:7.612.314.551.414.657.2]CYCLIN GENE REGULATION BY EIA 2213
fected with the ElA-deficient mutant d1312. High levels of cyclinEgene expressionwerealsofoundin 293cells.Similarly,
the addition of estrogen to quiescent IREE-1 cells led to a strongincrease ofcyclinE mRNA,detectable onlyafew hours
after the additionof the hormone.Takentogether,these data suggest that the cyclin E gene is rapidly turned on in an
EIA-dependent pathway, consistent with the assumption that
only veryfew intermediary events, ifany, are requiredbefore activationof the cyclin Egene. Furthermore, activation of this
genedoesnot merely reflect ElA-mediatedproliferation,since
it is observed also in growing cells.
(ii) Like cyclin E, cyclin A expression was also substantially increased upon activation of the EIA-ER protein in IREE-1 cells. However, incontrast totheresults obtained withcyclinE, expression of the cyclin A gene was not changed by EIA in growing fibroblasts infectedwith adenovirus. Since cyclin A is already expressed to a considerable extent in such cells, we
reasonedthat induction ofcyclin A by ETA maybe restricted
to quiescent cells. Consistent with this hypothesis, we found
that in NIH 3T3/13S cells the cyclin Alevel is similar to that found in control cells when cells are grown in 10% FCS.
However, unlike incontrol cells,cyclinAgeneexpressionwas not reduced by serum starvation in NIH 3T3/13S cells,
indi-cating that EIA can mimick serum-dependent cell cycle acti-vation.Weconclude that theElA-dependent increaseincyclin
Agene expression, observedselectivelyinquiescent cells,may representaconsequence ofcellcyclemodulationbyEIA(21),
rather than direct activation of the cyclin A gene by the viral
oncoprotein.
(iii) Incontrast totheresultsobtained withcyclinsAand E,
adenovirus infection of human fibroblasts led to a severe
reduction ofcyclin DI levels inthecells. Thiseffectapparently
depends on EIA expression, since it is also observed in 293
cells and NIH3T3/13Scells,whereasd1312-infected fibroblasts
expressnormal levels ofcyclinDI.
Cyclin
DIexpression
isalsodrastically reduced upon activation of the EIA-ER
protein
in IREE-1 cells. Takentogether,ourdatashow thatexpression
of cyclin DI is downregulated by ETA. Thisactivity
of ETA overridesthenormal regulationofcyclin DI bygrowth
factors(40) (Fig. 3C and 4B and C) and may reflect ETA's
ability
to specifically repress transcription from several genes(3).
(iv) ComplexesbetweenE2F andpRB were
disrupted
at 16hpi in the adenovirus infection (Fig. 6) aswell as in 293 cells (data notshown) (18). Disruption ofE2F-pRB
complexes
wasalso observed in IREE-1 cells grown in the presence of
estrogen (Fig.
5A).
These data confirm that also in IREE-1cells, complexesbetweenpRB andE2Fare
disrupted by
ETA.However, disruption ofcomplexes between E2F and
pRB
inthese cells occurs too
slowly
to account for therapid
ETA-dependentS-phaseentryobserved in
estrogen-treated
IREE-1cells. Consistent with this
interpretation, Wang
et al.(37)
hadpreviously demonstrated that ETA's
pRB-binding
domain isnotrequiredforthestimulation ofDNA
synthesis
inquiescent
rat fibroblasts. As proposed by theseinvestigators,
the inter-action ofETAwith pRBmayprevent immortalizedcellsfromreentering the Go phase. The
experiments
reported
hereextend these findings and raise the
question
whether thedisruption by EIA of
pRB-E2F
complexes
plays
any role inprogression to S phase.
Although
at present the conclusions relyonexperimentswith asingle
cell lineexpressing
ETA-ERchimericproteins,thedata
clearly
argueagainst
amodel which invokes disruption of E2F-pRBcomplexes
as akey
step forElA-mediatedS-phaseentry,atleast in the cellscharacterized
in this study. The absence of any detectable
changes
in thelevel ofE2F-pRB
complexes
inestrogen-treated
cells forupto72h,equivalent to at leasttwo
population
doublings
(Fig.
1),
is most
easily
reconciled with the notion that thestability
of suchcomplexes
isnotrelatedtotheposition
ofthecells in thecycle.
This view is furthersupported by
a recent reportby
Schwarzetal.(33a), demonstrating
thatE2Fremainsboundtohypophosphorylated
pRBthroughout
Sphase
insynchronized
humancells,
irrespective
ofthe overallphosphorylation
level ofpRB.
Similarly,
thepRB
species
found incomplexes
with E2F ingrowing
IREE-1 cells maycorrespond
to residualhypophosphorylated
pRB.Thisassumption
issupported by
ourfinding
that theaddition ofestrogen to IREE-1cells doesnotresult in
significant changes
ofpRB
phosphorylation,
a consid-erabledegree
ofpRB
phosphorylation being
detected also in the absence of estrogen(data
notshown).
We concludethat,
by
hormonewithdrawal,
IREE-1 cellgrowth
is arrested in astage at which
pRB
isalready
partially
phosphorylated.
Thisfinding
isunexpected
but mayactually
be correlated to theremarkably
high
level ofcyclin
DI inresting
IREE-1cells,
ascyclin
DI is known to contributesignificantly
topRB
phos-phorylation
(iSa).
The data
reported
here are consistent with thehypothesis
thatadenovirus
ETA
mediates itsmitogenic
effectby
activating
the
expression
of the genesforcyclin
E andcyclin
A,
both of whichwereshowntocontribute tocellcycle
progression.
The alternativepossibility,
that EIA-mediatedproliferation
wouldinvolve the known
capability
ofanotherG1cyclin,
cyclin
DI,
tofacilitate
progression through
G1(1, 33),
isnotcompatible
withour
findings.
On the contrary,ElA-dependent S-phase
entry coincides even withrepression
ofcyclin
Di,
a property thatETA
shares with anothertransforming
oncogene,c-Myc
(15).
While the
significance
of thisrepression
is notclear,
recentdata indicate that continuous
expression
ofcyclin
Dl at theGI-S
boundary
prevents the onset of DNAsynthesis
(25a),
indicating
thatcyclin
DI may blockprogression
through
thecell
cycle
at theG,-S
boundary.
Whereas suchfunctionmight
provide
a clue to theas-yet-unexplained
repression
ofcyclin
DI
by
transforming
oncogenes,furtherexperiments
areclearly
required
to address thisquestion.
Our data
provide
a newexample
for the use of theinacti-vation domain of steroid receptors to block the function ofa
heterologous
protein (reviewed
inreference30).
TheETA-ER
fusionprotein
describedinthisreport contains the functions of theoncoprotein
necessaryforimmortalization of rodentfibro-blasts,
including
thecapability
to modulateexpression
of thethree
cyclin
genesanalyzed
in thisstudy.
Thesefindings
mayprovide
a clue to understand the interference ofETA
with cellulargrowth
control, by
identifying
known cellcycle
regu-latory
genesaspotential
mediators ofElA'soncogenic
activity.
Theexperimental
systempresented
here should enableus torevealsomedetails of the molecular mechanism
underlying
the observedbiological
effects.ACKNOWLEDGMENTS
P.J.-D. and D.S. thankH. zurHausenforcontinuoussupport
and
members of the laboratory for critically
reading
themanuscript.
We thank Claude Kedingerforproviding
adenovirus5 anddl312 stocks. We thank Birgit Martin forassembly
of themanuscript
and Ulrike Ackermann for photography. We are grateful to Steve Reed forprovidingrabbitpolyclonalantiserato
cyclin
E. WethankH.Weidauer (HNO-Klinik, UniversitatHeidelberg)
forearly
passage of MS 107 fibroblasts. D.S. and P.S. made similarcontributionstothis work and should be consideredequalfirst authors.P.S.wassupported bygrantsfrom the RocheResearch Foundation and the Freiwillige Akademische Gesellschaft, Basel. Work in the
laboratory of M.E. was
supported by
the Bundesministerium furForschung und
Technologie (grant
BCT0381-5).
Collaborationbe-tweenM.P., M.E. andP.J.-D.was
supported
by
aresearchgrantfromthe Human Frontiers Science
Program
Organisation.
VOL.68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
REFERENCES
1. Baldin, V., J. Lukas, M. J. Marcote, M. Pagano, and G. Draetta. 1993. Cyclin DI is a nuclear protein required for cell cycle
progression inGl.Genes Dev. 7:812-821.
2. Bandara, L. R., and N. B. LaThangue. 1991. Adenovirus Ela
preventsthe retinoblastomageneproduct from complexing witha
cellular transcription factor. Nature(London) 351:494-497. 3. Berk, A. J. 1986.Adenovirus promotersandEIAtransactivation.
Annu. Rev. Genet.20:45-79.
4. Chellappan, S. P., S. Hiebert, M. Mudryj,J. M. Horowitz, and J. R.Nevins. 1991. The E2F transcription factor isacellulartarget
for the RBprotein. Cell 65:1053-1061.
5. Chittenden, T., D. M. Livingston, and W. J. Kaelin. 1991. The T/ElA-binding domain of the retinoblastoma productcan interact selectively with a sequence-specific DNA-binding protein. Cell 65:1073-1082.
6. Dou, Q. P., A. H. Levin, S. C. Zhao, and A. B. Pardee. 1993. Cyclin-E and Cyclin-A as candidates for the restriction point protein. Cancer Res.53:1493-1497.
7. Dyson, N., P. Guida, K. Munger, and E. Harlow. 1992.
Homolo-goussequencesin adenovirus ElA and humanpapillomavirus E7 proteins mediate interactions with the same set ofcellular
pro-teins. J.Virol. 66:6893-6902. 7a.Eilers, M. Unpublished results.
8. Eilers, M., S. Schirm, and J. M. Bishop. 1991. The MYC protein activates transcription of thealpha-prothymosin gene. EMBO J. 10:133-41.
9. Fanning, E. 1992. Simian virus 40 large T antigen: thepuzzle, the pieces, and the emerging picture. J.Virol. 66:1289-1293. 10. Goodrich, D. W., N. P. Wang, Y.-W. Qian, E. Y.-H. P. Lee, and
W.-H. Lee. 1991. The retinoblastoma gene product regulates progression through theGl phase of the cell cycle. Cell 67:293-302.
11. Hamel, P. A., R. M. Gill, R. A. Phillips, and B. L. Gallie. 1992. Transcriptional repression of the E2-containingpromotersEIIaE,
c-myc,andRBJ by the product of theRBIgene. Mol.Cell. Biol. 12:3431-3438.
12. Hiebert,S. W., S. P. Chellappan, J. M. Horowitz, and J. R. Nevins. 1992. Theinteraction of RB with E2F coincides withaninhibition of thetranscriptional activity of E2F. Genes Dev. 6:177-185. 13. Hinds, P. W., S. Mittnacht, V. Dulic, A. Arnold, S.I.Reed, and
R. A. Weinberg. 1992. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70:993-1006.
14. Jansen-Durr, P., H. Boeuf, and C. Kedinger. 1989. Cooperative binding oftwo E2FmoleculestoanEla-responsivepromoteris triggered by the adenovirus Ela, but not by a cellular Ela-like activity. EMBOJ. 8:3365-3370.
15. Jansen-Durr, P., A. Meichle, P. Steiner, M. Pagano, K. Finke, J. Botz, J. Wessbecher, G. Draetta, and M. Eilers. 1993. Differential modulation ofcyclingeneexpression by MYC. Proc. Natl. Acad.
Sci. USA 90:2547-2552.
15a.Kato,J.-Y., H. Matsushime, S. W. Hiebert, M. E. Ewen, and C. J. Sherr.1993. Directbinding of cyclin Dtothe retinoblastomagene
product (pRb) and pRb phosphorylation by the cyclin D-depen-dent kinase CDK4. GenesDev. 7:331-342.
16. Koff, A., F. Cross, A. Fisher, J. Schumacher, K. Leguellec, M. Philippe, and J. M. Roberts. 1991. Human cyclin E,anewcyclin
that interacts with two members of the cdc2 gene family. Cell
66:1217-1228.
17. Kumar, V., S. Green, A. Staub,and P.Chambon. 1986. Localisa-tion oftheoestradiol-binding and putative DNA-bindingdomains
of thehumanoestrogen receptor.EMBO J. 5:2231-2236. 18. Lees, E., B. Faha, V. Dulic,S.I.Reed, and E. Harlow. 1992. Cyclin
E/cdk2 and CyclinA/cdk2 kinases associate with p107 and E2F in
atemporally distinctmanner.Genes Dev. 6:1874-1885. 18a.Lukas, J. Unpublished data.
19. Matsushime, H., M. F. Roussel, R. A.Ashmun, and C. J. Sherr.
1991. Colony-stimulating factor 1 regulates novel cyclins during theG, phase of the cell cycle. Cell65:701-713.
20. Meyerson, M., G. H. Enders, C. L. Wu, L. K. Su, C.Gorka, C. Nelson, E. Harlow, and L. H. Tsai. 1992. A family of human cdc2-relatedprotein kinases. EMBO J. 11:2909-2917.
21. Moran, E., and M. B. Mathews. 1987. Multiple functional domains in the adenovirus ElA gene. Cell 48:177-178.
22. Murray, A. W., M. J. Solomon, and M. W. Kirschner. 1989. The roleofcyclin synthesis and degradation in the control of matura-tion-promotingfactoractivity. Nature (London) 339:280-286. 23. Nevins, J. R. 1992. E2F: a link between the Rb tumor suppressor
protein and viral oncoproteins. Science 258:424-429.
24. Nevins, J. R. 1993. Transcriptional activation by the adenovirus ElAproteins.Semin. Virol. 4:25-31.
25. Ohtsubo, M., and J. M. Roberts. 1993. Cyclin-dependent regula-tion of Gl in mammalian fibroblasts. Science 259:1908-1912. 25a.Pagano, M.Unpublishedresults.
26. Pagano, M., and G. Draetta. 1991.Cyclin A, cell cycle control and oncogenesis. Prog. Growth Factor Res. 3:267-277.
27. Pagano, M., G. Draetta, and P.Jansen-Durr. 1992. Association of cdk2 kinase with the transcription factor E2F during S phase. Science 255:1144-1147.
28. Pagano, M., M.Durst, S. Joswig, G. Draetta, and P. Jansen-Durr. 1992. Binding of the human E2F transcription factor to the retinoblastoma proteinbut not to cyclin A is abolished in HPV-16-immortalizedcells. Oncogene 7:1681-1686.
29. Pagano, M., R. Pepperkok, F. Verde, W.Ansorge, and G. Draetta. 1992. Cyclin A isrequired at twopoints inthehuman cell cycle. EMBO J. 11:961-971.
30. Picard, D.1993.Steroid-binding domains forregulatingthe func-tionsofheterologousproteinsincis. Trends Cell. Biol. 3:278-280. 31. Picard, D., S. J. Salser, and K. R Yamamoto. 1988. Amovable and regulable inactivationfunctionwithin thesteroid bindingdomain of theglucocorticoidreceptor. Cell 54:1073-1080.
32. Pines, J. 1993. Cyclinsandcyclin-dependent kinases-take your partners.TrendsBiochem. Sci. 18:195-197.
33. Quelle, D. E., R. A. Ashmun, S. A.Shurtleff, J.Y.Kato,D.Barsagi, M. F.Roussel, and C. J. Sherr. 1993.Overexpression ofmouse D-typecyclins acceleratesG(1)phasein rodentfibroblasts.Genes Dev.7:1559-1571.
33a.Schwartz, J. K., S. H. Devoto, E. J. Smith, S. P. Chellapan, L. Jakoi, and J. R. Nevins. 1993. Interactions of the p107and Rb proteins with E2Fduring the cell proliferation response. EMBO J. 12:1013-1020.
34. Sherr, C. J. 1993. Mammalian G(1)-cyclins.Cell 73:1059-1065. 35. Tommasino, M., J. P. Adamczewski, F. Carlotti, C. F. Barth, R
Manetti, M. Contorni, F.Cavalieri, T. Hunt, and L. Crawford. 1993. HPV16 E7 protein associates with the protein kinase p33CDK2 and cyclin A. Oncogene 8:195-202.
36. Tsai, L. H., E. Harlow, and M. Meyerson. 1991. Isolation of the human cdk2 gene that encodes the cyclin A- and adenovirus ElA-associatedp33kinase. Nature(London)353:174-177. 37. Wang, H. H.-G., G. Draetta, and E. Moran. 1991. ElAinduces
phosphorylation of the retinoblastoma protein independently of directphysical associationbetween the ElAand retinoblastoma products.Mol. Cell.Biol. 11:4253-4265.
38. Whyte, P., K. J. Buchkovich, J. M. Horowitz, S. H. Friend, M. Raybuck, R. A. Weinberg, and E. Harlow. 1988. Association between an oncogene and an anti-oncogene: the adenovirusElA proteins bind to the retinoblastoma gene product. Nature (Lon-don)334:124-129.
39. Whyte,P., N. W. Williamson, and E. Harlow. 1989. Cellulartargets for transformationby the adenovirusElAproteins. Cell 56:67-75. 40. Won, K.-A., Y. Xiong, D. Beach, and M. Z. Gilman. 1992. Growth-regulated expression of D-type cyclin genes in human diploid fibroblasts.Proc.Natl. Acad. Sci. USA89:9910-9914. 41. Zamanian, M., and N. B. La Thangue. 1992. Adenovirus Ela
prevents the retinoblastoma gene product from repressing the activity ofacellulartranscription factor. EMBO J.11:2603-2610. 42. zurHausen, H. 1991.Viruses in human cancers. Science
254:1167-1173.