transcribed in vitro from cloned cDNA. Both approaches yielded data consistent with a stable stem-loop
structure within the 3*-terminal 46 bases. In contrast, the 5*52 nucleotides of this 98-base element appear to
be less ordered and may exist in multiple conformations. Under the experimental conditions tested, interaction
between the 3* 98 bases and upstream HCV sequences was not detected. These data provide valuable
infor-mation for future experiments aimed at identifying host and/or viral proteins which interact with this highly conserved RNA element.
Hepatitis C virus (HCV) is a major cause of human liver disease worldwide (11). Persistent infections often result and can lead to cirrhosis and possibly hepatocellular carcinoma (reviewed in reference 16). Since the molecular cloning of HCV in 1989, HCV has been classified with flaviviruses (e.g., yellow fever virus) and pestiviruses (e.g., bovine viral diarrhea
virus) in the family Flaviviridae (12). HCV is an enveloped
virus with a positive-strand RNA genome of approximately 9.4 kb with a single, long open reading frame (ORF). This ORF encodes a polyprotein of approximately 3,000 amino acids which is proteolytically processed by a host signal peptidase and two viral proteinases to yield at least 10 structural and nonstructural proteins (34, 37).
For other RNA viruses, conserved sequences or structures at the termini of viral RNAs often modulate translation, replica-tion, and/or packaging via interactions with protein(s) and/or other RNA elements (reviewed in reference 40). The highly
conserved 59nontranslated region (NTR) of HCV is 341 to 344
nucleotides (nt) in length. Multiple stem-loop structures con-tribute to an internal ribosome entry site, mediating cap-inde-pendent translation of viral RNA (14, 15, 46, 47). On the other
hand, the 39NTR commences with a short sequence (;28 to
42 nt) that is poorly conserved among different genotypes followed by a poly(U)/polypyrimidine tract of variable length and composition. Recently, an additional 98 bases which
ap-pear to represent the authentic 39terminus of HCV genome
RNA were identified. Sequence data from divergent HCV genotypes revealed a high level of conservation within the 98-base element (21, 42, 43, 49). Unfortunately, progress in understanding the functional importance of this sequence has been hindered by the lack of convenient animal models and/or in vitro culture systems for HCV replication.
Secondary structures of this 98-base sequence element have been predicted by computer programs (21, 42, 43). Although such programs can be useful for structure prediction, limita-tions exist, especially when one is dealing with complex RNA
structures. The goal of this study was to use enzymatic and chemical probing techniques to experimentally determine the secondary structure of the 98-base element, both alone and in the context of upstream HCV sequences.
MATERIALS AND METHODS
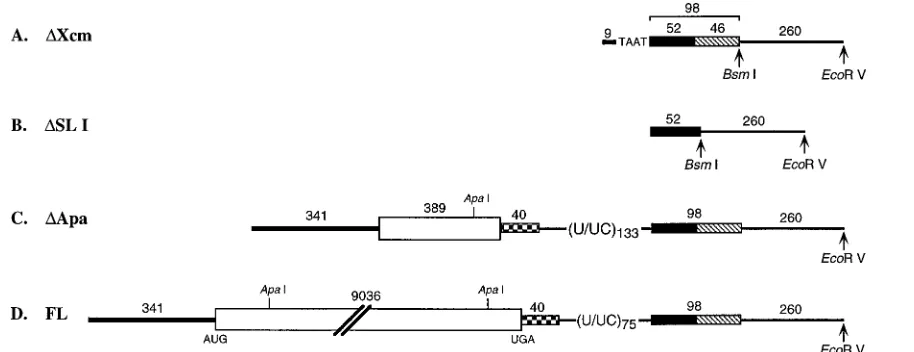
Construction of plasmids for RNA synthesis.The full-length HCV consensus sequence (genotype 1a) was cloned downstream of the T7 RNA polymerase promoter in a pBR322 derivative, pTET, where the sequences betweenDra I-EcoRI were deleted and replaced with a polylinker (20) (Fig. 1). The sequence between theApaI sites in the ORF were deleted, leaving the entire 59and 39
NTRs (DApa [Fig. 1]). A deletion betweenXcmI andMseI in theDApa plasmid created a construct containing 9 nt of the 59NTR and 4 nt of the poly(U/UC) tract upstream of the 98-base element (DXcm [Fig. 1]). The fourth construct contained only 52 nt of the 98-base element (DSL I [Fig. 1]). Plasmid DNAs were linearized with eitherBsmI orEcoRV such that transcribed RNA would pre-sumably have the same 39terminus as HCV RNA or additional nonviral se-quences to serve as a primer binding site during cDNA synthesis, respectively (Fig. 1). RNA was transcribed according to standard procedures, using T7 RNA polymerase (Epicentre).
Radioactive end labeling.Dephosphorylated in vitro-transcribed RNA and gel-purified oligonucleotides were 59end labeled by using T4 polynucleotide kinase (Gibco-BRL) and [g-32P]ATP (6,000 Ci/mmol; Amersham). Dephospho-rylated RNA and oligonucleotides were incubated in a 20-ml reaction mixture containing 70 mM Tris-HCl (pH 7.6), 10 mM MgCl2, 100 mM KCl, 1 mM 2-mercaptoethanol, 100mCi of [g-32P]ATP, and 10 U of T4 polynucleotide kinase for 30 min at 37°C. The reaction mixture was phenol-chloroform extracted and ethanol precipitated. Labeling at the 39end of RNA was performed by ligation of [a-32P]pCp (3,000 Ci/mmol; Amersham) with T4 RNA ligase (Gibco-BRL) as described by England and Uhlenbeck (10). All radiolabeled RNA was purified by denaturing polyacrylamide gel electrophoresis, eluted in 100 mM Tris-HCl (pH 8.0)–1 mM EDTA, phenol-chloroform extracted, and ethanol precipitated.
Structure-specific enzymatic probing. Enzymatic probing of in vitro-tran-scribed RNAs was performed at 0°C for 30 min with the following RNases: cobra venom RNase V1(0.01 and 0.1 U; Pharmacia), RNase CL3 (0.1 U; U.S. Bio-chemical [USB]), and RNases T1(USB), T2(Gibco-BRL), Phy M (Gibco-BRL), andB. cereus(USB) (each at 1 U). RNase digestion of 105cpm of32P-labeled RNA was carried out in a 5-ml reaction volume containing 30 mM Tris-HCl (pH 7.5), 20 mM MgCl2, 300 mM KCl, 1 mM dithiothreitol (DTT) and 4mg of yeast tRNA. Following RNase digestion, an equal volume of urea-dye mix (10 M urea, 2 mM EDTA, 0.06% each bromophenol blue and xylene cyanol) was added, and the cleavage products were analyzed on denaturing polyacrylamide gels. In parallel, RNA sequencing ladders were generated by digestion of end-labeled RNA with RNases Phy M, T1,B. cereus, and U2(each at 1 U) for 15 min at 55°C in urea-citrate buffers. Alkaline hydrolysis was performed at 90°C for 10 min in 50 mM NaHCO3(pH 9.5) to produce an RNA ladder.
Chemical probing.Chemical modification reactions were carried out with 1 to 2 pmol of in vitro-transcribed RNA, in the presence of 0 to 4mg yeast tRNA as a carrier, in 70 mM HEPES-KOH (pH 7.8)–10 mM MgCl2–270 mM KCl–1 mM
* Corresponding author. Mailing address: Department of Molecular Microbiology, Washington University School of Medicine, 660 S. Eu-clid Ave., Box 8230, St. Louis, MO 63110-1093. Phone: (314) 362-2842. Fax: (314) 362-1232
7345
on November 9, 2019 by guest
DTT. Modifying agents included 0.25% dimethyl sulfate (DMS; Fluka), 2.5% diethyl pyrocarbonate (DEPC; Sigma), and 21 mg of 1-cyclohexyl-3-[2-morpho-linoethyl] carbodiimide metho-p-toluene sulfonate (CMCT; Sigma) per ml. Re-actions were performed at 0 and 30°C for times ranging from 5 to 90 min. RNA cleavage was performed by incubation of 1 to 2 pmol of RNA and 0 to 4mg of yeast tRNA in 2 M imidazole (Sigma) buffer (pH 7.0) containing 40 mM NaCl, 1 mM EDTA, and 10 mM MgCl2for 9 h at 37°C. All chemically treated RNA was ethanol precipitated prior to primer extension analysis.
Primer extension analysis.In vitro-transcribed RNA (1 to 2 pmol) with 0 to 4
mg yeast tRNA was digested with RNases as described above, phenol-chloroform extracted, and ethanol precipitated prior to primer extension analysis. Modified or digested RNA was mixed with a 59-end-labeled primer (59-GACTCTAGTT AATTAATTCG-39), and the primer was extended at 37°C for 50 min in 50 mM Tris-HCl (pH 8.3)–75 mM KCl–3 mM MgCl2–10 mM DTT–100 U of Superscript II RNase H2reverse transcriptase (Gibco-BRL). The locations of specific cleav-age/modification sites were determined by polyacrylamide gel electrophoresis of the primer extension products. DNA sequencing reactions obtained by using ddATP or all four dideoxynucleoside triphosphates were run in parallel.
Temperature-dependent UV spectroscopy. Absorbance-versus-temperature profiles (melting curves) were measured at 260 nm in 10 mM sodium cacodylate (pH 7.0)–0.5 mM EDTA–NaCl (10 mM, 100 mM, 300 mM, or 1 M) or KCl (300 mM), using a Gilford 260 spectrophotometer. The temperature was scanned at a heating rate of 1°C/min at temperatures between 5 and 95°C.
RESULTS
RNA secondary structure prediction for the 98-base
ele-ment.RNA folding programs (18) were used to predict
ther-modynamically favored secondary structures for the 98-base element. Multiple structures of similar thermodynamic
stabil-ity were predicted for nt 1 to 52 (the 59 52 bases). The 39
terminal 46 nt (nt 53 to 98) form a stable stem-loop structure, designated SL I, which has a single-stranded loop of 6 nt (nt 73 to 78). This numbering scheme and nomenclature will be used to describe the structure probing experiments presented below. The most stable computer predicted structure for the 98-base element is shown as Fig. 5 in Discussion
RNase structure probing of the HCV 3*-terminal 98 bases.
Initially, the structure of the 98-base sequence element (DXcm
RNA [Fig. 1A]), labeled at either the 59or 39end, was probed
for RNase sensitivity. Nucleotides in single-stranded
confor-mations were monitored with RNases T1(G specific), T2(low
specificity with preference for A),B. cereus(U and C specific),
Phy M (U and A specific), and CL3 (C specific). RNase CL3
can also cleave after single-stranded A and U residues (6).
Nucleotides cleaved by cobra venom RNase V1are considered
to be involved either in base-pairing interactions or in a helical single-stranded arrangement (23). In parallel, sequence lad-ders were generated under denaturing conditions with RNases T1, B. cereus, Phy M, and U2 (A specific) and by alkaline
hydrolysis.
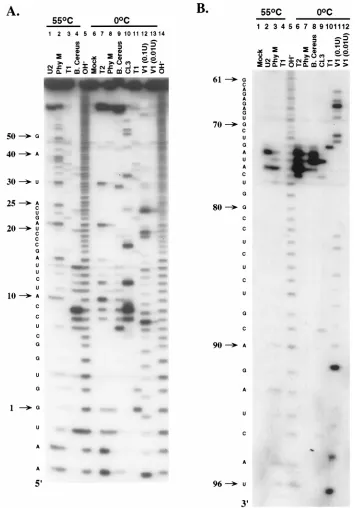
Analysis of the cleavage products by denaturing polyacryl-amide gel electrophoresis revealed different RNase cleavage
profiles for the 59and 39regions of the 98-base element (Fig.
2). These profiles remained unchanged following denaturation and slow cooling to room temperature, suggesting that the pattern of cleavage was not related to stacking interactions
caused by ethanol precipitation (data not shown). TheDXcm
RNA, transcribed from plasmid DNA linearized with BsmI
(Fig. 1), was labeled at the 59end to allow the cleavage pattern
for nt 1 to 52 to be examined. Cleavage with both
single-strand-specific nucleases and cobra venom RNase V1was observed in
some locations. For example, the C residues at positions 8 and
9 were susceptible to RNases CL3,B. cereus, and T2as well as
cobra venom RNase V1(Fig. 2A). On the other hand, cleavage
within SL I (see Fig. 5) produced a distinct profile (Fig. 2B). Nucleotides 73 to 77 were susceptible to digestion with the single-stranded RNases only. In this single-stranded region,
cleavage after G and A residues by RNaseB. cereus(C and U
specific) was observed. The remainder of the nucleotides in the terminal 46 bases were either not cleaved (nt 53 to 63, 70, 73 to 82, 85, 92, 93, and 96), weakly digested (nt 65, 68, 71, 83, 84, 86 to 90, and 94), or completely digested (nt 64, 66, 67, 69, 72,
91, 95, and 97) with cobra venom RNase V1 (Fig. 2B).
[image:2.612.84.541.70.246.2]Fur-thermore, the majority of these nucleotides were resistant to cleavage with the single-strand-specific RNases in the RNA sequencing ladders (Fig. 2B, lanes 2 to 4) where RNA diges-tion was performed under condidiges-tions thought to be denaturing. Although end labeling of short RNA transcripts allowed us to study the structure of the 98-base element, it is not suitable for analyzing the structure of this element by using many of the available modifying chemicals or in the context of longer RNA molecules, such as genome-length HCV RNA. Thus, sites of
FIG. 1. Schematic representation of the RNA transcripts used for the determination of the secondary structure of the 3998-base element. The 59NTR is shown as a thick solid line, and the polyprotein coding region commencing with the AUG at nt 342 is shown as an open box. The 39NTR is represented by (i) the hypervariable region (checkered box), (ii) the poly (U/UC) tract, and (iii) the 98-base element divided into the 5952 bases (solid box) and SL I (hatched box). (A)DXcm RNA consists of 9 nt of the 59NTR fused to 4 nt from the poly (U/UC) tract and the 98-base element. (B)DSL I RNA contains the 5952 bases. (C)DApa RNA contains the complete 59and 39NTR sequences, as well as an internal deletion in the ORF between theApaI sites at nt 461 and 9109. (D) Full-length HCV RNA (FL) has the complete ORF flanked by the 59and 39NTRs. Plasmid DNA was linearized with eitherBsmI orEcoRV such that RNA transcripts would terminate in the required 39nucleotide or contain additional downstream sequences to facilitate primer annealing, respectively.
7346 BLIGHT AND RICE J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
RNase digestion and chemical modification within the 98-base element were identified by primer extension in which cDNA
was synthesized by reverse transcriptase from a 59-end-labeled
downstream oligonucleotide. To create a primer binding site
such that the 39-terminal HCV sequences could be analyzed,
runoff transcripts were synthesized from plasmid DNA
linear-ized at the EcoRV site (Fig. 1). Initially, transcript RNA
(DXcm [Fig. 1A]) was digested with the panel of RNases
de-scribed above and subjected to primer extension, and the
di-gestion profile was compared with those obtained for 59- and
39-end-labeled RNA. Polyacrylamide gel electrophoresis of the
primer extension products (Fig. 3A) revealed a digestion
pat-FIG. 2. Enzymatic probing of the 98-base element obtained by using either 59-end-labeled (A) or 39-end-labeled (B)DXcm transcript RNA. Labeled RNA was digested at 0°C with single-strand-specific RNases T2, Phy M,B. cereus, CL3, and T1or with the double-strand-specific cobra venom RNase V1, and the cleavage products were separated on 20% polyacrylamide sequencing gels. Lanes 1 to 4 represent RNA digested under denaturing conditions with RNases U2, Phy M, T1, and B. cereus(omitted in panel B) to generate A-specific, U- and A-specific, G-specific, and C- and U-specific sequence ladders, respectively. Mock, control reaction in which the RNase was omitted; OH-, the alkaline hydrolysis ladder. Note that cobra venom RNase V1cleavage produces 39-terminal hydroxyl groups, and therefore 59- and 39-end-labeled products migrate slightly faster and slower than the alkaline hydrolysis ladder, respectively. Numbers at the left are nucleotide positions; nt 1 denotes the first base of the 98-base element.
on November 9, 2019 by guest
[image:3.612.129.484.68.576.2]tern similar to that for end-labeled RNA (Fig. 2). For example,
some positions within the 5952 bases were cleaved by
single-strand-specific RNases and cobra venom RNase V1 as
ob-served for 59-end-labeled RNA (Fig. 2A). Furthermore, as
noted for 39-end-labeled RNA (Fig. 2B), nt 73 to 77 were
susceptible to single-strand-specific RNases, while the remain-ing bases involved in SL I (see Fig. 5) had variable reactivities
to cobra venom RNase V1. Hence, these results validated the
primer extension method for studying the structure of the 98-base sequence element in the context of upstream se-quences.
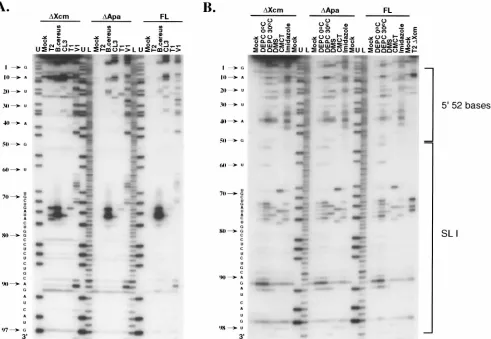
Chemical reagents as probes for structure determination. Regions resistant to all of the RNases might be sterically hin-dered from access by nucleases. To overcome the potential problem of nuclease accessibility, chemicals were used as struc-tural probes for the RNA transcript representing the 98-base
element (DXcm [Fig. 1A]). The chemicals DMS (G-N7 .
A-N1.C-N3), DEPC (A-N7.G-N7), and CMCT (U-N3.
G-N2) were used to modify specific unpaired bases (7, 39). In addition, imidazole-induced cleavage within single-stranded regions was used (45). The sites of modification and imidazole cleavage were identified by primer extension as described
above. Methylation at the N7 position of G by DMS does not terminate reverse transcriptase and as a result is not detected in these experiments. Chemical treatment of RNA confirmed the RNase cleavage pattern for the 98-base sequence. Those nucleotides susceptible to single-strand-specific RNases in SL I (nt 73 to 77 [see Fig. 5]) were cleaved by imidazole and mod-ified with DEPC, DMS, and CMCT, although DEPC tended to lack sequence specificity. In regions previously shown to be
susceptible to cobra venom RNase V1, but not the
single-stranded nucleases, nucleotide modification/cleavage was neg-ligible (Fig. 3). In contrast to the RNase digestion profile, chemical probing identified three additional nucleotides sus-ceptible to the single-strand-specific chemical reagents (Fig. 3): (i) nt 69 and 92, predicted to be bulges in SL I and; and (ii) nt 78, predicted to be the sixth nucleotide of the single-stranded loop in SL I (see Discussion and Fig. 5). On the other hand,
despite their susceptibility to cobra venom RNase V1,
nucleo-tides within the 5952 bases were heavily modified by the
single-strand-specific chemicals (Fig. 3), providing additional evi-dence suggesting the absence of a single defined structure within this region.
[image:4.612.64.555.70.409.2]To investigate potential interactions between the 5952 bases
FIG. 3. Primer extension analysis for RNase (A)- and chemical (B)-treatedDXcm,DApa, and full-length (FL) in vitro-transcribed RNAs, containing additional downstream vector sequences. RNA transcripts were digested with RNases T2,B. cereus, CL3, T1and V1at 0°C followed by primer extension with an end-labeled downstream oligonucleotide. The single-strand-specific chemicals DEPC (0 and 30°C), DMS (30°C), CMCT (0°C), and imidazole (37°C) were used to modify the RNA transcripts. Mock represents those reactions performed in the absence of RNases or chemical reagents. U and L represent DNA sequencing ladders with ddATP only (nucleotide U) and all four dideoxynucleoside triphosphates, respectively. Note that cDNA products synthesized from digested and modified RNA are displaced one nucleotide relative to the DNA sequencing lanes, since the last nucleotide incorporated by reverse transcriptase is complementary to the one on the 39side of the modified/digested position in the RNA template. Nucleotide positions, corresponding to the relative position within the 98-base element, are shown to the left of the gel and have been corrected for the one-nucleotide displacement.
7348 BLIGHT AND RICE J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
and SL I, RNA structural analysis was performed by chemical
probing of transcript RNA comprising nt 1 to 52, with (DXcm)
and without (DSL I) SL I. RNA was transcribed with additional
downstream vector sequences to facilitate primer binding dur-ing reverse transcription (Fig. 1). Primer extension was per-formed on the modified RNA, and the labeled cDNA products were analyzed by denaturing polyacrylamide gel
electrophore-sis. Modification profiles for the 5952 bases were similar
re-gardless of the presence or absence of SL I (data not shown). RNase digestions were performed at 0°C in order to main-tain even unstable RNA structures. Nevertheless, similar re-sults were obtained when chemical modification was per-formed at higher temperatures such as 25, 30, and 37°C (data
not shown). The possibility that Mg21might stabilize tertiary
interactions led us to determine the effect of Mg21 on the
structure of the 98 bases. Since some RNases are dependent on
Mg21 for activity, chemical treatment was used to monitor
unpaired bases in the absence of Mg21. Those nucleotides
susceptible to chemical modification in the presence of Mg21
were equally susceptible when Mg21 was omitted from the
reaction (data not shown).
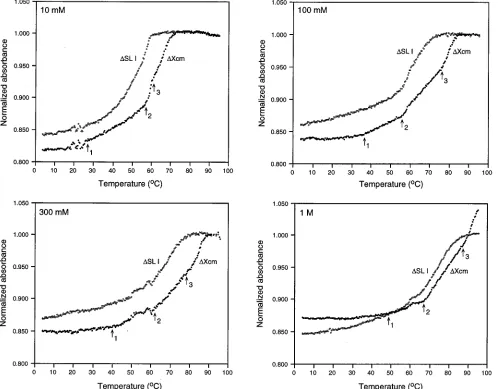
Thermal melting spectroscopic analysis of the 98-base
ele-ment.Due to the ambiguous structure probing data for the 59
52 bases, we sought to further understand the structure of this region by thermally unfolding gel-purified in vitro-transcribed RNA. This process was monitored by the increase in UV absorbancy of two RNA transcripts: (i) the complete 98-base
sequence element (DXcm [Fig. 1A]) and (ii) the 59 52 bases
(DSL I [Fig. 1B]). The salt concentration was varied including
300 mM to mimic the conditions of RNase digestion and chem-ical modification. Since structure probing was performed in KCl, we initially verified that identical melting curves were obtained with NaCl and KCl (data not shown). Figure 4 shows a representative set of the four melting profiles normalized to the absorbance values at 90°C. For the 98-base element, three transitions were evident in all salt concentrations with
mid-points, termed melting temperatures, of 58, 75, and.90°C in
[image:5.612.61.559.73.462.2]1 M NaCl (Fig. 4). The upper melting transition (transition 3),
FIG. 4. Normalized absorbance at 260 nm versus temperature for transcript RNA containing the 98-base element (DXcm) and a truncated version lacking the terminal 3946 nt (DSL I). Absorbances are normalized to the value at 90°C. Panels compare the melting profiles of the two RNA transcripts in 10 mM, 100 mM, 300 mM, and 1 M NaCl. Arrows denote the beginning of the main melting transitions 1, 2, and 3 inDXcm RNA. The occasional minor fluctuations in the absorbance values, also observed in the reference cell, were caused by air bubble formation during heating.
on November 9, 2019 by guest
which was stable in all salt concentrations, probably represents SL I since this transition was absent in the RNA containing
only the 5952 bases (DSL I [Fig. 4]). Corresponding transitions
(transition 2) were observed for theDXcm andDSL I RNAs,
indicating a structure(s) within the 5952 bases (Fig. 4).
How-ever, the first transition in the complete 98 bases was absent in
theDSL I RNA at salt concentrations less than 1 M. Instead,
a linear melt characteristic of unstructured (single-stranded) RNA was observed (Fig. 4), suggesting a weak structure within
the 59 52 bases that maybe stabilized by stacking interactions
with SL I.
Search for potential interactions between the 98-base
ele-ment and HCV sequences. Cross-talk between the 59 and 39
NTR sequences in RNA viruses is important for the modula-tion of translamodula-tion and replicamodula-tion (reviewed in reference 40). Hence, it was of interest to examine potential interactions
between the 98-base element with local sequences in the 39
NTR (e.g., poly[U/UC] tract) and/or more distant sequences
such as the coding region and the 59 NTR. We made two
constructs allowing the transcription of positive-sense RNA
containing (i) the 59 NTR, an internal deletion in the ORF
followed by the complete 39NTR (DApa [Fig. 1C]); and (ii)
full-length HCV cDNA (FL [Fig. 1D]) to produce transcripts representative of the native genome. RNAs, transcribed from
plasmid template DNAs linearized withEcoRV, were digested
with RNases or modified by chemicals as described above, and the sites of cleavage and modification were identified by cDNA synthesis primed by an end-labeled downstream
oligonucleo-tide as described above. Addition of the complete 59 and 39
NTR sequences (DApa) or the full-length HCV genome to the
98-base element did not alter the pattern of cleavage/modifi-cation from that observed for the 98 bases alone (Fig. 3). The
only exception noted was the weak cleavage with RNase T2for
the DApa and full-length RNA transcripts. However, after
purification of these RNAs by using Sephadex G-50 columns,
we observed RNase T2 cleavage profiles for the DApa and
full-length RNA transcripts (data not shown) similar to those shown for the 98 bases alone (Fig. 3A).
These data suggest that no stable base pairing occurs be-tween the 98 bases and upstream HCV sequences in the ab-sence of protein factors. However, it is possible that interac-tions may be destabilized by the additional vector sequences at
the 39end or are too weak to be detected by these methods.
DISCUSSION
In this report, we studied the structure of the 39-terminal
98-base sequence of HCV RNA by using a combination of chemical and enzymatic probing techniques and temperature-dependent UV spectroscopy. Due to the low level of virion RNA in naturally infected patients, this study used synthetic transcript RNA. The results reveal a stable stem-loop structure
(SL I) within the 39-terminal 46 bases with a 6-nt
single-stranded loop and 2 nt representing bulges (Fig. 5). In contrast, no definitive chemical modification or RNase
(single-strand-specific and cobra venom RNase V1) cleavage profile was
ob-served for the 5952 bases, suggesting this sequence folds into
more than one structure (Fig. 5; note that only the most ther-modynamically favored structure is shown). No dramatic dif-ferences in the structure were observed when RNA was (i) heated and cooled slowly to room temperature, (ii) modified
without Mg21, or (iii) modified at various temperatures.
Tem-perature transition profiles of the 98-base element confirmed a
stable structure at the 39terminus and a structure(s) of lower
thermodynamic stability within the 5952 nt. These
experimen-tal data are consistent with the thermodynamically predicted
secondary structures generated by using the mfold program (18). SL I was always predicted to form within the terminal 46
bases (DG5 226.5 kcal/mol [Fig. 5]) with a melting
temper-ature of 85°C in 1 M NaCl, which is comparable to the results obtained in the melting experiments. Analysis of the sequence of the 98-base element in different genotypes reveals that all base changes identified thus far appear within SL I. These mutations occur either in the single-stranded loop or when they arise in the double-stranded stem; the base changes are compensatory, resulting in the preservation of the predicted
SL I structure. No phylogenetic data are available for the 5952
bases due to the invariant nature of this sequence. However,
many alternative structures of similar stabilities (DG5 211.5
to215.7 kcal/mol) can be predicted for this region.
It is likely that the 98-base element is involved in multiple RNA-protein interactions. The stability and structural conser-vation of SL I suggest it may represent a recognition site for viral and/or cellular proteins. For other RNA viruses, con-served terminal structures play critical roles in the initiation of minus-strand and plus-strand RNA synthesis. Such processes
can be mediated via interactions with trans-acting proteins
encoded by the virus or host. The specificity of protein binding can depend solely on the presence of RNA secondary and tertiary structures (9, 17, 26, 27, 35). For example, essential stem-loop structures at the termini of rubella virus (26, 27, 38) and West Nile encephalitis virus (WNV) (5, 36) represent recognition sites for multiple cellular proteins. In rubella virus, one cellular protein binds to the stem-loop structure located at the termini of positive and negative strands (27). Cellular
pro-teins have also been shown to specifically bind to the 39end of
mouse hepatitis virus minus-strand RNA (13), measles virus plus-strand RNA (22), and poliovirus (17) and rhinovirus (44) genomic RNAs. A further source of binding site complexity comes from protein-induced conformational changes in the RNA as proposed for WNV (36). For alfalfa mosaic virus, binding of a viral protein (coat protein necessary to establish
infection in plants) to the 39NTR is accompanied by an
alter-ation in the RNA conformalter-ation, which has been speculated to generate the replicase recognition signal (2).
In contrast, the 5952 bases are invariant without the
forma-tion of an obvious single stable structure. It is possible that this conserved sequence is responsible for multiple protein inter-actions mediated through sequence-specific recognition involv-ing either the entire sequence or subdomains. Recognition of RNA by proteins in a sequence-specific manner, independent of structure, has been described for Tat/Tar in human immu-nodeficiency virus (8, 19, 24) and U snRNP proteins (24). In
addition, multiple structures formed by the 5952 bases (or its
complement near the 59end of the negative strand [42, 43])
may be necessary for binding to different viral and/or cellular proteins, each fulfilling distinct roles in the viral life cycle. It seems likely that sequence-specific and/or multiple structural determinants contribute stringent selective constraints to
ac-count for extreme conservation of this region of the 39NTR.
For rubella virus, WNV, and mouse hepatitis virus, different
cellular proteins have been found to bind to the 39end of the
minus-strand RNA and to the complementary 59end of
posi-tive-strand RNA (13, 33, 36). In the case of Sindbis virus, the
La protein binds to multiple sites within the 39-terminal
nucle-otides of the negative-strand RNA and the 59terminus of the
positive-strand RNA with variable affinities (29, 30).
Another possibility tested in our study was that the 98-base element base pairs with upstream HCV sequences. Although enzymatic and chemical probing showed no evidence for base-pairing interactions with upstream HCV RNA sequences,
communication between the 59and 39NTR sequences is
cru-7350 BLIGHT AND RICE J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
cial in other RNA viruses. For instance, initiation of positive-strand synthesis in the negative-positive-strand viruses such as influenza virus and vesicular stomatitis virus is mediated through
inter-actions between the conserved sequences at the 59 and 39
termini that can base pair. This constitutes thecisregulatory
elements not only for transcription and replication but also for RNA encapsidation (28, 31). On the other hand, for the pos-itive-strand viruses alphaviruses (41), nodaviruses (3, 4), and picornaviruses (48), terminal sequences play essential roles in replication and packaging. Although we found no evidence for cross-talk between the 98-base sequence and upstream HCV sequences, we cannot exclude the possibility that weaker ter-tiary (“kissing”) interactions as described for poliovirus (32) and coxsackie B virus (25) occur, which may not be detectable by the analyses described in this report. Furthermore, binding of host and/or viral proteins to the 98-base element and up-stream RNA sequences may be necessary to stabilize potential RNA-RNA interactions. Recognition of the poly(U/UC) tract by polypyrimidine tract binding proteins (PTB) could also be important for the stabilization of the interactions between the
59and 39NTRs, especially since PTB binding sites have been
identified within the 59NTR (1).
Although a functional assay is required for studying the role
of the 39 end in the viral life cycle, this work provides an
essential starting point for characterizing the interaction of
viral and/or host proteins capable of selective binding to this element. This highly conserved 98-base sequence element could represent an attractive therapeutic target. Identification of compounds which block interaction of this element with its cognate host and/or viral factors may prove useful in control-ling chronic HCV infections.
ACKNOWLEDGMENTS
We thank Kathleen Hall and Jeremy Williams for help with the thermal melting analysis and Michael Zucker for assistance with com-puter predictions. We are also grateful to Sean Amberg, Brett Lin-denbach, John Majors, and Tina Myers for critical reading of the manuscript.
This work was supported in part by grants CA57973 and AI40034 from the Public Health Service.
REFERENCES
1.Ali, N., and A. Siddiqui.1995. Interaction of polypyrimidine tract-binding protein with the 59noncoding region of the hepatitis C virus RNA genome and its functional requirement in internal initiation of translation. J. Virol.
69:6367–6375.
2.Baer, M. L., F. Houser, L. S. Loesch-Fries, and L. Gehrke.1994. Specific RNA binding by amino-terminal peptides of alfalfa mosaic virus coat pro-tein. EMBO J.13:727–735.
3.Ball, L. A.1995. Requirements for the self-directed replication of flock house virus RNA 1. J. Virol69:720–727.
[image:7.612.64.554.70.412.2]4.Ball, L. A., and Y. Li.1993. cis-acting requirements for the replication of FIG. 5. Computer-predicted secondary structure of the 39-terminal 98 bases superimposed with results from RNase digestion (A) and chemical modification (B). The 98-base element commences at the second nucleotide (G). Colored nucleotides represent cleavage by cobra venom RNase V1(A) and imidazole (B). Asterisks represent those bases susceptible to single-strand-specific RNases and chemical modification. The intensity of the asterisk and nucleotide color reflects the level of cleavage (A) and modification (B).
on November 9, 2019 by guest
flock house virus RNA 2. J. Virol.67:3544–3551.
5.Blackwell, J. L., and M. A. Brinton.1995. BHK cell proteins that bind to the 39stem-loop structure of the West Nile virus genome RNA. J. Virol.69:
5650–5658.
6.Boguski, M. S., P. A. Hieter, and C. C. Levy.1980. Identification of a cytidine-specific ribonuclease from chicken liver. J. Biol. Chem.255:2160– 2163.
7.Christiansen, J., and R. Garrett.1988. Enzymatic and chemical probing of ribosomal RNA-protein interactions. Methods Enzymol.164:456–468. 8.Cullen, B. R.1990. The HIV-1 tat protein: an RNA sequence-specific
pro-cessivity factor? Cell63:655–657.
9.Dildine, S. L., and B. L. Semler.1992. Conservation of RNA-protein inter-actions among picornaviruses. J. Virol.66:4364–4376.
10. England, T. E., and O. C. Uhlenbeck.1978. 39-terminal labelling of RNA with T4 RNA ligase. Nature275:560–561.
11. Esteban, J. I., J. Genesca´, and H. J. Alter.1992. Hepatitis C: molecular biology, pathogenesis, epidemiology, clinical features and prevention, p. 253–282.InJ. L. Boyer and R. K. Ockner (ed.), Progress in liver diseases. W. B. Saunders, Philadelphia, Pa.
12. Francki, R. I. B., C. M. Fauquet, D. L. Knudson, and F. Brown.1991. Classification and nomenclature of viruses: fifth report of the international committee on taxonomy of viruses. Arch. Virol. Suppl.2:223.
13. Furuya, T., and M. M. C. Lai.1993. Three different cellular proteins bind to complementary sites on the 59-end-positive and 39-end-negative strands of mouse hepatitis virus RNA. J. Virol.67:7215–7222.
14. Honda, M., E. A. Brown, and S. M. Lemon.1996. Stability of a stem-loop involving the initiator AUG controls the efficiency of internal initiation of translation on hepatitis C virus RNA. RNA2:955–968.
15. Honda, M., L.-H. Ping, R. C. A. Rijnbrand, E. Amphlett, B. Clarke, D. Rowlands, and S. M. Lemon.1996. Structural requirements for initiation of translation by internal ribosome entry within genome-length hepatitis C virus RNA. Virology222:31–42.
16. Houghton, M.1996. Hepatitis C viruses, p. 1035–1058. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology. Raven Press, Philadelphia, Pa.
17. Jacobson, S. J., D. A. M. Konings, and P. Sarnow.1993. Biochemical and genetic evidence for a pseudoknot structure at the 39terminus of the polio-virus RNA genome and its role in viral RNA amplification. J. Virol.67:2961– 2971.
18. Jaeger, J. A., D. H. Turner, and M. Zuker.1989. Improved predictions of secondary structures for RNA. Proc. Natl. Acad. Sci. USA86:7706–7710. 19. Karn J., M. J. Gait, M. J. Churcher, D. A. Mann, I. Mikae´lian, and C.
Pritchard.1994. Control of human immunodeficiency virus gene expression by the RNA-binding proteins tat and rev, p. 192–220.InK. Nagai and I. W. Mattaj (ed.), RNA-protein interactions. IRL Press, Oxford, England. 20. Kolykhalov, A. A., E. V. Agapov, K. J. Blight, K. Mihalik, S. M. Feinstone,
and C. M. Rice.1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science277:570–574.
21. Kolykhalov, A. A., S. M. Feinstone, and C. M. Rice.1996. Identification of a highly conserved sequence element at the 39terminus of hepatitis C virus genome RNA. J. Virol.70:3363–3371.
22. Leopardi, R., V. Hukkanen, R. Vainionpa¨a¨, and A. A. Salmi.1993. Cell proteins bind to sites within the 39noncoding region and the positive-strand leader sequence of measles virus RNA. J. Virol.67:785–790.
23. Lowman, H. B., and D. E. Draper.1986. On the recognition of helical RNA by cobra venom V1 nuclease. J. Biol. Chem.261:5396–5403.
24. Mattaj, I. W.1993. RNA recognition: a family matter? Cell73:837–840. 25. Melchers, W. J. G., J. G. J. Hoenderop, H. J. Bruins Slot, C. W. A. Pleij, E. V.
Pilipenko, V. I. Agol, and J. M. D. Galama.1997. Kissing of the two pre-dominant hairpin loops in the coxsackie B virus 39untranslated region is the essential structural feature of the origin of replication required for negative-strand RNA synthesis. J. Virol.71:686–696.
26. Nakhasi, H. L., X.-Q. Cao, T. A. Rouault, and T.-Y. Liu.1991. Specific binding of host cell proteins to the 39-terminal stem-loop structure of rubella
virus negative-strand RNA. J. Virol.65:5961–5967.
27. Nakhasi, H. L., T. A. Rouault, D. J. Haile, T.-Y. Liu, and R. D. Klausner.
1990. Specific high-affinity binding of host cell proteins to the 39region of rubella virus RNA. New Biol.2:255–264.
28. O’Neill, R. E., and P. Palese.1994. Cis-acting signals and trans-acting factors involved in influenza virus RNA synthesis. Infect. Agents Dis.3:77–84. 29. Pardigon, N., E. Lenches, and J. H. Strauss.1993. Multiple binding sites for
cellular proteins in the 39end of Sindbis alphavirus minus-sense RNA. J. Virol.67:5003–5011.
30. Pardigon, N., and J. H. Strauss.1996. Mosquito homolog of the La autoan-tigen binds to Sindbis virus RNA. J. Virol.70:1173–1181.
31. Pattnaik, A. K., L. A. Ball, A. LeGrone, and G. W. Wertz.1995. The termini of VSV DI particle RNAs are sufficient to signal RNA encapsidation, rep-lication, and budding to generate infectious particles. Virology206:760–764. 32. Pilipenko, E. V., K. V. Poperechny, S. V. Maslova, W. J. G. Melchers, H. J. Bruins Slot, and V. I. Agol.1996.Cis-element,oriR, involved in the initiation of (2) strand poliovirus RNA: a quasi-globular multi-domain RNA structure maintained by tertiary (’kissing’) interactions. EMBO J.15:5428–5436. 33. Pogue, G. P., X. Q. Cao, N. K. Singh, and H. L. Nakhasi.1993. 59sequences
of rubella virus RNA stimulate translation of chimeric RNAs and specifically interact with two host-encoded proteins. J. Virol.67:7106–7117.
34. Rice, C. M.1996.Flaviviridae: the viruses and their replication, p. 931–960. InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology. Raven Press, Philadelphia, Pa.
35. Romaniuk, P. J., P. Lowary, H.-N. Wu, G. Stormo, and O. C. Uhlenbeck.
1987. RNA binding site on R17 coat protein. Biochemistry26:1563–1568. 36. Shi, P.-Y., W. Li, and M. A. Brinton.1996. Cell proteins bind specifically to
West Nile virus minus-strand 39stem-loop RNA. J. Virol70:6278–6287. 37. Shimotohno, K., Y. Tanji, Y. Hirowatari, Y. Komoda, N. Kato, and M.
Hijikata.1995. Processing of the hepatitis C virus precursor protein. J. Hepatol.22:87–92.
38. Singh, N. K., C. D. Atreya, and H. L. Nakhasi.1994. Identification of calreticulin as a rubella virus RNA binding protein. Proc. Natl. Acad. Sci. USA91:12770–12774.
39. Stern, S., D. Moazed, and H. F. Noller.1988. Structural analysis of RNA using chemical and enzymatic probing monitored by primer extension. Meth-ods Enzymol.164:481–489.
40. Strauss, J. H., and E. G. Strauss.1988. Evolution of RNA viruses. Annu. Rev. Microbiol.42:657–683.
41. Strauss, J. H., and E. G. Strauss.1994. The alphaviruses: gene expression, replication, evolution. Microbiol. Rev.58:491–562.
42. Tanaka, T., N. Kato, M.-J. Cho, and K. Shimotohno.1995. A novel sequence found at the 39terminus of hepatitis C virus genome. Biochem. Biophys. Res. Commun.215:744–749.
43. Tanaka, T., N. Kato, M.-J. Cho, K. Sugiyama, and K. Shimotohno.1996. Structure of the 39terminus of the hepatitis C virus genome. J. Virol.70:
3307–3312.
44. Todd, S., J. H. C. Nguyen, and B. L. Semler.1995. RNA-protein interactions directed by the 39end of human rhinovirus genomic RNA. J. Virol.69:3605– 3614.
45. Vlassov, V. V., G. Zuber, B. Felden, J.-P. Behr, and R. Giege´.1995. Cleavage of tRNA with imidazole and spermine imidazole constructs: a new approach for probing RNA structure. Nucleic Acids Res.23:3161–3167.
46. Wang, C., S.-Y. Le, N. Ali, and A. Siddiqui.1995. An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 59noncoding region. RNA1:526–537.
47. Wang, C., and A. Siddiqui.1995. Structure and function of the hepatitis C virus internal ribosome entry site. Curr. Top. Microbiol. Immunol.203:99– 115.
48. Wimmer, E., C. U. Hellen, and X. Cao.1993. Genetics of poliovirus. Annu. Rev. Genet.27:353–436.
49. Yamada, N., K. Tanihara, A. Takada, T. Yorihuzi, M. Tsutsumi, H. Shimo-mura, T. Tsuji, and T. Date.1996. Genetic organization and diversity of the 39noncoding region of the hepatitis C virus genome. Virology223:255–261.
7352 BLIGHT AND RICE J. VIROL.