Function of
an Internal
Bacteriophage
T7
Core During
Assembly of
a
T7 Procapsid
PHILIPSERWER* AND ROBERT H. WATSON
DepartmentofBiochemistry, The UniversityofTexasHealth Science Center, San Antonio, Texas 78284
Received 28 October 1981/Accepted 13 January 1982
ADNA-free, proteinaceous procapsid of bacteriophage T7 (capsidI)hasbeen
shown in previousstudies toconsist of an
external,
spherical shell (envelope) and aninternal, cylindrical corewith fibrous projections that connect the core to theenvelope.Todeterminetherole of the core in assembly of the envelope of capsid
I, the kinetics of appearance of capsid I and possibleintermediates in capsid I
assembly (AG particles)weredetermined in the presence and absence of the core.
Forobtaining these data,agarose gel electrophoresis was used and appeared to be atechniquemoreaccurate and efficient than techniques used for obtaining similar
datainthe past. The results of these experiments were: (i)in thepresence of the core,AGparticles behavedkineticallyasintermediatesin theassembly of capsid
I;(ii)in the absence ofthecore, assembly of capsid I terminated prematurely and
AG particles accumulated. These and other data have been interpreted by
assuming that: AG particles are breakdown products of precursors of capsid I; theseprecursorshaveuncorrectederrorsin the assembly of their envelopes; anda
function
of thecoreistocorrect theseerrors.
Ithas been shown that proteinaceous subunits in the envelopes of spherical, viral capsids are
arranged with icosahedral symmetry to
mini-mize thefreeenergyofthe envelope (3-5, 7, 9). Todetermine the mechanismsby whichsubunits
of envelopes of viral capsidsassemble,attempts are made to isolate incomplete capsids from
virus-infectedcells. Results withbacteriophages
that containdouble-stranded DNApackaged in
a capsid with a spherical envelope, including
bacteriophages
X, T3,T7,
P2, P4, and P22,indicate thataspherical, DNA-free capsid
(pro-capsid)
is assembled and undergoes structuraland compositional alterations before or (and) during
packaging
of DNA (reviewed inrefer-ences2, 6, 10, 11,22).
Asdescribed in the above-referenced
reviews,
theprocapsids of
bacteriophages X,
P22, T3,andT7 have internal
proteins
that assistassembly
of theirouterenvelope
and that insome cases areejected
from theenvelope
ordigested
before orduring
DNApackaging (see
Fig.
2of reference19 for modelsof thebacteriophageT7
procapsid
[capsid I] and the
capsid
to whichcapsid
I converts[capsid II]
during
packaging
ofDNA).
It has been
proposed
that these internalpro-teins, sometimes referred to
collectively
as acore, assist
assembly
ofprotein
in the outerenvelope
by:
(i)
converting
theenvelope
protein
from a
nonassociating
to anassociating
form(P22
[8]), (ii)
forcing
theproteins
of theenvelope
to the orientation needed to
produce
anenve-lope with the needed curvature
(T7
[15];
P22[8]), (iii) forminganucleus forpolymerization of
other subunitsof the capsid (reviewed for
sever-al
bacteriophages
in reference 11), or (iv)pre-venting host proteins from entering the capsid
(6). Hypotheses (i), (ii), and (iii) predictthat in
theabsence of the procapsidcore, assembly of
the procapsid eitherdoes not occur oris slower
than in thepresenceof thecore. Hypotheses(i)
and (iii) also predict thatin the absence of the
core, proteins of the capsid envelope
accumu-late in unpolymerized form. Therefore, the above hypotheses can be tested by measuring thekinetics ofassembly ofprocapsids and pre-cursors of procapsids in the presence and ab-senceofcores. Measurementof the kinetics of assembly ofprocapsids intheabsence ofacore
protein has apparently been
performed only
once (for P22 [1]), and in thisexperiment
only
twotimesweresampled;theresultssuggest thatprocapsidassembly is slowedin theabsenceofa
functional core.
Impediments
toobtaining
the above dataaredifficultiesinidentifying
procap-sidprecursors and thelargeinvestment oftimeand resources needed if the
procedure
forquan-titating
procapsids
is(asitusually
is)
centrifuga-tion.
Therefore, to testtheabove
hypotheses,
it is advantageous todevelop:
(i)
efficient(in
timeand cost)
techniques
foridentifying
andaccu-rately
quantitating
eitherprocapsid
precursors or at least breakdownproducts
of precursors(such precursors and theirbreakdown
products
will both be referred to asprocapsid assembly
595
on November 10, 2019 by guest
http://jvi.asm.org/
intermediates) and (ii) techniques more efficient thanthe techniques ofcentrifugationused inthe past for accurately quantitating procapsids. Agarose gel electrophoresis has been demon-strated to be an accuratetechnique for identify-ing T7capsid I, and it meets the above criteria for a technique to quantitate capsid I during
kinetic labeling experiments (16, 17). In addi-tion, agarose gelelectrophoresis is used in the accompanying communication (19) to identify particles (AG particles) whichareproduced dur-ingassembly of capsid I andarepossibly capsid Iassemblyintermediates (AG particlesare iden-tifiedby their failure to form asharpbandduring agarosegelelectrophoresisand theirpossession ofseveral characteristics ofcapsid II; most AG
particles are converted by trypsin to particles that migrate with capsid II during agarose gel
electrophoresis). Therefore,inthepresent
com-munication,the kinetics of appearance of capsid I and AG particles were determined in the presence and absence of the T7 core, using
agarosegelelectrophoresistoquantitate capsidI and AG particles. The results indicate that hypotheses (i) to (iii) do notaccuratelydescribe
the role of the T7 core and suggest another
hypothesis for the role ofthe T7 coreduringthe
assembly of capsidI.
MATERIALSAND METHODS
Bacteriophageand bacterial strains.Wild-type
bacte-riophageT7andT7ambermutants(20)werereceived
from F. W. Studier. The following amber mutants
were used: gene 5-28 and gene 8-11. The host for
bacteriophageT7and thenonpermissivehostfor am-bermutants wasEscherichiacoliBB/1;thepermissive
hostfor ambermutants wasE.coli 0-11'. Lysates of
the nonpermissive host infected with a T7 amber
mutantwill be referredtobythe number of themutant
T7 gene;particles isolated from such lysates will also
be referredtobythe number of themutantgene.
Buffers and media. Standard/G buffer is 0.15 M
NaCl,0.05 M Tris-chloride, pH 7.4, 0.005 M EDTA,
and 100 ,gof gelatin per ml. Electrophoresis buffer is
0.05 M sodium phosphate, pH 7.4, and 0.001 M
EDTA. Sample buffer is 0.005 M sodium phosphate,
pH 7.4, 0.001MEDTA, 100 ,ug of gelatin perml, 4%
sucrose, and 400 ,Lg of bromophenol blue per ml.
Cultures used for radiolabeling were grown in M9 medium (14).
Stocksofambermutants. Stocks of amber mutants
and wild-type phage T7 were grown and purified as
previously described (19).
Kinetic radiolabeling and artificial lysis of amber
mutant-infected E.
coil.
Log-phase cultures (1.25 ml) ofE.coli BB/1 were grown in M9 medium with aeration
at 30°C to 4 x 108/ml and were then infected at a
multiplicity of 15. At 14 min after infection,
14C-labeled algal hydrolysate (Schwarz/Mann;12 ,Ci/ml)
wasadded to the infected culture, and1min later 100
,lof the culture waschilled,as previouslydescribed
(13);simultaneously with chilling, 15
pl
of20%Casa-mino Acids (freshly prepared) was diluted into the
culture,and aeration was continued at 30°C. Further
100-p,l samples were chilledat15.5, 16.3, 17, 18, 20,
22.5,and 27 minafter infection.
Allchilled samples were centrifuged in 1-ml
cellu-loseacetatebutyratecentrifuge tubes (Sorvall)at4°C
and 11,000 rpm for 10 min. Pelleted bacteria were
suspendedandlysed withlysozyme and the nonionic
detergent Brij 58 aspreviously described (13), except
that volumes used were one-half those previously
used.
Kinetic labeling, lysis, andprefractionation of
wild-typeT7-infected E. coHl. Wild-type Ti-infected E. coli
were subjected to kinetic labeling, lysed (with
lyso-zymeandBrij58), andfractionatedbysedimentation
in a biphasic gradient of sucrose and metrizamide
(sucrose-metrizamide gradient), as previously
de-scribed (16).
Digestionwithtrypsin.Samples weredigested with
trypsinaspreviously described(19).Trypsin-induced
conversion of AG particles in unfractionatedor
pre-fractionatedlysatestoparticlescomigratingwith
cap-sid IIduring agarose gelelectrophoresis (19)wasnot
decreasedbydecreasingtheamountoftrypsinusedby
afactor of 10, indicating thatthis conversion wasat
completion for allexperimentsreported here.
Electrophoresis in agarose gels. Samples were
broughtto13p.l with standard buffer andwerediluted
with 17 ,ulof sample buffer. Of thismixture,20,ulwas
layered beneath4 to6mmofelectrophoresisbuffer in
sample wells ofa0.9%oME agarose(MarineColloids,
Rockland, Maine) slab gelin electrophoresisbuffer,
andelectrophoresis wasperformedas previously
de-scribed(19) for16h.Afterelectrophoresis,gelswere
dried undervacuumandsubjectedtoautoradiography.
Densitometry. Theamountof 14C in selectedregions
ofgels wasdetermined by densitometric scanning of
autoradiograms (anR and Ddensitometer of Helena
Laboratories, Inc., Beaumont, Tex., was used) and
convertingareas thusobtainedto countsperminute,
usinganempiricallydeterminedcalibration factor.
RESULTS
Infections with T7 amber mutants. Tomeasure the kinetics ofappearance ofcapsid I andAG
particles,the amount of radiolabel in these parti-cles is measured as a function of time during
kinetic labeling (pulse-chase) experiments.
Agarose gel electrophoresis of unfractionated lysates can be used to quantitate 14Cin capsid I and also in those AG particles which are con-verted by trypsin to particles migrating with capsid II (19) (all references to AG particles madebelowwillrefer only to those AG particles converted by trypsintoparticles migratingwith
capsid II during agarose gel electrophoresis).
Advantageousaspectsof agarose gel
electropho-resisoflysates without prefractionation by
cen-trifugation are: (i) avoidance of the possible
selective loss of particles during a procedure that includes prefractionation by centrifugation and (ii) requirement for 1/10 to 1/100 the time
and cost of aprocedure which includes
centrifu-gation. However, if 14C-labeled capsid II is
on November 10, 2019 by guest
http://jvi.asm.org/
present in lysates, theseparticles, becausethey comigrate with trypsinized AG particles, will decrease theaccuracywith whichthe amount of 14C in AGparticles can bemeasured; the
pres-ence of
14C-labeled
phage T7 and T7capsid-DNA complexes would further decrease the accuracyof such measurements (17, 18).
Therefore,tomeasurethe kinetics of appear-anceof capsidIandAGparticlesinthepresence
of the
17 core, 5am 17 were used toinfect
anonpermissive host(no DNA synthesized [21])
because capsid I, but neither capsid IInorphage
T7 nor capsid-DNA complexes, is produced
during such aninfection (16, 19, 21; P. Serwer andR. H. Watson, unpublished data). To mea-surekinetics ofappearance of AG particles and capsidIin the absence ofafunctionalcore, an
8am mutant was used to infectthe nonpermis-sive hostbecausean8ammutantis the onlyone
of thecore-defective T7 mutantsthatmakes no
capsid
II,capsid-DNA complexes,
orbacterio-phage T7 during such an infection (12, 19;
Serwer and Watson, unpublisheddata). Howev-er, the 8am mutant makes some capsid I in a
nonpermissive host; the envelope of 8am capsid
Iisindistinguishable from the envelope of
wild-type capsid I by electron microscopy (12) and
agarose gelelectrophoresis (19).
Alog-phase culture ofE.coli BB/1was infect-ed with the5ammutant,andasecondportion of the same culture was infected with the 8am
mutant at 30°C. Kinetic labeling (with
14C-la-beled amino acids) of both cultures was
per-formedas described in theMaterials and Meth-ods; samples takenateight times after thestart
of
labeling
weresubjected
toagarosegel
electro-phoresis aftertreatment withtrypsin and
with-outtreatment with
trypsin.
No band at the
position
ofcapsid
II waspresentinanyof the
untrypsinized
5amlysates,
as expected (Fig. la, channels3-10).
In thetrypsinized
5amlysates
(Fig. la,
channels11-18), a band at the
position
ofcapsid
II wasobserved;
this band is formedby
AGparticles
(19). In Fig. la,
14C appeared
in AGparticles
earlier than it
appeared
incapsid
I;
the amountof
14C
inAGparticles
became maximalat16.3to17 min after infection and
subsequently
de-creased(shown
quantitatively
inFig.
2).
These kineticssuggest that theAGparticles
arecapsid
Iassembly intermediates.
Initially, cells infected with the 8am mutant
(core
negative)
incorporated
14C intocapsid
I and AGparticlesatroughly
thesamerateasthe cells infected with the 5am mutant(core
posi-tive). However, in contrast toresults obtained with the5ammutant,withthe 8ammutant
entry
of
14C
intocapsid
I ceasedby
1.3 min aftertermination of
labeling
with14C(Fig.
lb and2).
The prematurecessation oftheentryof 14C intocapsid I during infection with the 8am mutant was accompanied by an accumulation of14C in
AG particles, an accumulation not seenduring infection with the Sam mutant.
Thepremature cessation of the entry of 14C-labeled protein into capsid I during infection with the8am mutant could be caused byeither (i)adecrease inthecapabilityof theinfectedcell
forassembling capsid I withincreasing time or
(ii) thepresenceof twopathways forassembling capsid I, one in which capsid I is assembled
morerapidly than in theother, and the
require-mentforafunctional core in only the pathwayin which capsid I is assembled more slowly. If possibility (ii) iscorrect,itmustalso beassumed
that no
14C-labeled
protein can enter thepath-way of more rapid capsid I assembly laterthan 1.3 minaftercompletion of labeling.
To test the possibility of a time-dependent
cessation inthe capability of8am-infected cells forassembling capsid I, the experiment of Fig.1 was performed with labeling starting at 19 in-stead of14 min after infection. The amount of
14C thatentered8amcapsidIwas1.5times the
amountthat enteredintheexperimentof Fig. 1,
andaprematuretermination ofthe synthesisof capsidIoccurredat 1.3min afterthecompletion
oflabeling. Thus,it isconcludedthatthe
prema-turecessation ofthe assembly of capsid I does notresultfromadecreaseinthe capability of the
infected cell forassembling capsid I. It is likely,
therefore, that possibility(ii) above is correct.
Infectionwith wild-type 17. Although, as de-scribed above, kinetic labeling ofwild-type T7-infectedE. colidoes not yield data as accurate as data obtained with the above mutants, a
kinetic labeling experimentwas alsoperformed with wild-type 17-infected cells. To remove
interference
ofphage T7 and capsid-DNAcom-plexes, determination of the amount of
14C
in AG particles was performed with lysatespre-fractionated by sedimentation in sucrose-metri-zamidegradients, asdescribed in Materials and Methods. The amount of 14C in AG particles
wasdetermined bysubjecting 14C-labeled
parti-cles
cosedimenting
withcapsid
II toagarosegelelectrophoresis with and without
prior
treatmentwithtrypsin, and
subtracting
the amountof14Cmigrating
ascapsid
IIin theuntrypsinized
sam-ple from theamountof 14C
migrating
ascapsid
IIin the trypsinized sample.
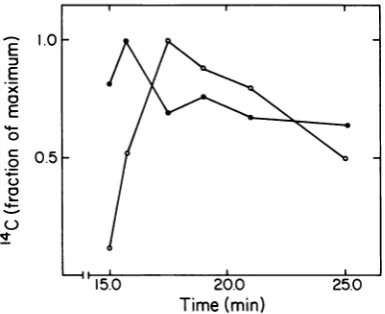
Figure 3 showsthe results of kinetic
labeling
ofwild-type 17-infectedE. coli. As found with
5am17-infectedE.coli in
Fig. 2,
the kinetics ofappearanceofAG
particles
suggested
thatmostof the AG
particles
wereintermediates incapsid
I
assembly.
However,
atthe later times inFig.
3,14C
left AGparticles
lessrapidly
than it leftcapsid
I.Although
the reason for this effect isnot
known,
itcould result from theproduction
ofon November 10, 2019 by guest
http://jvi.asm.org/
598 SERWER AND WATSON
0.
Capsid I
*4.
4) W81;W d W
* .. 1 Xq. ... ..~~~~~AL
;.irie...t:: .:
IS%
I15~
10
15
20
-
Capsid
E
it
b.
--
Capsid
I
-4
I 10
5
10
1520
FIG. 1. Kineticlabeling of5am-and8am-infectednonpermissive host. Akinetic labeling experiment was
performed, and infected bacteriawerelysedasdescribed in the text,usingastheinfecting bacteriophage: (a)
5amT7and (b)8am T7.Thesameculture ofE.coliBB/1wasused for both mutants. Aportion of eachlysatewas
digestedwithtrypsin,asdescribed in thetext.Trypsinized and equaluntrypsinized portions of the lysateswere
subjectedtoelectrophoresis,as werecapsidIandcapsidIIisolated bysedimentation (16), as described in the
text. Thechannels have,respectively: (1) capsid II; (2) capsidI;(3-10) untrypsinized samples taken at 15, 15.5,
16.3, 17, 18, 20, 22.5,and27min after infection; (11-18) the same samples as in channels 3 through 10, but with
trypsinization; (19) trypsinized capsid II; (20) trypsinized capsidI.
D-
Ca
psid
I
,.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.505.103.405.103.575.2]4
x
C
E
-C,n
0
0
3
2
15.0 20.0 25
Time
(min)
FIG. 2. Kinetics of appearance of capsid I and AG particles after infection with 5am and 8am T7.
Amounts of 14C in capsid I were determined by
densitometry ofproffles ofuntrypsinized lysates in
Fig. 1; amounts of"Cin those AG particles converted
bytrypsin to particles comigrating during agarose gel
electrophoresis with capsid II were determined by
densitometry ofprofilesof trypsinizedlysates in Fig.
1.Anempirical calibration factor was used to convert
integrated optical densities to counts per minute.
Plot-ted as a function of time after infection are the
amountsof"Cin:(@)SamcapsidI; (0)8am capsidI;
(U)
5amAG particles;(0)8am AGparticles.a
comparatively
small amount of AG particlesafterassembly of capsidI.
DISCUSSION
The datapresented here indicate that thereare twopathways for theassembly of
capsid
I:(i)
acore-independent pathway in which AG
parti-clesare not an
assembly
intermediate and(ii)
acore-dependent
pathway
in whichassembly
ofcapsid
Iis slower than in thecore-independent
pathway
and in which AGparticles
are anas-sembly intermediate. Furthermore, in the ab-senceofafunctionalcore,AG
particles
accumu-late,
apparently
as abortive endproducts
ofassembly. If so, two
questions
concerning
as-sembly of
capsid
Icanbeasked: Howmuch has the structure of AGparticles
changed
during
cellular
lysis
andsubsequent
fractionation? What is thefunction ofthecorein thepathway
of slower
capsid
Iassembly?
Postlysischangesin AG
particles.
It has been shown that 9am 17lysates
and 10am T7lysates
contain noAGparticlesorcapsids of any
recog-nizable kind (19). Thissuggeststhat P9 andP10,
the two proteins of the capsid I envelope (14), must bind to each other before either of these twoproteinscan assemblein a capsid-like
com-plex. The final product of assembly, capsid I,
also has P9 and P10, apparentlybound to each other. Unless P9 andP10disassociate andthen
reassociateduring assembly, an unlikely occur-rence,both ofthese proteins must be present in
all intermediates in capsid I assembly.
There-fore,it islikelythatAGparticles (capsidII-like
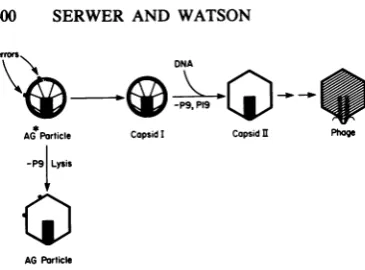
particles without P9), shown here to be interme-diates in capsid I assembly, are a breakdown product of a capsid I-like particle (AG* particle) thathas P9; these ideas are illustrated in Fig. 4. Possiblefunction of the T7 core during capsid I
assembly.
The presence of an apparentlyunal-tered pathwayofmore rapid capsid Iassembly in a nonpermissive host infected with the 8am mutantindicates that the core does not affect the rate ofcapsid I assembly in this pathway. The data inFig. 2 also suggest that in the absenceof
the core the rate of AG* particle assembly is not
1.0 E
E
>C
0
c)
0
j..
u
v
0.5F
15.0 20.0 25.0
Time
(min)
FIG. 3. Kinetics of appearance ofcapsidIandAG
particles after infection with wild-type T7. Kinetic
labeling ofwild-type T7-infectedE. coli and
fraction-ation oflysatesby sedimentationwereperformed as
described in Materials and Methods. The amountof
"CincapsidIwasdeterminedbyagarosegel
electro-phoresis of unfractionatedlysatesanddensitometryof
the resultant profiles. The amount of 14C in AG
particleswasdeterminedbysubjectingsamplesfrom
thecapsidIIregionsofgradients(usedto
prefraction-ate thelysates) to agarose gelelectrophoresis, with
and withoutdigestionwithtrypsin,andthen
subtract-ing the amountof
"4C
migratingascapsidII withouttrypsinization from the amount of "C migrating as
capsidII aftertrypsinization.The ratio of theamount
of14Ctothemaximalamountisplottedas afunction
of time. Symbols: (-)AGparticles,maximalamount
of
"IC
=51,101 cpm;(0) capsid I,maximalamountof14C =122,305 cpm. Thesamplesused in this
experi-ment arethe sameasthesamplesusedin reference 16.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.505.66.259.59.305.2] [image:5.505.271.464.338.495.2]errors
DNA
-P9.9
pi-g
AGParticle CapsidI Capsid Phoge
-P9{Lysis
AGParticle
FIG. 4. Pathway ofslowercapsidIassembly. The
pathway of slower capsid Iassembly, asdeduced in
thetext,is illustrated. The models ofcapsidsI andII
werededuced from observationsin references14and
15(seealso reference19).
decreased; the onlyprocessincapsidIassembly whose rate appears to be decreased by the absence of the core is the conversion of AG* particles (capsid I-like particles) to capsid I. Therefore, hypotheses (i), (ii), and (iii)
men-tionedatthebeginning allappeartobeincorrect for the assembly of T7 capsid I. (The data presented herearenotatestofhypothesis [iv].) Knowledge of the difference in structure
be-tweencapsid I andAG*particlesshouldprovide
cluestothe function of the T7coreduring capsid
Iassembly. Because theAG*particleshave not yetbeenisolated, deductionof this difference in
structure must be made from the properties of
their breakdown products, AG particles. The
only property of AG particles that appears to
provide aclue is the sensitivity to trypsin ofa smallpercentage ofP10 moleculesinAG parti-cles and the lack of thistrypsin-sensitive P10in capsid 11(19). This suggests that the
trypsin-digestible molecules of P10 are incorrectly
as-sembled in AG and presumably AG* particles. Therefore, it is proposed here that: (i) the func-tion of the T7coreduring capsid I assembly isto
correct errors in the assembly of the capsid I
envelope and (ii) AG* particlesareparticles that
haveuncorrectederrorsinthe assembly of their
envelopes (error correction hypothesis) (see Fig. 4). The error correction hypothesis explains
particles of capsid I assembled in the pathway of
more rapid assembly as particles which, by chance, didnotmakeerrorsduring assembly of theirenvelopes and, therefore, did notneed the
coreforassembly. This hypothesis explains the
conversion of AG* particles to capsid I in the
pathway of slower assembly as a process of
errorcorrection, possibly performedby
proteol-ysis of incorrectly assembled P10and (or) P9. Thedatathatnowexistsuggestsomeaspects
of howaprocessoferrorcorrection mightwork. Theinternalcoreof capsid I has beenshownto
consist of:(i)acylinder connectedatits baseto
the capsid I envelope and (ii) fibers connecting
the cylinderto thecapsid Ienvelope (14, 15;see Fig. 4). The fibers may be designed to fit the envelope in complementary fashion (the binding of the fibers to the envelope would be analogous tothe fitting together of two separately
assem-bled halves of ajigsaw puzzle). If the correct binding of envelope and fibers does not occur afterassemblyof the capsid Ienvelope,anerror correction process is activated.
Some core-deficient particlesofcapsidI pack-age DNA and the particles of bacteriophage
formed are noninfectious (12). Therefore, a
core-promotedcorrection of errors in the
assem-bly of the capsid I envelope may increase the
efficiency of productionof infectiveparticles by reducing the probability of the packaging of DNA by core-deficient particles. This would occurbecause a particle with a defective core
wouldnotbe ableto correct errorsin the assem-bly of its envelope, and the resultant particles
with uncorrectedassembly errorsin their
enve-lopeswould beincompetenttopackageDNA.
Other capsid I assembly intermediates. Inter-mediates in capsid I
assembly
less assembled than AG*particlesmustexist, but havenotbeen analyzed in the presentstudy.
In aprevious
study (16), it was shown that unassembled T7
capsid proteins sediment either more
slowly
than capsids (i.e., with sedimentation coeffi-cients less than 20) or more
rapidly;
the morerapidly sedimenting
capsid proteins
areboundtoparticles that haveproperties of membranes of
E. coli. Further studies of
capsid
Iassembly
intermediates less completely assembled than
AGparticles areinprogress.
ACKNOWLEDGMENTS
Fortechnical assistance we thank Elena S. T. Moreno. For secretarialassistance we thank Brenda Moylan.
Support was received from the National Institutes of Health (PublicHealthService grantAl16117).
LITERATURECITED
1. Casjens, S., and J. King. 1974. P22 morphogenesis. I: catalytic scaffolding proteinincapsidassembly. J. Supra-mol.Struct. 2:202-224.
2. Casjens, S., and J. King. 1975. Virus assembly. Annu.
Rev.Biochem. 44:555-611.
3. Caspar, D. L. D. 1980. Movement and self control in protein assemblies: quasiequivalence revisited. Biophys.
J.32:103-138.
4. Caspar, D. L. D., and A.Klug.1962.Physical principles in the construction of regular viruses. Cold Spring Harbor Symp. Quant. Biol. 27:1-24.
5. Crowther, R. A., and A. Klug. 1975. Structural analysis of macromolecularassemblies by imagereconstruction from electronmicrographs. Annu. Rev. Biochem. 44:161-182.
6. Earnshaw, W. C., and S. R.Casjens.1980. DNA packag-ing by the double-stranded DNA bacteriophages. Cell 21:319-331.
7. Elserling,F. A. 1979. Bacteriophagestructure, p. 543-580.
InH.Fraenkel-Conrat and R. R.Wagner (ed.), Compre-hensive virology, vol. 13. PlenumPublishingCorp., New
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.505.54.237.53.188.2]York.
8. Fuler, M. T., and J. King. 1980. Regulation of coat
proteinpolymerization by the scaffolding protein of bacte-riophage P22. Biophys. J. 32:381-401.
9. Harrison, S. C. 1980. Proteininterfaces and intersubunit bonding: thecaseoftomatobushystuntvirus. Biophys. J. 32:139-153.
10. Hoel,T., andI.Katara. 1977.Structure and assemblyof
bacteriophage lambda. Curr. Top. Microbiol. Immunol. 78:69-110.
11. Murlaldo, H.,andA.Becker.1978. Headmorphogenesis ofcomplexdouble-stranded deoxyribonucleic acid bac-teriophages. Microbiol. Rev. 42:529-576.
12. Roeder, G. S.,andP.D.Sadowsld. 1977.Bacteriophage
T7morphogenesis: phage-related particles inceils
infect-edwithwild-type andmutant17phage.Virology 76:263-285.
13. Serwer, P. 1974. Fastsedimenting bacteriophage T7 DNA from 17-infectedEscherichiacoli.Virology59:70-88. 14. Serwer, P. 1976. Internal proteins ofbacteriophage T7. J.
Mol.Biol. 107:271-291.
15. Server, P. 1979. Fibrousprojections from thecoreofa
bacteriophage 17 procapsid. J. Supramol. Struct.
11:321-326.
16. Serwer,P. 1980. A metrizamide-impermeable capsidin
the DNApackaging pathwayofbacteriophageT7. J.Mol. Biol.138:65-91.
17. Serwer,P.,andM. E.Pichler. 1978.Electrophoresisof
bacteriophageT7 andT7capsidsinagarosegels.J. Virol.
28:917-928.
18. Serwer, P.,andR.H.Watson. 1981.Capsid-DNA
com-plexesinthe DNApackaging pathwayofbacteriophage
T7:characterizationofcapsidsboundtomonomeric and concatemeric DNA.Virology108:164-176.
19. Serwer,P.,R. H.Watson,andS.J.Hayes.1982.
Detec-tion andcharacterizationofagarose-binding,capsid-like
particlesproducedduringassemblyofabacteriophageT7
procapsid.J. Virol.42:583-594.
20. Studier, F. W. 1969. The genetics and physiology of
bacteriophageT7.Virology39:562-574.
21. Studier,F.W.1972.BacteriophageT7. Science
176:367-376.
22. Wood, W. B., and J. King. 1979. Genetic control of
complex bacteriophage assembly, p. 581-633. In H. Fraenkel-Conratand R. R.Wagner (ed.),Comprehensive
virology,vol. 13. PlenumPublishing Corp.,NewYork.