0022-538X/81/120635-10$02.00/0
Dual Functions of Bacteriophage
T4D
Gene 28 Product:
Structural Component of the Viral Tail
Baseplate Central
Plug and
Cleavage Enzyme for Folyl
Polyglutamates
I.
Identification of T4D
Gene
28
Product in the Tail
Plug
LLOYD M.
KOZLOFFt*
AND JORGE ZORZOPULOStDepartmentof Microbiology and Immunology, Universityof Colorado Health SciencesCenter,
Denver, Colorado 80262
Received 18
July
1980/Accepted14July
1981The T4Dbacteriophagegene 28product isacomponentof thecentralplug of the tailbaseplate,asshown by thefollowingtwoindependent lines of evidence. (i)Ahighlysensitive method for radioactive labeling ofonly tailbaseplate plug
components was developed. These labeled plugcomponents were incorporated
byacomplementation procedure intonewphageparticles andwereanalyzed by radioautography after sodium dodecylsulfate-polyacrylamide gel electrophoresis. Threenewstructural proteinswerefound in additiontothe three known tailplug proteins(i.e., gP29, gP27, and gP5). One of the three newly identifiedcomponents
hadamolecular weight of24,000 to 25,000and appeared to be a product of T4D
gene28. (ii)Characterization ofmutantsofEscherichia coli bacteriophageT4D
whichproducedalteredgene 28products alsoindicated that thegene 28product
was aviral tailcomponent. T4D
28'
phageparticles producedatthepermissivetemperaturehadaltered heat labilities compared withparentT4Dparticles. We
isolated a single-step temperature revertant of T4D
28'
and found that itproduced phage particles which phenotypically resembled the original T4D
particles. Since the properties of the phage baseplatecomponents usually
deter-mine heatlability, these two changes inphysical stability after twosequential single mutations ingene 28supported the other evidence that thegene 28product
was aviralbaseplatecomponent.Also, compared withparent T4Dparticles,T4D
28'
andT4D 28amviralparticles adsorbedatdifferentrates tovarious typesofhost cells. In addition, T4D
28'
particles exhibited adifferent host range thanparentT4D particles. ThisT4D mutant formed plaques with anextremely low efficiency onall E. coliK-12 strains tested. We found that although T4D
28'
particles
adsorbedrapidly
andirreversiblytothe E. coliK-12strains, asjudgedby gene rescue
experiments,
these particles were not ableto inject their DNAinto theE. coli K-12 strains.
On
the otherhand, the T4D28'
revertant had aplatingefficiency onE. coli K-12 strains that was quite similar to the plating efficiency of the original parent,T4D. These properties ofphage particles
con-taininganalteredgene 28product supported the analyticalfinding that the gene 28product isastructuralcomponent of the central plug of the T4D tail baseplate.
They also indicated that thiscomponentplaysaroleinboth hostcellrecognition
and viralDNAinjection.
The first characterization ofthe role ofthe the long tail fibers) were observed. The gene
Escherichia coli Bbacteriophage T4Dgene 28 examinedmappedinan areaof severalgenes,all
productwas by Epstein etal. in 1963 (8). The of which appeared to be involved in tail mor-onephagemutantwhich these authors described phogenesis. Later, Edgar and Lielausis (7) and
was a
conditionally
lethal amber mutant; upon King et al. (13-15) showed clearly that the T4Dinfection of nonpermissive host bacteria viral gene 28 product was involved in forming the
DNAsynthesisoccurred andempty heads were complex tail baseplate upon which the rest of
formed,
but novisibletailstructures(exceptfor the tail structure was assembled. Using anin-tPresentaddress:DepartmentofMicrobiology,University genious indirect genetic method, Snustad con-of
California,
SanFrancisco, CA 94143. cluded in 1968 (29) that the gene 28 product635
on November 10, 2019 by guest
http://jvi.asm.org/
636
fuilfiled
some unknown "catalytic" function intail morphogenesis rather than functioning as
"stoichiometric" componentof thetail.
In 1965Kozloff and Lute (17) found that the
phage baseplate contained a folic acid as an
untMsual low-molecular-weight structural
com-ponent. Thefolatewasidentified lateras
dihy-dropteroylhexaglutamate (16, 18, 20-22, 26, 28).
Since the formation of thiscompound in infected cells might have involved catalytic reactions
necessary for phage baseplate morphogenesis,
Kozloff and Lute investigated the possibility
thatthe gene 28 product or some other phage
catalytic gene product was involved in folate metabolism (18). Baugh and Krumdieck (2) had developed methods to synthesize pteroyl hex-aglutamate, and this compound andsome
ana-logs of itwere tested for activity inphage tail morphogenesis. Complementation experiments
wereperformed byusingmixtures of folate-de-pleted extracts from host cells infected with different T4D tail mutants, such as T4D
7-extract complemented with T4D 10- extract.
Considerable increases in
phage
formationwereobserved when
synthetic pteroyl hexaglutamate
wasadded butnotwhen
pteroyl
penta- or hep-taglutamatewasadded (21).If the T4Dgene 28productwas anenzymewhich
functioned
solelyin theformation of the
pteroyl hexaglutamate,
then it seemed possible that the addition ofsynthetic pteroyl hexaglutamate
to extracts of nonpermissive bacteria infected with the T4Dgene 28 amber mutant
might
lead tophage
formation.
Subsequently,
itwasfound that smallamountsofphagewereformed whenthe
phage-specific folate was added to extracts of cells infected with the T4D gene 28 ambermutant.
Nootherextractof bacteria infected with
only
onephagemutant
responded
totheaddition of thisfolate compound, and it seemed clear thatthegene 28productwas involved infolate
me-tabolism; however, itwas notapparentwhy the
response to the added
pteroyl
hexaglutamatewas solow. Inview of the evidence(19)
concern-ing the dual role of thegene 28product,
only
aminimalresponsetothe addedfolatecompound
ingene 28- extracts wouldbeexpected.
Later,
experiments
(18, 28) showedthatwild-type T4D infections caused host cell folatesto
beconverted frompopulations of molecules
con-tainingmostlythreeglutamate residuesto
pop-ulations of molecules in which the number of
folates containing six
glutamate
residues wasincreased
substantially.
However, in T4D28--infectedcells,up to7% of thefolate compounds
were in forms containing 12 to 14 glutamate
residues. Suchlarge folyl polyglutamates could
notbe detected ineither uninfected cellsor
wild-type T4D-infected cells. Kozloff and Lute
sug-gested (18) that the catalytic function of the
gene 28 product involved the cleavage of the
high-molecular-weight folates to hexaglutamyl forms.
In1975, Kikuchi andKing (10-12) described
the sequential roles of the gene products
in-volved intail baseplate formation. They found that the gene 28 product acted early in the pathway which ledtotheformation of the
cen-tralplug of the tail plate. The role of thegene 28 product was not defined further, and since theseinvestigatorswere notabletofind thegene 28 product as a tail plug constituent, they thought that it acted catalytically in some
un-known fashion by altering the properties of the
gene 29product.
These observations caused us to continue
in-vestigationsontherole of thegene 28product. The results described in this paper and in the accompanyingpaper (19) ledtothe conclusion
thatthe gene 28 product has two functions in
forming phage
particles.
We present evidence that the gene 28 product is not only a folylpolyglutamate cleavage
enzyme but also is astructural
component of theplug
of the tailbaseplate.
In this paper we presentanalytical
evidence that thegene 28product
isatailplug
component. Inaddition, T4D gene 28 mutants
which producedaltered gene products showed
both in vivo and in vitro characteristics which indicated that the gene 28 product has these two
functions and also indicated that the gene 28
productis localized on the distalportionofthe
central plugofthebaseplate.Inthe
accompany-ingpaper(19),Kozloff and Lute describe folate
metabolism
in infected cells, present apartial
characterization of theenzymatic
natureofthe folatepolyglutamate cleavage reaction,
andcon-firn thelocalization of this
enzymatic
activity
onthe distal
portion
of thebaseplate.
MATERIALS AND METHODS
Bacteriophage and bacterial strains. We used
standard methods forphageassays(1).The bacterial
strains used includedE.coli B anda mutantof E. coli
B, strain OK305 (9). The E. coli K-12 strains used
included prototype strain AB259(furnished byA. L.
Taylor) and anumber of otherE. coli K-12 strains
from theColi Genetic StockCenter,includingCGSC
4617 containingan
Su2+
gene(glutamine)andCGSC2597containinganSu3" gene(tyrosine);wealso used
E. coli K-12 strain CR63, which contains an Sul+
(serine) gene. The E. coli bacteriophage strains used
havebeendescribedpreviously; these included
wild-typeT4D, the ambermutantof T4D in gene28(A452)
(18), other T4D ambermutantsin gene7(B16),gene
26(N131), gene27(N69), gene29(B7), and gene51
(529), and themultipleT4 ambermutantX6,which
containsamber mutations in gene23(B17), gene34
(B25 and A455), gene36(El),gene37(N52), and gene
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
38 (C290). In addition,we obtaineda new (31) T4D
temperature-sensitivemutantin gene 28(A61).Since
the gene 28phagemutants wereof critic lixnportance
inthiswork,wepickedasingle plaqueiromthe T4D
28amand T4D28'mutantstocks received from
Cali-fornia Institute ofTechnology collection. The T4D
28amstock has been characterizedpreviously(18, 20,
28).The T4D28'stockwasbackcrossed twiceto
wild-type T4D (10 T4D 28+/1 T4D 28k) to reduce the
possibilityofmultiplemutations. A total of88plaques
werepicked from the secondbackcross,andonly2of
the 88 phage stocks produced temperature-sensitive
plaques. Thesetwo stockswerecomparedwith each
otherand with theoriginal A61stock. Allproperties
testedwere identical for the original A61 stock and
thetwostocks obtainedby backcrossing. These
prop-ertiesincluded thefollowing: (i) complementationat
39°C,asdeterminedbyspottests onplates containing
E.coli B(negativefor T4D28ambutpositivefor T4D
26am,T4D 27am, T4D 29am, and all othermutants
tested); (ii) temperaturesensitivity (plating efficiency
was 1.0 at 250C,0.7 at 32°C, and <0.01 at 370C or
higher); (iii) all stocksplated with identical low
effi-cienciesonall of the E.coli K-12 strains listedabove,
butthey plated equally wellonE. coli B and strain
OK305,aderivative of E. coli B.
One of thetwostocks obtained afterbackcrossing
was then used in the experiments described below
with the assumption that it differed from our
wild-typestrain of T4Donlyin gene28.
Media. We used M9 mediumpreparedasdescribed
by Kikuchi and King (10-12) to prepare 14C-labeled
extracts. Tryptonebroth containing 8 gof tryptone
(DifcoLaboratories)perliter and5gofNaCl per liter
wasusedtoprepareunlabeledextractsand the other
phage stocks.
Preparation of
"4C-labeled
extracts. Anover-night culture of E. coli B in tryptone brothwasdiluted
30-fold in M9 medium and grownto adensity of4x
108cells per mlat370C.400
pi
ofasolutioncontaining40,uCiof
14C-labeled
amino acidswasaddedto25mlof theculture,and the incubationwascontinuedfor5
min; then the cellswere infected with a T4 amber
mutant at amultiplicityof4phageperbacterium. The
culturewassuperinfectedwith thesamemultiplicity
ofphage 8 minlater. At 45 min after the firstinfection,
thecellswerecollectedby centrifugationat40Cfor 10
minat1,500xg, and theresulting pelletwasfrozenat
-70°C. To prepareanextract, 1 mlofasolution of
M9 mediumlacking glucose (1),whichpreviouslyhad
been diluted 10-fold with water, was added to the
frozenpellet,andasmallamountof DNasewasalso
added. This mixturewasincubatedat300Cuntil the
pellet melted; then the mixturewasfrozenat-60°C
and then incubatedfor15minat300C.Then1ml of
M9 medium diluted 10-fold and containing 10 mM
EDTA andasmallamountof RNasewasadded. The
resulting mixture wasincubated for20min at 300C
and thencentrifugedat27,000xg for1htoeliminate
cell debris andlargephage structures, suchasheads
and sheaths. Thesupernatantsolutionwasusedasthe
sourceoflabeledphage protein.
Preparationof unlabeledextracts.E. coli Bwas
grown withvigorous aeration to adensity of4 x 108
cells per ml in2.5liters of tryptone broth mediumat
370C.
This culturewasinfected withanarrbermutantat amultiplicity of infection of4andsuperinfected8
min lateratthesamemultiplicity.
The cellswerecollectedby centrifugation at 1,500
xg for 10min at 2 h after the first infection, and the
pelletwasfrozenat-70°C. Then2ml of M9 medium
lackingglucose, diluted 10-fold, and containing small
amountsof DNase wasadded, and the mixture was
incubatedat300C until the pelletwas nolonger frozen.
This mixturewasfrozen andmeltedtwice, incubated
for15minat300C,and usedasthe crudeextractin
thecomplementation reactions.
Sucrose gradient centrifugation of extracts
containingbaseplates. A 2-mlamountofalabeled
extractwasplaced inadialysistube and surrounded
with dry Sephadex G-200 to reduce the volume to
about 400
pl.
Then theextract waslayeredontop ofa 5-ml 15 to 30% sucrose gradient in M9 medium lacking glucose diluted 10-fold. After centrifugation
for3hat45,000 rpmand5°C in an SW50.1 rotor of a
Spinco ultracentrifuge, 0.25-ml fractions were col-lected from the top of the gradient with a Buchler
autodensiflow apparatus andaperistaltic pump.
Complementation reaction to identify
base-plate tail plugs. A
25-pl
amount of each fractionobtained after sucrosegradient centrifugation was
in-cubated overnight at 300C with 50,A of unlabeled
extractfrom E. coli infected with T4D 29- (a tail
plug-negative extract). Themixtureswerethen diluted and
platedontoE.coli CR63tomeasurephage formation.
The fractions identifiedasthe fractions thatcontained
central plugs by both sedimentation properties and
complementation activity were pooled, mixed with a large excess of the unlabeled gene 29- extract, and
incubated overnight at300C.Before this incubation,
10
pl
ofasuspension containing unlabeled T4D phage(5 x 1012 particles per ml) was added in order to
saturateanypossible host membrane pieces
contain-ing T4 receptors, to avoid inactivation of the newly
formedphageparticles, and to act as a carrier in the
purification of the labeled new phage. After incubation
the phagewere purified by four standard cyclds of
differential centrifugation. Four cycles were enough to
obtaina highly purified phage preparation, as
indi-catedby electron microscopy and by the absence of
anycontaminantlabeled E. coli outer membrane
pro-teins, whicharesynthesizedevenafter infection. The
phagewerethen concentrated once more by
centrifu-gation and suspended in 100 to 200,l of a buffer
containing 2% sodium dodecyl sulfate, 10 mM
'tris-hydrochloride (pH 6.8), 10% glycerol, 0.7% 2-mercap-toethanol, and bromophenol blue dye.
Acrylamide gel electrophoresis and
autora-diography. The slab gels, buffers, and autoradiogra-phy techniques used have been described previously byKikuchi andKing(10-12).
Phage heat lability. Heat lability experiments
wereperformedasdescribed previously (24,25).Stnall
temperaturefluctuationsfromday to day made precise
comparisonsdifficult,and only samples heated
simul-taneouslywerecompared.
Chemical and other methods. All procedures
used have beendescribed previously (23).All
chemi-calswereanalytical grade. The radioactive amino acids
werepurchased fromNewEngland NuclearCorp.
on November 10, 2019 by guest
http://jvi.asm.org/
KOZLOFF AND ZORZOPULOS
RESULTS
Preparation and isolation of"C-labeled
baseplate tail plugs. We obtainedlabeled
cen-traltail plugs by usinganambermutantof T4D
ingene7whichwasnotabletoform the "wedge"
outer structuresof the baseplate under
nonper-missive conditions.
14C-amino
acidswereaddedat5min before host E. coli B cellswereinfected
to obtain early and late viral labeled proteins.
The labeled central plugswereseparated from
other large viral substructures bycentrifugation
(see above), and the plugswerepurifiedby
su-crosegradient centrifugation (10-12). The
frac-tion containing the central tail plugswas
iden-tified by a complementation reaction with an
extractfromanambermutantdefective in
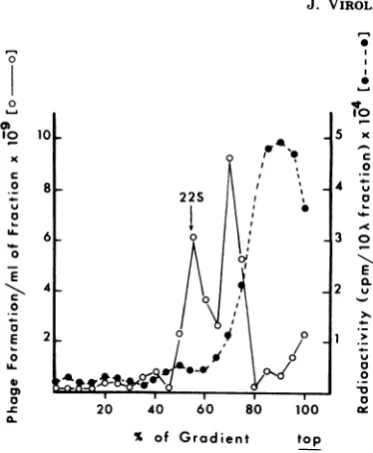
cen-tral plug formation (T4D 29-). Figure 1 shows
thatwefound two peaks ofcomplementing
ac-tivity. The peak containing the centralplug had
asedimentationconstantof22S,asreportedby
Kikuchiand King (10-12). However, we found
that this fraction also containedalarge number
of labeled proteins and could not be used to
identify central plug components. To identify
thespecific central plug proteins,weperformed
a complementation reaction between the 22S
gradient fraction andalargeexcessofunlabeled
gene29-extract.Since the 22Ssucrosegradient
fraction contained preformed labeled central
plugs, thenewviralparticles formedduring the
complementation reaction should havebeen
la-beled only in this substructure. Subsequent
steps involved the addition of unlabeled T4
phage as a carrier and purification of phage
particles by differential centrifugation. The
pu-rifiedphage particleswerethenheated at95°C
in the presence of sodium dodecyl sulfate, and
theproteinswereanalyzed by 1% sodium
dode-cyl sulfate-polyacrylamide gel electrophoresis.
Thesubsequent radioautography reflected only
theprotein compositionof thebaseplate central
plug. Figure 2 shows the densitometrictraceof
agel obtainedasdescribed above. We foundsix
labeled proteins; based on their sizes, three of
these were known components of the central
plug, namely, the product of gene 29 (80,000
daltons), the product ofgene27(49,000daltons),
and theproduct ofgene5 (44,000 daltons)
(10-12). The other threecomponents (designated A,
B, and C) havenotbeen reportedpreviously.
Toidentify these three unknown central plug
components,weusedasimplified version of the
method described above. Sucrosegradient
sep-aration of the central plugwas notperformed,
butan extremely large excessofthe unlabeled
complementing extract was added. We hoped
that this wouldnotallowsignificant
incorpora-tionofanylabeledprotein into the newly formed
0
0 0
10x c
0
E0 41
01
0E
c
.
20 40 60 80 100
% of Gradient top
.
rI0
0
0
x
0
E
._
o 0 -Q
u 0
1-u
tY
FIG. 1. Sedimentation of baseplate central tail
plugs in a 15 to 30% sucrose gradient. Radioactive
lysateswerepreparedfrom cells infected with aT4D
ambermutantdefective in gene7. Centrifugation was
performedfor3hat45,000 rpmwith anSW50.1 rotor
at5°C.A
25-gIl
amountof eachfraction was mixedwith50ulofaT4D 29- extract, and the mixture was
incubatedat37°C overnight. The viable phage were
assayed for plaque-forming activity after the
incu-bation. The centralplugwaslocalized by comparing
the complementation pattem with the
complementa-tion patternobtainedbyKikuchi and King(10-12).
viral
particles,
with themajor exception
ofthe viral componentsmissing
from the unlabeledextract. To test this
method,
weprepared
thesame labeled extract of bacteria infected with the T4ambermutantdefective in gene 7.
Any
possible intacttails dueto evensmallamountsof leakiness in gene 7 and other
large phage
structures
(such
asphage heads)
wereremovedby
centrifugation,
and the supernatant extract wascomplemented
with alarge
excess(more
than100-fold)ofanunlabeledextractfrom
bac-teriainfectedwith T4 am29.Carrier
phage
wasadded,andthenewviral
particles
werepurified
andanalyzed. Figure3ashows that thelabeled proteins
incorporated
into thenewviralparticles
werethe same
proteins
obtainedpreviously (Fig.
2).Thismodified
technique
permitted
theuseofvarious T4D ambermutantsdefectiveinone or
more of the central
plug
components in theprepar4tion of both labeled and unlabeled
ex-tracts. The unlabeled extract contained the
othercentral
plug
components,which couldin-corporate the labeled missing gene
product
toform atail
plug
and thenacomplete
newviralJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.500.263.450.62.289.2](a) (b)
I
fI
(c) (d) (e) (f)
gP29 gP27
7-+29- 4 gP5
A B
FIG. 2. Densitometric traceshowing the
distribu-tion of radioactive proteins incorporated into new
viral particles thatwereobtained by
complementa-tion of labeled tailplugs withanunlabeledT4D
29-extract. The fractions obtained from sucrose
gra-dients thatcontained the central plugsweremixed
with largeexcessesof unlabeled T4D29-extract,and
unlabeledphagewasaddedas acarrier. After
over-night incubation, the newparticles werepurified,
mixed, and boiled with buffer containing 2% sodium
dodecyl sulfate, 10mM Tris (pH 6.8), 10%glycerol,
0.7 Mmercaptoethanol, and bromophenolblue dye.
Theradioactive proteins wereanalyzed by 10%
so-diumdodecylsulfate-polyacrylamide gel
electropho-rests. The densitometrictraceofthe autoradiogram
obtainedfrom the dry gel is shown. The arrowsat
thetopindicate the positions of themolecular weight
standards,asfollows:arrow a,phosphorylaseb
(mo-lecularweight,94,000);arrowb, bovineserum
albu-min (67,000); arrow c, ovalbumin (43,000); arrowd,
carbonicanhydrase (30,000);arrow e, soybean
tryp-sininhibitor (20,100);arrowf,a-lactalbumin(14,400).
particle. Usingdifferent combinations ofmutant
extracts, we identified the unknown proteins.
Under these circumstances more than one
la-beled protein could be incorporated in
signif-icantamountsinto thevirusparticleifasecond
labeledprotein forned a complexor
substruc-turewith the labeledproteinwhichwasabsent
in the unlabeled extract. Figure 3b shows the
results obtained after complementation of
la-beled extracts from bacteria infected with the
T4D 51- amber mutant with an unlabeled
ex-tract of bacteria infected with the T4D 28-
am-bermutant.Peak C and thepeak corresponding
togene product27 werethe onlytwo proteins
incorporated insignificant amounts intophage
particles. The simplest explanation of these
re-sults was to assume that protein C was the
product of gene 28 and that gP27 formed a
strongcomplex with gP28,sothat this complex
was incorporated into the new viral particles
without undergoing dilution by the unlabeled
gP27 in the gene 28- extract. All ofthe other
viral proteins appeared to have been diluted successfully by the unlabeled products. (It should be pointed out that Kikuchi and
King
[12]
showed that in a gene 28- extract only asmallamountofgP27 formedatailplug precur-sor and thatmostof thegP27 remained at the
top of the
gradient.
This observation supportsthehypothesis that gP27canformastable
com-plex with gP28.) Figure 3c shows the results obtained by using a
complementation
mixture containingaT4D51-labeledextractandaT4D26-
unlabeled
extract. Inthiscase,only protein
AandgP5were
incorporated
into thenewviral particles. Byusing
similarreasoning,
protein
Ashould
correspond
totheproduct
ofgene26, anid(a) 7 * +29- gP29 gP27
(b)Sl- +28- gP
llC
9P5
A
(c)51*+265
gP27gPS
[image:5.500.52.239.58.223.2](d)28-*+51 S
;A
FIG. 3. Densitometric traces showing the
distri-butionsofradioactiveproteins incorporatedintonew
viralparticlesthat wereobtained by
complementa-tionbetweenlabeled andunlabeledextractsofE.coli
B infected with different T4 amber mutants. The
analyticalprocedures usedweretheprocedures
de-scribedin thelegendtoFig.2. (a) 7- labeled+
29-unlabeled. (b) 51- labeled+ 28- unlabeled. (c)
51-labeled+26-unlabeled.(d)28-labeled+51-
unla-beled.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.500.254.441.245.577.2]it appears that gP26 formed a complex with gP5.
Figure 3d shows the results forcomplementation
of a gene 28- labeled extract with a gene
51-unlabeled extract. In this case, the three labeled
proteins incorporatedweregP27, gP5, and
(ten-tatively) gP26, andnoother labeledproteinwas
incorporated. With regard to peak C, this
com-ponent waslabeledwhen the 14C-labeled extract
containedintact gP28 (Fig. 3b); however, in the
reciprocal experiment, when intact gP28 was not
presentin the labeledextract (Fig. 3d), peak C
was not present in the new phage particles.
Theseresultsstrongly supportedthehypothesis
thatpeakC wasthe gene 28product. Fromthe
mobilityofgP28duringsodiumdodecyl
sulfate-polyacrylamide gel electrophoresis, the
molecu-lar weightof this compound (peak C)appeared
tobe24,000to25,000.
Peak A had an apparent molecular weight of
41,000; based on the results shown in Fig. 3c,
this peak appeared to be gP26. However, this
identification needs confirmation. The
molecu-lar weight ofpeak B was29,000 to 30,000, and
peak B appeared to be the monomer of
thymi-dylate synthetase (3, 4, 26), which has been
reported to be abaseplate structural component
based on a variety of evidence (4, 14). The
assembly pathway for the central plug of the
baseplate involves not only more components, butalsoasomewhat differentsequence of
mor-phogenetic steps than originally proposed by
Kikuchi andKing (10-12). This scheme has been
discussed elsewhere inapaperdealingwith the
assembly of thymidylate synthetase into the
centralplugandtheuseof thefolatecompound
to link the central plug to the outer wedge
components (Proceedings ofthe Seventh
Bac-teriophageAssemblyMeeting, in press).
Heatlabilities ofT4D, T4D 28t8, and T4D
28tsrevertants. Kozloffetal. have shown that
the complex multicomponent tail baseplate of
T4D is the most labile portion of the phage
particleand that thestructure of thisbaseplate determines the heat lability ofa particle (23).
This isnotsurprisingsincethis structure
under-goesaradical conformational change during
in-fection. When temperature-sensitive
bacterio-phage gene products are incorporated into the
baseplates ofphageparticles, theheatlabilities
of these phage particles are changed and are
usually,butnotnecessarily,increased(23). The
temperature lability ofT4D
28'
particles wascompared with that of parent wild-type T4D
particles at 62°C (Fig. 4). Under these
condi-tions,T4D28' particleswereconsiderablymore
heatlabile thanT4Dparticles.
To be certain that itwas achange in gene28
protein that was the cause of the altered heat
stability of the T4D 28ts particles, we isolated
MINUTES
FIG. 4. Heat labilities of T4D, T4D2818,and aT4D
28'S revertant. Purifiedphage particles were treated
with DNase and diluted to a concentration of106
cells per ml in 0.1Mphosphate buffer(pH 7.0).After
equilibration at room temperature for 30 min, the
phage were diluted 1:10 into preheated buffer at 62°C
and assayed at differenttimes.
T4D
28"
revertants. Theserevertants wereiso-lated by plating independent plaque-derived
stocks at 44°C; 1 heat-resistant phagewas
ob-tained per 106phage plated.The phenotype of
one isolate (designated
a,)
was consistent with thehypothesisthatit wasa true revertantofapoint mutation; Fig.4shows that a, phage
par-ticleswere moreheatresistantthan their parent T4D
28"
particles andwereessentially
similartowild-type T4Dparticles.
HostrangesandadsorptionratesofT4D
mutantsin gene 28. Since the gene28
product
was located on the phage
baseplate,
weexam-ined the effects ofchangesinthe gene28product
onhost range,adsorptionrates,and DNA
injec-tion. We found that the change in the gene 28
product which made this protein heat labile
affected theabilityof thisphagemutant toform
plaquesonall strains of E. coli K-12 examined. For example, onE. coli K-12strain AB259 the
efficiency
ofplaqueformationat30°Cwas<0.3%of the efficiencyonE. coliB, and itwas1% on
E. coliK-12 strainCR63
(Su"+)
and 0.1% onE.coli K-12
(Su2+)
(Table1).OnE.coliK-12strainJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.500.262.454.72.346.2]CGSC
2597(Su3+),
theefficiency
ofplaque
for-mationwasless than 0.1%compared with E. coli B. These plating properties suggested that thegene 28productwasinvolved sinceafteraT4D
28tsspontaneous revertantlost itstemperature
sensitivity, it
regained
theability
toform
plaquesonE. coli K-12 strains(Table 1).
Pugsly and Schnaitman (28) have shown that thecell walls ofE. coliBand E. coli K-12are
different with respect to protein composition andfunction.
Recently,
work inourlaboratory
(32) hasfocusedonthe role ofcell wall
protein
b instabilizingphagereceptors.Since therewasno reason to suspectthat thechange inthe gene
28protein which made the phagetemperature
sensitive when it was grown on E. coli B in-hibited the biosyntheticprocessesin E. coli
K-12 hosts even at permissive temperatures, we
examined the initialstepsofthe interaction
be-tween T4D mutants and E. coli K-12 strains.
Cultures of the E. coli K-12
Su2+
strain and E. coli Bwere grown,washed in saline, and then heatedat650C
for1h. Heat-killed bacterial cell preparationsarequite stable and havebeen usedwidely to measure phage adsorption rates (1).
After treatment with DNase, the heated
cells
werewashed in saline and storedin thecoldat a concentration of 4 x
108
cells per ml. For adsorption ratedeterminations,
the heat-killed bacteriawereusedat afinal concentrationof2.5 x10"
cells perml, and the phage preparation wasaddedsothat therewere twophageparticlesperkilledcell. This bacterial suspension contain-ing the added phagewasincubatedat
270C,
andsampleswereremoved andassayed for live (i.e., unadsorbed) phage.
Theratesofadsorption of three phage stocks
differing only in thegene 28protein are shown
fora
typical
experiment in Fig.5.Allthreetypesofphageparticles readily attachedtotheE. coli K-12
Su2`
strain, but thereweresignificantdif-ferences in the initial adsorptionrates as
mea-sured by 50%
adsorption.
T4D28's
particles,
TABLE 1.
Platingproperties
of T4D,T4D28'8,andaT4D28'8revertant onvarious bacterialstrainsa
Plating efficiencyon:
Phage
~~~~~~~E.
coliPhage E.coi E.coliK- K-12 E.
col
K-B 12AB259 CR63 12(Su2+)(Sul+)
T4D 1.0 0.9 1.0 1.0
T4D28ts 1.0 <0.003 0.01 <0.001
T4D28'sre- 1.0 0.9 0.9 1.0
vertant a,
5The
plating efficiency for each virus strain wasdefinedas1.0onE. coliB grownonOK305 medium
(6)andincubatedat300C.
z
U
uJ
100.11 100.0
80.0 0 50.0 TAD
TAD 28S '-. TAD 28am
1 2 3I
10.0 a
TAD
5.0 TA0D 28S
TAD 28am
i.e1%I
MINUTES
FIG. 5. Adsorption of T4D, T4D
28",
and T4D28am to heat-killed E. coli K-12SU2+ cells. Phage
particles were added to heat-killed E. coli K-12SU2+
cellsataconcentration of 2.5 x 108 cells per ml in
broth. There weretwophage particles per cell, and
the suspension was incubated at 27°C. Samples
re-movedatdifferent times and assayed for live
unad-sorbedphage.
whichdidnotform
plaques
whenplated
onthis host, adsorbedmorerapidly than wild-typeT4Dparticles. Furthermore,
T4D 28am which wasprepared
by growth
onthe E.coliK-12 permis-sive strainCR63,
carriedadifferent
mutation ingene 28than T4D
28',
andproduced
adifferentgene 28
product
than T4D 28X, adsorbed evenmore
rapidly
thanT4D28's.
Sixseparate adsorp-tionexperimentswereperformed by using either this strain ofE. coli K-12orheat-killed
E. coliB. Inthedifferentexperimentsthetotalamount
ofphage adsorption varied from60 to97%, but therelative initial
adsorption
ratesfor thethree phage mutants werealways
similar andinde-pendent
of thetype ofhostcell. Intheexperi-mentshown in the insert inFig.5, theadsorption
rate (1) forT4D
28'
was 1.9 times faster thanthe rate forwild-type T4D,whereas the
adsorp-tion ratefor T4D 28amwas2.6timesfaster than
the rate forwild-type T4D. Forthe six
experi-ments, theadsorptionratefor T4D
28'
was 1.8±0.3times faster than therateforT4D,whereas
theadsorptionrate for T4D 28am was 2.2 + 0.4
timesfaster than the rateforwild-typeT4D.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.500.248.446.56.344.2] [image:7.500.47.240.541.634.2]DNA injection efficiency of T4D 28t'into
E. coli K-12. Since the reactions leading to
irreversible adsorption on E. coli K-12 strains
appeared to be functioning efficiently, we
ex-aminedthe DNAinjection efficiency of T4D28t"
particles by gene rescue experiments. Cultures of either E. coli K-12 strain AB259 (which
con-tains no tRNA suppressor and is infected by
T4D
28'
at anefficiency of only<0.3%) and E.coli B were grown to densities of 2 x 108 cells
per ml and were simultaneously multiply
in-fected with T4D 28ts and various T4D amber
mutants. Both typesof phage quickly adsorbed
to the E. coli K-12 strain and to the E. coli B
strain. After incubation at30°C for 1 h, sodium
azide was added to lyse the cultures, and the
lysates were assayedonE. coli B at permissive
andnonpermissive temperatures. Based on the
knowndistances between T4D gene 7, T4D gene
10, orthe multiple genes in T4D amX6 and gene
28 (8, 32), recombination would be extremely
likely in all three cases, leading to the production
of allpossiblerecombinants, including the
wild-type parent, T4D. In all cases, the amount of
wild-type T4D formed afteramixedinfection of
E.coliK-12strain AB259wasonlyafew percent
of theamountformed afteramixed infection of
E. coli B (Table 2). We concluded that the
efficiency of DNA injection of the T4D
28'
phageparticlesinto this E.coliK-12strain was
very low, so that verylittle rescue of the T4D
28ts genes, such as gene 7+ and gene 10+,
oc-curred.
TABLE 2. Rescueof variousT4D mutant amber genesby simultaneousinfection withT4D28ts in
varioustypesofhostcellsa
Titerofwild-type T4Dphage produced
in: Ratio of
Expt Phagecross titersb
E. coli E.coli B K-12
1 T4Dam7 x 1.0x106 1.2 x108 0.008 T4D28tS
2 T4D amlOx 4.0x 106 1.0x108 0.04 T4D28LS
3 T4DamX6 9.0X 106 1.6x108 0.05 xT4D28L
In these experiments E. coli K-12 strainAB259
andE. coli Batfinal concentrations of2 x 108cells perml in separate tubes of brothweresimultaneously
multiply infectedwithaT4D ambermutantand T4D
28'.Each type ofphage was presentat afinal concen-tration of5 x 108 phage per ml. The cultures were incubatedat30°Cfor1h,and thensodium azide was addedto a final concentration of 0.01%. The titer of
wild-typeT4Dproducedineachcrosswasdetermined
by measuring the plaque titer under nonpermissive
conditionsfor both parents(i.e.,onE.coli Bat44°C).
bRatio oftiter ofwild-typeT4Dproducedin E.coli K-12totiter ofwild-typeT4Dproducedin E. coli B.
DISCUSSION
Thedata in this paper and in the
accompany-ing paper (19) support the hypothesis that the
T4D gene28productactscatalyticallyand also
forms a
bU
uctural component of the tail plate.Thiscatalyticfunction is in agreement with the
proposals ofSnustad(29) and Kikuchi andKing
(10-12), whohavesuggestedacatalytic function
for the gene 28 product, and with previous
re-sults from this laboratory on the absence of a
folyl polyglutamate cleavage activity in T4D
28--infected cells.
The conclusion that the gene 28 product is a
structuralcomponentof theplug portionof the
baseplate and plays a role in phageadsorption
and DNAinjection dependsonanalyticalresults
and the properties of phages with chemically
different gene28products. The analytical
tech-niques described hereseem to be useful in the
identification of viral structural components
even if these componentsare synthesized early
duringinfectionoriftheyareminor components and therefore difficult to detect with
conven-tionaltechniques.Wefound threenewproteins
which arestructural componentsof the central
plug ofthe T4baseplate. Basedontheir
migra-tion distances during sodium dodecyl
sulfate-polyacrylamide gel electrophoresis,the apparent
molecular weights of theseproteins are 41,000,
30,000, and 24,000 to 25,000. The smallest
pro-tein (24,000 to 25,000daltons) wasidentifiedas
theproduct ofgene28by
reciprocal
complemen-tationexperiments, usingachase
technique.
Theothertwocomponentswere identifiedonly
ten-tativelyasproductsof gene26(the
41,000-dalton
component) and thymidylate synthetase (the 30,000-dalton component). Detailson theiden-tity of these two componentswill be
published
elsewhere. It isinterestingthat thesize of gene
28 product on the genetic map of Wood and
Revel(31) isrelatively small,in agreementwith our finding that it has a molecular weight of
24,000to25,000.
Thepropertiesof T4Dparticles havingaltered gene 28 products support the analytical
evi-dence. It may be that T4D 28'
particles
pro-duced even atpermissive temperatures do not
contain the normalamountsofstructural folate
in theirtailplatesand that thisdeficiencyleads
toanincreasedheatlabilitycomparedwiththe
parent, T4D. Measurements of the folate
con-tents ofphageparticles are notprecise
enough
(18, 22) to answer this question since values
varying from three to six folates per
particle
havebeen obtained. However, thepropertiesof
therevertantin gene 28argue againstthis
pos-sibility. The mutant obtained by selecting for
particles which plated at 44°C was more heat
stable than the parentT4D
28'
and hadaheaton November 10, 2019 by guest
http://jvi.asm.org/
sensitivity
similar to thatof
theoriginal
wildtype, T4D. The data in Table 1 and in the
accompanyingpaper(19) showthatrevertant
a,
simultaneously regainednotonly its heat
stabil-itybut alsoto asignificant extent its resistance to sulfadiazine and pyrimethamine; it also
re-gained its
ability
toinfect E.coli K-12 strains. Theinfluence of changes in the gene 28pro-tein on the adsorption rate indicated that the
gene 28 product itselfparticipates in host cell
adsorptionorinteracts directlyor indirectly with
the
baseplate
componentsthat affect adsorption. Furthermore, there is the direct demonstration that the failure of T4D28'
toinfectatypical E. coli K-12 strain, such as AB259, is not in the adsorptionstepbutin the DNAinjectionmech-anism of the phage particle. We believe that
componentsof the central tail plugareinvolved
intheinjectionprocess, asthisprocessinvolves release of viral DNA.
There remain the related questions of how
many molecules of the gene 28
product
arein the tail plug and why previous polyacrylamidegel
analyses of tail structures (6, 7, 10-15, 31)havenotrevealed apolyacrylamide gel
electro-phoresisprotein band correspondingtothisgene
product. Basedonthetracings shown in Fig. 2
and3 for the
plug
componentsandthe quanti-tationgiven by Crowtheretal.(5)
for other tail components,itappearsthat thereareonly
about sixmolecules ofgP28
percentralplug.
Astothe failuretodetect thisplug
componentpreviously,
itshould be noted thatnotonly
isgP28
presentin small amounts, but it would have been ob-scured in
previous
conventionalanalyses by pY,
another smallbaseplate wedge
componentre-ported by Kikuchi and King, which is quite similar in size
(10-12).
Itshould also be noted that acomplementation analysis
ofbaseplate
plug
precursors(10-12)
failedtodetect thegene28productas astructuralcomponent.This
neg-ative result now is understandable because of thedual role of thegene 28
product.
Inextractsof E.coliBinfected withT4D28am,not
only
is there very little of the requiredhexaglutamyl
form offolicacid, but significant concentrations ofdihydropteroyl glu12
14 arealso present (28). Thiscompound
shouldcompetewithanyof the phage folate compounds and blockbaseplate
morphogenesis. Based on these considerationsand others discussed elsewhere(16), we propose
that thephage dihydropteroylhexaglutamateis
necessaryto join or link the
baseplate
centraltailplugtothe sixouterwedgelikestructuresof
the
baseplate.
ACKNOWLEDGMENT
This research wassupported by Public Health Service researchgrant AI06336from the NationalInstitute ofAllergy andInfectious Diseases.
LITERATURE CITED
1. Adams, M. H. 1959. Bacteriophages. Interscience Pub-lishers, Inc., New York.
2. Baugh, C. M., and C. L. Krumdieck. 1971. Naturally occurring folates. Ann. N.Y. Acad. Sci. 186:7-28. 3. Capco, G. R., J. R. Krupp, and C. K. Mathews. 1973.
Bacteriophage-coded thymidylatesynthetase: charac-teristicsof the T4 and T5 enzymes. Arch. Biochem. Biophys. 158:726-735.
4. Capco,G.R., and C. K.Mathews. 1973. Bacteriophage-coded thymidylate synthetase. Evidence that the T4 enzyme is acapsidprotein. Arch. Biochem.Biophys. 158:736-743.
5. Browther,F.A.,E. V.Lenk,Y.Kikuchi,and J.King. 1977.Molecularreorganizationinthehexagonto star transition of thebaseplate of bacteriophage T4. J. Mol. Biol. 116:489-523.
6. Dickson, R. C., and S. L Barnes. 1970. Structural proteinsofbacteriophageT4.J. Mol. Biol. 53:461-473. 7. Edgar,R.S.,and I.Lielausis. 1968.Some steps in the assemblyofbacteriophageT4. J.Mol. Biol. 32:263-276. 8. Epstein, R.H.,A.Bolle,C. N.Steinberg,E. Kellen-gerger, E.Boyde laTour,R.S.Edgar,M.Susman, G. H.Denhardt,and A. Lielausis.1963.Physiological studies of conditional lethal mutants ofbacteriophage T4D.ColdSpringHarbor Symp. Quant. Biol. 28:375-394.
9. Hall, D.H., L. Tessman,and0.Karlstrom.1967. Link-age of T4 genescontrollingaseriesofsteps in pyrimi-dinebiosynthesis. Virology31:442-448.
10. Kikuchi, Y.,and J.King.1975.Genetic control of bac-teriophage T4 baseplate morphogenesis. I. Sequential assemblyof themajor precursor in vivo and in vitro. J. Mol.Biol. 99:645-672.
11.Kikuchi, Y.,and J.King.1975.Genetic control of bac-teriophage T4 baseplate morphogenesis. II. Mutants unable to form the central part of thebaseplate.J.Mol. Biol.99:.673-694.
12.Kikuchi, Y.,and J.King.1975.Geneticcontrol of bac-teriophageT4baseplate morphogenesis.III.Formation of the centralplug andoverall assembly pathway. J. Mol. Biol. 99:695-716.
13.King, J.1968.Assemblyof the tail ofbacteriophage T4. J. Mol.Biol. 32:231-262.
14.King, J.,and U. D.Laemmli.1973.Bacteriophage T4 tailassembly:structural proteins and their genetic iden-tification. J. Mol. Biol.75:315-337.'
15.King,J., and N.Mykolajewycx. 1973.Bacteriophage T4 tail assembly: proteins of the sheath, core and baseplate.J. Mol. Biol.75:339-358.
16.Kozloff, L.M. 1980.Folyl polyglutamate and
folate-re-quiringenzyme asbacteriophageT4Dbaseplate
struc-tural components.Biosystems 12:239-247.
17.Kozloff,LM.,and M. Lute. 1965.Folicacid, a structural component of T4bacteriophage.J. Mol. Biol. 12:780-792.
18.Kozloff, LM., and M. Lute. 1973.Bacteriophagetail components. IV. Pteroyl polyglutamate synthesis in T4D-infectedEscherichia coli B. J.Virol. 11:630-636. 19.Kozloff, L M., and M. Lute. 1981. Dual functions of
bacteriophage T4Dgene 28 product: structural compo-nent of the viraltailbaseplate central plug and cleavage enzyme for folyl polyglutamates.II.Folatemetabolism andpolyglutamatecleavage activity of uninfected and infected Escherichia coli cells andbacteriophage par-ticles. J. Virol.40:645-656.
20. Kozloff,L. M., M. Lute, and C.Baugh. 1973. Bacterio-phage tail components. V. Complementation ofT4D gene28-infectedbacterial extractswith pteroyl hexag-lutamate. J. Virol. 11:637-641.
21.Kozloff, L.M., M. Lute, andL. K.Crosby. 1970. Bac-teriophage tail components. III. Use of synthetic pteroyl hexaglutamate forT4Dtail plateassembly. J. Virol. 6: 754-759.
on November 10, 2019 by guest
http://jvi.asm.org/
22. Kozloff, L. M., M. Lute, and L. K. Crosby.1975. Bac-teriophage T4 baseplate components.I. Binding and location of the folic acid. J. Virol. 16:1391-1400. 23. Kozloff, L M., M. Lute, and L K. Crosby. 1977.
Bac-teriophage T4 virion baseplate thymidylate synthetase and dihydrofolate reductase. J. Virol. 23:637-644. 24. Kozloff, L M., M. Lute,andL.K.Crosby.1978.Folate
polyglutamate and folate enzymes, includinga folyl
polyglutamate cleavageenzyme,asbacteriophageT4D
baseplate structuralcomponents,p.309-314.InR. L. Kisliuk and G. M. Brown (ed.), Chemistry and biology ofpteridines. Developments in biocheniistry, vol. 4. Elsevier/North-Holland Publishing Co., New York. 25. Kozloff, L. M.,M.Lute,L K.Crosby,N.Rao,V.A.
Chapman,and S. S. DeLong. 1970. Bacteriophage tailcomponents. I.Pteroylpolyglutamates inT-even bacteriophages. J.Virol.5:726-739.
26. Krauss,S. W., B. D.Stollar,and M.Friedkin. 1973. Genetic andimmunologicalstudiesofbacteriophage T4 thymidylate synthetase. J.Virol.11:783-791. 27. Nakamura, K., and L M. Kozloff.1978. Folatepolyglu.
tamatesin T4DbacteriophageandT4D infected
Esch-erichia coli. Biochim. Biophys.Acta540:313-319. 28. Pugeley, A. P., and C. A. Schnaitman. 1978. Outer
membraneproteins of Escherichia coli. VII. Evidence thatbacteriophage-directed protein functionsas a pore.
J. Bacteriol. 133:1181-1189.
29. Snustad,D. P. 1968.Dominance interactionsin Esche-richia colicells mixedly infected with bacteriophage
T4D. Wiild-type and ambermutantsand their possible implications astothetypeof gene-product function: catalyticvs.stoichiometric.Virology 25:550-563.
30. Vanderslice, R. W., and C.D.Yegian. 1974. The iden-tification of late bacteriophage T4 proteinsonsodium
dodecyl sulfate polyacrylamide gels. Virology 60:265-275.
31. Wood, W. B., and H. R.Revel. 1976. Thegenome of bacteriophage T4. Bacteriol. Rev. 40:847-868. 32. Zorzopulos, J., L. M. Kozloff, V. A. Chapman, and S.
DeLong.1979.BacteriophageT4Dreceptorsandthe Escherichia coli cell wall structure.Role ofspherical particles and proteinb ofthecell wallinbacteriophage
infection. J. Bacteriol. 137:545-555.