0022-538X/81/050539-09$02.00/0 Vol.38, No.2
Molecular
Genetics of Herpes
Simplex
Virus
VI.
Characterization
of a
Temperature-Sensitive Mutant Defective in the
Expression
of
All
Early Viral Gene
Products
DAVID M.KNIPE,' WILLIAM
BATTERSON,'
CATHYNOSAL, BERNARDROIZMAN,2* AND ALEXANDERBUCHAN3Department ofMicrobiologyandMolecularGenetics,HarvardMedical School,Boston, Massachusetts
021151; The Marjorie B.KovlerViral OncologyLaboratories, University of Chicago, Chicago, Illinois
606372;andDepartmentofMicrobiology, University of
Birmingham,
Birmingham,England3
Received14April1980/Accepted19January 1981
The herpes simplex virus 1 (HFEM) mutant tsB7 failed to express any detect-able viral polypeptides and did not significantly inhibit hostcellprotein synthesis in infectedcellsmaintained at the nonpermissive temperature. The mutant could complement the growth of a coinfecting temperature-sensitive mutant virus differing in plaque phenotype and thus appeared capable of penetrating doubly infectedcells.The yield of tsB7 wasenhanced by the coinfecting virus but not to
theextentthat the coinfecting virus was enhanced. Coinfection studies suggested
that the tsB7 defect was complemented in trans, butpoorly, by the wild-type parent and other
viruses.
Marker rescue of tsB7 by transfection with herpes simplex virus 2XbaI DNA fragments mapped the mutation between 0.45 and 0.70mapunits.Analysis of the DNA structure of the ts+ intertypic recombinantsgenerated bythis rescue showed that the herpes simplex virus 2 DNA substitu-tions all contained the region between 0.46 and 0.52 map units, thus further
definingthe mappositionofthe mutation. Analyses of the polypeptides expressed by these intertypic recombinants defined the genome location of the genes
specifying polypeptides2, 6, 10, 32, 43, and44 and indicated that the mutation
mapsinorneargenescoding for virion structuralpolypeptides.Thisregion of the
genome is represented as stable transcripts and cytoplasmic mRNAonly after viral DNAreplication (P. C. Jones and B.
Roizrnan,
J. Virol.31:299-314, 1979), and thus this gene appearstobealatefunction.These resultsareconsistent with the ts mutation in tsB7 being in a gene coding for a virion component which functions before expression of the alpha genes early in infection. Themostlikely explanationis that themutantisblocked at astageofuncoatingandthe defect iscomplemented, althoughpoorly, byacoinfecting virus gene product.Inthis paper,wereportonthe characteristics of theherpessimplexvirus1 (HSV-1) tempera-ture-sensitivemutanttsB7. Thismutantfailsto
produce any detectable viralpolypeptides and hasnoappreciablelong-termeffectonthe
syn-thesis of hostproteinsatthenonpermissive
tem-perature.Pertinenttothis reportarethe follow-ing.
(i) HSV-1 andherpessimplexvirus2(HSV-2)
aregenetically closelyrelated(14). Both HSV-1
and HSV-2 specify approximately 50
polypep-tides (12, 23) which formatleast three groups whose synthesis is coordinately regulated and
sequentially ordered in a cascade fashion (12,
25). The apolypeptidesmadeimmediatelyafter
infection ofpermissive cells with HSV-1 attain
peakratesofsynthesisbetween2and4h
post-infection andrequirenoviralproteinsynthesis
for thesynthesisandprocessingof their mRNA. Thesepolypeptidesarerequiredfor the
synthe-sisof, polypeptides,and these in turn shut off the synthesis ofapolypeptides;the ,B
polypep-tidesareinvolved in thesynthesisof viral DNA and induce thesynthesisof structuraly
polypep-tides.
(ii) Inthe course of their
replication,
herpessimplex viruses shut off the synthesis of host proteins andDNA and reduce the
synthesis
ofhost RNA (22, 29, 30). The shutoffof the
syn-thesis of hostproteinsappearsto occur intwo
stages. Thefirstoccurs
immediately
afterinfec-tion and is mediated by a structural protein
inasmuchasitdoesnotrequirethe
transcription
of the viral genome (6). The second stage
re-quires the expression of viral protein and ap-pears to coincidewith the synthesis ofJ8
poly-539
on November 10, 2019 by guest
http://jvi.asm.org/
peptides (12, 21). In general, HSV-2 isolates inhibithost proteinsynthesismorerapidlyand more completely than HSV-1 during the first
stage ofinhibitionofhostmacromolecular
syn-thesis(2,22).AnalysisofHSV-1 x HSV-2
inter-typic recombinants showed that the HSV-2
gene(s)responsiblefor the accelerated shutoffof
host protein synthesis maps between 0.52 and
0.59mapunits(5, 20).
MATERIALS AND METHODS
Viruses. The isolation andpropertiesof HSV-2(G) and HSV-1 (HFEM) ts+ syn are described in refer-ences4and 9,respectively.HSV-1(HFEM)tsB7 syn+ was isolated by bromodeoxyuridine mutagenesis of HSV-1 (HFEM)-infectedcells.HFEMoriginally con-tainedamixture of syn and syn+viruses,andthetsB7 mutantapparentlyarosefromasyn+virus. HSV-1(F)
is a primaryisolate usedextensively intheChicago
laboratoryand has beenpassagedno morethanfour
times in cell cultureat340C (4, 11). Wehave found that this strainreplicatespoorlyat39°Candthat the
efficiency ofplatingat 390Crelative to 34°C is less
than l0'. It doesreplicate well at 370C, asits effi-ciency ofplatingat370Crelativeto34°Cis0.5.This phenotype is not unusual for fresh isolates ofHSV-1, since many other isolates testedshowed similar prop-erties (D. M. Knipe and B. Roizman, unpublished
observations). The temperature-sensitive marker in
HSV-1 (F) maps in the repeated sequences of the S component, 0.83 to 0.865 and 0.965 to 1.0map units (P.J.Godowski andD.M.Knipe,unpublisheddata). The virus expresseslargely a polypeptides at 39°C, andtherefore thewild-typeHSV-1(F)strainproduces atemperature-sensitiveaproduct similartothose of ts mutantsincomplementationgroup1-2(28).
HSV-1 (F) ts502 synl,2 is the designation of a recombinant produced by marker transfer of the syn loci from HSV-1 (1061) to HSV-1 (F) (27). At the permissive temperature it fuses bothHEp-2and Vero cells.
Virus stockswereprepared andtitrated in Vero cell cultures. The infected cells were maintained in
me-dium 199supplemented with 1% inactivated calf
se-rum.
Complementation tests.Complementation tests
wereperformed by a modification of theprocedure of Schaffer et al. (28). After addition of virus to the cultures, theflasksweresubmerged ina390Cwater bath or a 390C convection incubator. After the 1-h
absorption period, the virus inoculum wasremoved
and 5 ml ofmedium 199-1% inactivated calf serum was added. After 18 h ofincubation, the virus was harvested and titrated. Equivalent results were ob-tainedinexperiments in which thecellswere or were notwashedattheend of theabsorption period.
Markerrescuemapping.Mappingofthets lesion in tsB7wasperformedby thecotransfectionofmutant DNA andindividual fragments of HSV-2 (G) DNA generatedbycleavageofthe DNAwithXbaI restric-tionendonuclease (6, 15). The flasks were incubated for 3 to 4 days at 340C, and the progeny virus was harvested and titrated at 34 and390C. The rescued
ts+ cloneswereisolatedbyfourcyclesofplaque puri-fication underagaroseoverlayat390C(19).
Purification ofviral DNA andanalysis of
re-combinant genomes. Viral DNA for the marker
rescue studies and for analysis ofrecombinant
ge-nomes waspurified byNaldensitygradient
centrifu-gationofinfected cell extracts(31).HsuI,BgllI, XbaI,
andHpaIrestrictionendonucleaseswerepreparedas describedpreviously(19).BamHI andKpnIrestriction endonucleases were purchased from New England
Biolabs.
Analysisofpolypeptidesininfected and mock-infected cells. Cells were labeled with
L-U-'4C-la-beled amino acids(leucine, isoleucine,andvaline)at
timesspecified in the text andunder conditions
de-scribedbyMorseetal.(20).At the end of thelabeling interval,the cellswereharvested, disruptedwith
so-diumdodecylsulfate,andsubjectedtoelectrophoresis
inpolyacrylamideslabgelsasdescribed(20).
RESULTS
Replication
ofHSV-1 (HFEM) tsB7. Theefficiency
ofplaque
formationby
tsB7was 1 x105-
to3x105-fold
lowerat39thanat340C. Theyield
ofinfectious virus from cultures infectedata
multiplicity
of 5PFU/cell
andmaintained at390C
was 102_ to 103-fold lower than thatob-tained from
replicate
cultures maintained at340C.
The viral progenyobtained from thein-fected culturesincubatedat390Cwerestill
tem-peraturesensitiveanddidnotrepresent revert-ants.
Therefore,
athigh
multiplicities ofinfec-tion themutantshowedsomeleakiness.At
340C
the virus formed plaques which were much
smaller than those of its parent, HFEM, and whichincreasedin size
slowly.
Transient temperature fluctuations below
390C allowed the formation of very small
plaques and expression of viral polypeptides;
thus,
thephenotype of tsB7 wasvery sensitiveto temperature fluctuation. Forthis reason all
experimentsweredone inflaskssubmerged ina waterbath maintained at390Corin a
convec-tion-heated incubator with strict temperature
control.
Polypeptide expression by tsB7. To
ex-amine theproteins specified bythemutantvirus
at the permissive and nonpermissive
tempera-tures, weinfected cultures ofcells withHSV-1
(HFEM) andtsB7, incubatedthe culturesat 39 or330C,andlabeledthem with
[35S]methionine.
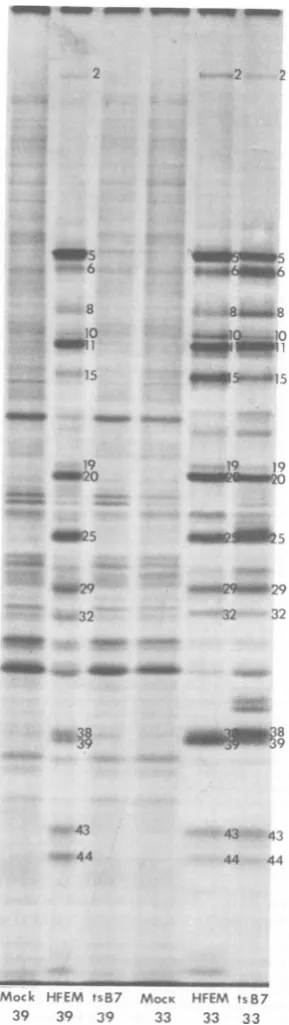
Thelabeledcell extracts were then subjected togel electrophoresis (Fig. 1). At 330C, tsB7
ex-pressedapatternof polypeptidessimilar to that
ofHFEMexceptthat certain early proteins
(in-fectedcellpolypeptides[ICP]6,-8, and -38) were
overrepresented and certain late proteins
(ICP10,-15, -19,and-39) wereunderrepresented
in the mutant profile, presumably because the
mutant infection had not progressed as far as J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
MOCK HFEM tsB7
39 39 39 33 33 33
FIG. 1. Autoradiographic images ofpolypeptides extracted from HEp-2 cells infected with HSV-1
(HFEM) tsB7orHSV-1 (HFEM) or mock infected
andelectrophoretically separatedinsodiumdodecyl
sulfate-polyacrylamidegels. Theinfectedcells were
incubatedattemperatures shown and were labeled
with "4C-amino acidsfrom 18 to 20 hpostinfection.
the wild-type infection. In
all
infectionsper-formedat
330C,
theinfections initiatedbytsB7progressed moreslowlythan thoseinitiated by
HFEM.This isconsistentwith theslowplaque
formation
bythemutant virus.At
390C,
HFEMexpressed apattemofpoly-peptides equivalenttothat at
330C,
exceptthat some late proteins (ICP10, -19, and -39) weremadeatlower levelsat
390C.
However,at390C
tsB7didnotexpress anydetectable viralpoly-peptides.The
pattern
ofproteins labeledincellsinfectedwith tsB7at
390C
was verysimilar
tothe
pattern
ofpolypeptides from mock-infected cellsat390C,
withatmost a veryslightdecrease inintensity ofeachcellular
polypeptide band.Thus,tsB7 did notsignificantly inhibithost
cell
protein synthesisat
390C.
Complementation of other mutants by
tsB7.
Schaffer et al. (28) previously reported thattsB7couldcomplementother temperature-sensitive mutants, but in those studies thevi-ruses were absorbed for 1 h at
370C.
Wehavefound that, under these conditions of infection,
tsB7willexpress manyof the lateviralproteins
and is therefore leaky (W. Batterson and B.
Roizman,
unpublished
data). Wethereforereex-amined the question of the ability of tsB7 to
perform
complementation under conditions where it expresses no proteins, i.e., continual maintenanceat390C.
Inthisseries ofexperiments, cultures ofVero
cells
wereindividually
or doubly infected withtsB7,
ts502,
orHFEM.CellscoinfectedwithtsB7showeda 100-fold increase in the yield ofsyn
plaques (ts502 marker) andatwofold increase in the syn+plaques (tsB7 marker) relative tothe singlyinfected cultures (Table 1). Theincrease in yield was not due to recombination, since
ts+recombinantsconstitutednomorethan1% of
the virus
yield
from thedoublyinfectedcells.Itshould be noted thatmostof the ts+
recombi-nants weresyn+, liketsB7. Ahigh frequency of
the ts+ recombinants wouldbe expected to be
syn+
in that the synl,2 mutations (0.70 to0.83 mapunits)areclosertots502
(0.83to0.865and0.965 to1.0 mapunits) thantothemaplocation
for tsB7, shown below to be 0.46 to 0.52 map
units.
Theobservation thatin the
complementation
test the increase in titer of ts502 was greater
than the increase in the tsB7 titer raised the
possibilitythattsB7 containedacis-acting
mu-tation thatcauses ittoreplicatepoorly. To
ex-amine thisquestion,wefirstcoinfectedcellswith
tsB7and ts502at
340C
andexamined theprog-Theinfectedcellpolypeptideswerenumbered
accord-ingtothenomenclaturepublishedelsewhere(12,20). VOL. 38,1981
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.500.68.213.73.587.2]TABLE 1. Testsfor complementation between tsB7 andts502a Complementationtest
390C 340C
Virus(PFU/cell) Yield(105) Virus (PFU/cell) Yield(106),340C
340C 390C
ts502 tsB7 ts502 tsB7 ts502 tsB7
ts502 tsB7 ts502 tsB7
5 0.64 10-5 0 1 49
10 1.33 10-5 0 5 150
5 11.7 1.4x 10-2 1 0 96
10 16.0 1.5x 10-2 5 0 153
5 1 278 20
5 5 207 32.0 0.19 1.85 5 5 172 62
1 5 56 117
aCells
wereinfected with eachmutantaloneorwithmixturesof bothatmultiplicities
shown and incubatedat39and340C.Theviralprogenyproducedin thesecellswastitratedattemperatures shown. The titers for
ts502and tsB7 are basedontheplaquemorphology typesof theprogeny.
eny at 340C. Again the syn or ts502 progeny
were favored, although by a lower ratio than from the cultures infected at 390C. Thus, the
poor growth of tsB7, even at the permissive temperature, couldexplain inpartthepooryield
oftsB7from thecomplementation experiments. We also examined the ability of HFEM ts+
syntocomplementtsB7syn+ at
390C.
Theyieldofsyn+ viruseswassignificantlygreaterin
cul-tures coinfected with HFEM ts+ syn than in
singlyinfectedcultures (Table 2).Thus, HFEM could complement the replication of tsB7 at
390C.
However, the yield ofsyn+ viruswasap-proximately 10% of the yield of the syn virus,
eventhough allcellswerecoinfected with both. Therefore, even the wild-type parent of tsB7
couldonlypartiallycomplementtsB7. This is in
contrast with the situation when HFEM and
ts502 coinfected cells. The progeny virus from cellsinfected with thesetwomutantsshowedan
efficiencyofplating
(39/340C)
ofapproximatelyone-half that of the progeny from a culture infected with HFEM alone (Table 2). Thus,
approximatelyone-half oftheprogeny from the
doublyinfected cellsweretemperaturesensitive.
Undertheseconditions ofinfection,theprogeny
contained approximately equal amountsof the
twoinfecting viruses.
The mutant tsB7 appearedto have a partial
cis-acting phenotype, although it could
appar-ently be complemented bycoinfecting viruses. As discussed further below, a partial
comple-mentation ofanuncoatingmutant would give a
partial cis-actingmutantphenotype.
MarkerrescueoftsB7 with HSV-2 DNA
fragments.The genomelocationof the ts lesion
in tsB7 was determined by the cotransfection
method ofmarker rescue (16). In these
experi-TABLE 2. MarkerrescueofHSV-1 (HFEM) tsB7by cotransfection with XbaI restriction endonuclease
fragments ofHSV-2(G)DNA
Ratio oftiters,
Transfection mixture
39/340C
(103)
tsB7DNA alone 0.01
+XbaI-A-C 1
+XbaI-D 25
+XbaI-G 1.8
+XbaI-H 0.3
+XbaI-I 0.1
+XbaI-J 0.5
ments,cellcultureswerecotransfected withtsB7
DNA andindividual XbaIrestriction
endonucle-asefragmentsofHSV-2 (G) DNA (Fig. 2).After
3to 4 daysofincubationat340C, the progeny viruswasharvested and assayedat34 and390C (Table 3). The most efficient rescue was
ob-tained withthe XbaI-D fragment, mapping from
0.45 to 0.71 map units. To further characterize
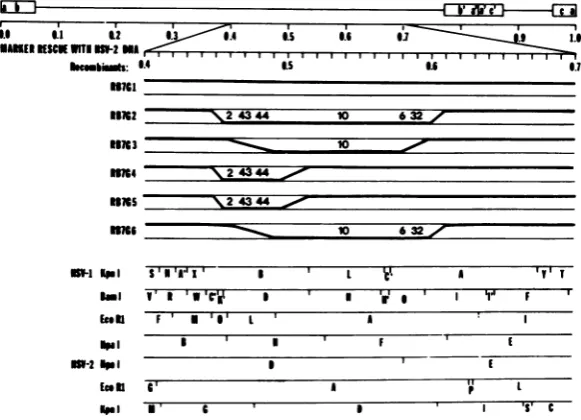
the site of therecombinational events,six
recom-binantsproduced bymarkerrescue with HSV-2
(G) DNA were analyzed with respect to the
domainsofthe HSV-2DNAsubstituted in the
recombinants.Theresults (Fig. 3 and 4) were as follows.
(i) Therecombinant RB7G1containedno
de-tectableHSV-2 sequences(Fig. 3) andexpressed
nopolypeptides which comigratedwith HSV-2
polypeptides (Fig. 4). Thus, RB7G1 was either
a revertant or a recombinant containing too
small areplacement of HSV-1 sequences tobe detected.
(ii) The recombinant RB7G2 retained the
HSV-1EcoRI-M-O and theHpaI-F-Ecleavage
sites but lost all intervening HSV-1 cleavage
sites. Therefore, the maximalleft boundary of
542
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.500.270.465.308.400.2]HSV-2(G)
4 L >o S >
ab
UL
e Us.aC D G H J I
45 26 12 I44..
45 26 12
I8.4
4.455 XbaI0 0.2 0.4 0.6 0.8 1.0
FIG. 2. Schematicdiagram ofthesequencearrangementandXbaI restrictionendonucleasemapsofDNA fragmentstested in markerrescuestudies. L and Srepresentthelongand shortcovalentlylinkedcomponents
ofHSV-DNA, and ULandUsrepresent theunique sequencesofeach component,respectively; ab and b'a'
represent the inverted repeatedsequences bounding the L component, whereas a'c' andca represent the inverted repeatedsequences boundingthe Scomponent. The lettersabove the line denote thenameofthe fragment; thenumber below the line is the molecularweight ofthe DNAfragmentin millions.
TABLE 3. Coinfection oftsB7 and HFEM ts+syna
34°Ctiter 390Ctiter
Infecting virus 39/34°C ratio
syn synf+ 8syn syn+
tsB7 <103 <1i3
HFEM syn 5x106 3.5x106 0.7
ts502 105 <103 10-2
tsB7+HFEM 6x 106 6 x105 3.5 x 10c 0.58
ts502+HFEM 6.3x10c 2.5 x 106 0.39
aCultureswereinfected with the indicated virusesat
390C.
After18hofincubation,the progenyvirus
was harvested and titrated at 34 and390C..6 .1 62
MAKER KSCe WITI 6-26M6
hm ut:
31763
66M6
6x6
T _A
fo-IIIII I I I I I I I I I I I I I I I I I I I I I I I;:io
1.4 .5 6 0.
-,24344 10 632 7
10 2 44
rzzz\zzzz zzz~zzzzzzz
_~~~~~1
63 ;ml61RPM S 1A'r
Sa1 v wlee
Ie I" i'1
I 'gi F
[image:5.500.101.399.62.151.2]Is.
FIG. 3. Summary oftherescueofthe HSV-1 (HFEM)tsB7 with HSV-2DNA.Diagramshows the location oftheHSV-2sequencesreplacingHSV-1sequencesin recombinantsproduced bymarkerrescueoftsB7 with HSV-2 DNA. The topandbottom lines next tothedesignation ofeach recombinantrepresentHSV-1 and HSV-2 DNA, respectively. The heavy line represents the sequences identified in each recombinant. The
numbers above theheavylineidentifythe HSV-2ICPs produced bythe recombinants andshown inFig.4.
Themapping ofthe DNAsequencesin the recombinantsis basedontherestriction endonucleasemapsfor HSV-1 and HSV-2 shown on the bottom. The HSV-1 KpnI and BamHIofHSV-1 (HFEM) tsB7 were
determinedfromthemapsforHSV-1(F) produced byLocker and Frenkel(18).
6.5 6.16 I I
I I l '
cll Eot
6662 MPa
feeII
A *Y* T
F' U'S'1 L 6
I I 6 F I I
I I
' A if L
38,1981
IJ
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.500.103.394.367.577.2]544 KNIPE AL.
FIG. 4. Autoradiographic images ofthe polypep-tidesextractedfromHEp-2cellsinfectedwith HSV-2 (G), HSV-1 (HFEM) tsB7, andrecombinants
pro-ducedby markerrescueoftsB7 with HSV-2(G)DNA andelectrophoreticallyseparatedin sodiumdodecyl sulfate-polyacrylamide gels. The cells were labeled with "C-amino acids from 10to 12 hpostinfection. Thepolypeptidesareidentified bynumberaccording tothe nomenclatureofHoness and Roizman(12)and Morseetal.(20).The HSV-2 ICPsareidentified bya
dash under the number. The insert on the bottom illustratesaportion ofagelshowingabetter sepa-rationofHSV-1 and HSV-2 ICP6.
the recombinational event was the HSV-1
EcoRI-M-O cleavage site,andthe minimal left
boundary was the HSV-1 HpaI-B-H cleavage site.Themaximal rightboundarywasthe
HSV-1HpaI-E-Fcleavage site, and theminimal right boundarywastheBamHI-O-Icleavagesite(Fig. 3). This recombinant expressed the ICP2, -6, -43, -44,-10,and -32 of HSV-2 (Fig.4).
(iii) The recombinant RB7G3 retained the HSV-1 EcoRI-O-L cleavage site and the BamHI-O-I cleavagesite but lost allintervening
HSV-1 cleavagesites. Themaximal left
bound-ary was the HSV-1 EcoRI-O-L cleavage site,
andthe minimal leftboundarywasthe
EcoRI-L-Acleavage site. The maximal right boundary
was the HSV-1 BamHI-O-I cleavage site, and
the minimal right boundary was the HSV-2
HpaI-D-Ecleavage site. This recombinant
spec-ified theHSV-2ICP10 (Fig. 4).
(iv) The recombinants RB7G4 and RB7G5
retained the HSV-1 EcoRI-M-O cleavage site
and the HSV-1 BamHI-D-H cleavage site but lost allinterveningHSV-1 cleavagesites.
There-fore, the maximal left boundary ofthe recombi-national events was the EcoRI-O-L cleavage site, and the minimal left boundary was the HSV-1 HpaI B-H cleavage site. The maximal right boundary was the BamHI cleavage site, and the minimalrightboundarywastheHSV-1 EcoRI-L-A cleavage site. These recombinants gained one new KpnI cleavage site consistent with theposition of the HSV-2KpnI-G-D
cleav-age site. These recombinants expressed the
HSV-2 ICP2,-43, and-44 (Fig. 4).
(v) The recombinant RB7G6 retained the HSV-1HpaI-B-HandHpaI-F-Ecleavage sites but lost all intervening HSV-1 cleavage sites.
The maximal left boundary was the HSV-1 HpaI-B-H cleavage site, and the miniimal left boundary was the HSV-1EcoRI-L-A cleavage site. The maximalrightboundarywasthe HpaI-F-Ecleavage site, and the minimal right
bound-ary was the HSV-1 BamHI-O-I cleavage site.
This recombinant expressed the HSV-2 ICP6,
-10,and-32 (Fig. 4).
In summary, the region of HSV-2 DNA
re-placementcommon toall recombinantswas the
region betweenthe HSV-1EcoRI-O-Lcleavage
site and theBamHI-D-H cleavage site,ormap
positions 0.46 to 0.52. This further defines the
map position of the ts mutation in tsB7. The
maximal boundaries for thetype-specific
deter-minants of thepolypeptideswereICP2, -43, and
-44(the EcoRI-M-O cleavagesiteto the EcoRI-L-A cleavage site); ICP10 (the EcoRI-L-A cleavage site totheBamHI-O-I cleavage site);
andICP6and -32(theHSV-2HpaI-D-E
cleav-age site totheHSV-1HpaI-F-E cleavage site).
The ts mutation in tsB7 was alsoclosely linked
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.500.66.254.83.544.2]1981
totheICP10 type-specific determinant(three of
sevenrecombinants) and the ICP2,-43, and -44
type-specific determinants (three of seven
re-combinants).
DISCUSSION
Wehavepresentedevidencethat the mutant
HSV-1 (HFEM) tsB7 expresses no detectable
viral polypeptides and doesnotsignificantly
in-hibit host cell protein synthesis in infectedcells maintained at the nonpermissive temperature.
Furthermore, the mutant cancomplement the
growth ofacoinfectingvirus. Thus, the mutant
viralgenome canpenetratedoublyinfected cells.
The block at the nonpermissive temperature
appears to occurafterentryof thevirus into the
host cell and before the expression ofthealpha
geneproducts.
Complementationstudieswith tsB7. The
mutanttsB7wasclearly showntobe capable of
complementing the replication ofacoinfecting temperature-sensitive mutant which maps
within the region of thegenomeencoding ICP4
(Godowski and Knipe, unpublished data) and which expresses largely alpha proteins at the
nonpermissive temperature (L. Pereira and B.
Roizman, unpublished data). Therefore, in order for tsB7tocomplement ts502, tsB7must express
ICP4. Because tsB7 does not express ICP4 in singly infected cells at any detectable level, it
appears that ts502 must supply the defective
genefunction in order for tsB7toexpressICP4. The mutated tsB7 gene function must be (at least in part)atrans-actinggeneproduct.
How-ever, we observed that the yield of tsB7 was
alwayslower than the
yield
ofthecoinfecting
virus when cellswere infectedat390C.
There-fore, it appears that the mutation in tsB7 is
partially cis-actinginnature; i.e.,the mutation
cannotbefully complemented byacoinfecting
virus. It should be noted that a defect in
un-coating which is partially complemented by a
coinfecting virus would give reduced yields of
themutantvirus defective inuncoatingbecause
theeffective number ofgenomesparticipatingin
replicationwould be less for theuncoating
mu-tant.Because of the time of theblock in
repli-cation intsB7-infectedcells,adefect in
uncoat-ing of the tsB7 genomeinside infected cells isa
possible explanation for the mutantdefect. An
alternative explanationthatwe cannotruleout
completely at the present time is that a very
small, undetectableamountofearlyproteinsis
made from the tsB7 genome whichcan
comple-mentthe growth ofts502. If theblockin
repli-cation of tsB7were inuncoating, this
explana-tion would also lead to higher yields of ts502
than tsB7.
ThetsB7mutation differsfrom thetwoother
MOLECULAR GENETICS OF HSV. VI. 545
classes ofviral mutantsthat show a complete
lack of viral gene expression under nonpernis-siveconditions, i.e., the ts3 mutantof polyoma
virus (3), the tsD mutants of simian virus 40
(24), and the group I host range mutants of
adenovirus (1, 10). The simian virus 40 and polyomamutantsmapinthe lateregion of the
genome (8, 17) and appear to be defective in
uncoating of the viral DNA (3, 24). Therefore, the DNAof thesemutants cannotbe transcribed
toyield early mRNA. Thesemutantsareunable
to complement other coinfectingmutants, and thusthisuncoatingfunction isacis-acting func-tion (3).
ThegroupIhostrange mutantsofadenovirus
map in the early regionI of the viral genome,
andonly the mRNA's for thatspecificregionare
expressed from themutant genome in
nonper-missive cells (1, 7). Thisappearsto be a "pre-early"genefunction whose synthesisis needed forexpression of theearlygenes.The mutation of tsB7 differs from these because it can be complemented by other viruses,and the existing
evidencestronglyarguesthatthe a gene prod-uctsrequirenoprior viral proteinsynthesis (12,
13).Several different inhibitors ofalpha protein
synthesis have been usedtoallowaccumulation
ofamRNA. There isnoevidenceto suggest that a"pre-early"protein gene product must be
syn-thesized beforeexpression of the agenes.
Map position of the tsmutation oftsB7.
The markerrescuemapping of thetsmutation of tsB7 by analysis of intertypic recombinants
determined themapposition of the lesionto be
within0.46 to 0.52 mapunits.This location has
been confirmedby fme-structure mappingusing
cloned HSV-1 (F) DNA fragments (Batterson
andRoizman, workinprogress).Therefore, the
tsmutationseemslikelytomap inthis region of the viral DNA. This region of the genome is represented in stable cytoplasmic mRNA only after viral DNA replication (13). It therefore
seemslikelythat the tsB7mutation is withina
geneexpressed afterDNAreplication.
Further-more,becausemostof thegenesexpressed after
DNAreplicationcodefor virion structural
poly-peptides, it alsoseemslikelythatthe tsB7lesion
iswithinastructuralpolypeptide gene.
We have used the intertypic recombinants generated by rescue of tsB7 with HSV-2DNA
to determine the viral polypeptides encoded
nearthetsB7lesion. None of the viral
polypep-tides which show type-specific mobilities on
polyacrylamide gelscorrelatedexactlywith the
tsB7lesion;thus,none canbe saidtocontain the
lesion.However,thetype-specific determinants
forICP2,-10, -43, and-44 weretransferredalong
with therescuingsequences in three of theseven
recombinants analyzed. Thus, the genes for
on November 10, 2019 by guest
http://jvi.asm.org/
546
these proteins are closely linked to the tsB7 locus.
As indicated in the beginning of this paper, theshutoff of hostprotein and DNA synthesis
occurs intwo stages.The mutanttsB7 did not
significantly inhibit host cell protein synthesis
as assayed by thepattern of host cell proteins displayedonpolyacrylamidegels.Inadditionto
theexperiments cited in the text, other
experi-mentsinvolving infection ofsparselyseededcell
cultures incubatedatthenonpermissive
temper-atureindicated that the infected cellswereable
to multiply foratleast3 days, but that
subse-quent shiftdowntothepermissivetemperature
resulted in the destruction of the cells(Batterson
andRoizman, work inprogress).Thus,the effect
onthe cells ofinfection with tsB7atthe
nonper-missive temperatureseems to be
minimal.
Pre-vious studies have mapped the accelerated shutoffby HSV-2 virionstotheregionbetween0.52and0.79mapunits(5,20) ornearthe tsB7 lesion. Therefore,it isconceivable that thetwo
functionsare encoded within thesamegene. It isstill unclear whether the lack of host shutoff
bytsB7isduetothe lack ofexpressionof viral
geneproductsin cellsinfectedatthe nonpermis-sive temperature.Thus,we cannotdetermineat
this time whether thetwofunctions arewithin the same gene, whether they are in different
genesandbotharelostby tsB7,orwhetherthe
twofunctionsareindifferentgenesandonlyone
islostby tsB7. Further studies are in progress
todiscriminate among thesepossibilities.
Nature ofthe tsB7 defective gene
prod-uct. The current evidence suggests that the
defectivegene product in tsB7is avirion
com-ponent that can be complemented by a gene
product contributed by anothercoinfecting vi-rus. Thisgeneproduct couldactuponthetsB7
capsidtouncoat theviral DNAor toallowit to
expresstheagenes.Alternatively,it could alter
the host RNA polymerase so that it can tran-scribe the viralgenomeefficiently. These func-tionswould be required only forinfections
ini-tiatedby virions because it is knownthat rigor-ouslydeproteinized HSV isinfectious, albeitat
lower efficiencies than virions. Therefore the
tsB7 gene function appears to be a late viral gene product that functions early in the viral
replicationcycle.
ACKNOWLEDGMENTS
These studies were aided by grants from the National
CancerInstitute,Public HealthService(CA98494, CA19264, and CA26345) and the American CancerSociety (MV-02).
D.M.K. was a special fellow of the Leukemia Society of
America,Inc.W.B. isapredoctoral trainee of the National Institute ofAllergyandInfectious Diseases (AI 07182).
LITERATURE CITED
1. Berk,A.J.,F.Lee,T.Harrison,J.Williams,andP.
A.Sharp. 1979.Pre-earlyadenovirus 5 geneproduct
regulatessynthesisofearlyviral messengerRNAs. Cell
17:935-944.
2. Courtney,R.J.,andK. L.Powell. 1975.Immunological
andbiochemical characterization ofpolypeptides in-ducedby herpessimplex virus types1 and2,p.63-73. In G.de-The,M. A.Epstein,and H. ZurHausen(ed.), Proceedingsof theSymposiumonHerpesvirusesand
Oncogenesis. International Agency for Research
Against Cancer, Lyon.
3. Eckhart, W.,and R.Dulbecco. 1974. Properties of the ts3 mutant of polyoma virus during lytic infection.
Virology60:359-369.
4.Ejercito, P.M.,E. D.Kieff, and B.Roizman. 1968.
Characterizationofherpes simplexvirus strains
differ-ingin their effectonsocialbehavior of infected cells. J. Gen. Virol.3:357-364.
5. Fenwick, M., L. S. Morse, and B. Roizman. 1979.
Anatomyofherpes simplexvirus DNA. XI.Apparent clusteringoffunctionseffecting rapidinhibition of host DNA andprotein synthesis.J.Virol. 29:825-827. 6.Fenwick,M.L,and M.J.Walker. 1978.Suppression
ofthesynthesisofcellular macromoleculesbyherpes simplexvirus. J. Gen. Virol.41:37-51.
7. Frost, E.,and J.Williams.1978.Mapping
temperature-sensitive and host range mutations ofadenovirustype
5bymarkerrescue.Virology 91:39-50.
8. Feunteun, J.,LSompayrac,M.Fluck,and T.
Ben-jamin.1976.Localization of gene functions inpolyoma virusDNA. Proc. Natl. Acad. Sci. U.S.A. 73:4169-4174. 9. Halliburton,L.W., R. E. Randall, R. A.Killington,
and D. H.Watson. 1977. Some properties of recom-binants between type 1 and 2 herpes simplex viruses. J. Gen. Virol.36:471-484.
10.Harrison, T.,F.Graham,and J.Williams. 1977. Host range mutantsof adenovirus type 5 defective forgrowth in HeLa cells.Virology77:319-329.
11.Hoggan, M. D., and B. Roizman. 1959. The isolation andproperties of a variant of herpes simplexproducing multinucleated giantcells in monolayer cultures in the presence ofantibody.Am. J.Hyg. 70:208-219. 12. Honess,R.W., and B. Roizman. 1974. Regulation of
herpesvirusmacromolecular synthesis. I.Cascade reg-ulation of thesynthesis of three groups of viralproteins.
J. Virol.14:8-19.
13. Jones, P. C., and B. Roizman. 1979. Regulation of
herpesvirus macromolecularsynthesis. IX. The tran-scription programconsists of 3phases during which both extent of transcription andaccumulation of RNA in thecytoplasm are regulated. J. Virol. 31:299-314. 14. Kieff,E.D.,B.Hoyer, S. L.Bachenheimer,andB.
Roizman. 1972. Geneticrelatedness of type 1 and type 2herpessimplex viruses. J. Virol. 9:738-745. 15. Knipe,D.M.,W. T.Ruyechan,R. W.Honess, and B.
Roizman. 1979. Molecular genetics of herpes simplex virus. IV. The terminal a sequence of the L and S components areobligatorily identical and constitute a part of a structural genemapping predominantlyin the S component. Proc. Natl. Acad. Sci. U.S.A. 76:4534-4538.
16. Knipe,D.M.,W. T.Ruyechan, B. Roizman, and I. W. Halliburton. 1978.Molecular genetics of herpes sim-plex virus.Demonstration of regions of obligatory iden-tity in diploid regions of the genome by sequence re-placement andinsertion. Proc. Natl. Acad. Sci. U.S.A. 75:3896-3900.
17. Lai, C. J., and D.Nathans. 1975. A map of temperature-sensitive mutants ofSV40.Virology 66:70-81. 18.Locker, H., and N. Frenkel. 1979.BamHI, KpnI and
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
Sallrestriction enzyme maps of the DNAs ofherpes simplexvirus strains Justin and F: occurrence of het-erogeneities in defined regions of the viral DNA. J. Virol. 32:429-441.
19. Morse,L.S.,T.G.Buchman,B.Roizman,andP.A.
Schaffer.1977.AnatomyofherpessimplexvirusDNA. IX.Apparent exclusion of some parental DNA arrange-mentsin the generation ofintertypic(HSV-1xHSV-2)
recombinant. J. Virol.24:231-248.
20. Morse,L.S.,LPereira,B.Roizman,and P. A. Schaf-fer. 1978. Anatomy ofherpes simplex virus (HSV) DNA.X.Mapping of viral genesbyanalysis of
polypep-tidesandfunctionsspecified by HSV-1xHSV-2 recom-binants.J.Virol. 26:389-410.
21. Nishioka, Y.,and S.Silverstein. 1978.Requirementof proteinsynthesisfor thedegradationof host mRNA in
Friend erythroleukemiacellsinfected withherpes sim-plex virus type1.J. Virol. 27:619-627.
22. Pereira,L,M.Wolff,M.Fenwick,andB.Roizman. 1977.Regulation of herpes-virus synthesis. V. Proper-ties of apolypeptidesspecified by HSV-1andHSV-2. Virology77:733-749.
23. Powell, K.L.,and R. J.Courtney.1975.Polypeptides
synthesized in herpes simplex virus type 2 infected HEp-2cells. Virology 66:217-228.
24.Robb,J.A.,and R.G.Martin.1972.Geneticanalysisof
simian virus 40. HI.Characterizationofa temperature-sensitive mutant blocked atanearlystageofproductive
infection inmonkeycelLs. J.Virol.9:956-968.
25. Roizman,B.,M.Kozak,R. W.Honess,andG.
Hay-ward. 1975.Regulationofherpesvirusmacromolecular synthesis:evidenceformultilevelregulation ofherpes simplex 1 RNA and protein synthesis. Cold Spring
HarborSymp. Quant.Biol.39:687-702.
26. Roizman, B.,and P.R.Roane,Jr. 1964.The
multipli-cation ofherpessimplexvirus.II. The relation between
protein synthesisand theduplicationofviral DNA in infectedHEp-2cells.Virology22:262-269.
27. Ruyechan,W.T.,L S.Morse,D. M.Knipe,andB.
Roizman.1979.Moleculargenetics of herpessimplex
virus. H.Mappingof themajorviral glycoproteins and ofthe genetic loci specifyingthe social behavior of infected cells. J. Virol.29:677-697.
28.Schaffer, P.A., V. C. Carter, and M. C.Timbury.
1978.Collaborative complementation study of temper-aturesensitive mutants ofherpessimplex virus types1 and 2. J.Virol.27:490-504.
29. Sydiskis,R.J., andB. Roizman. 1967. The disaggre-gationofhostpolyribosomesinproductive and abortive infection withherpessimplex virus. Virology 32:678-686.
30.Wagner, E. K., and B. Roizman. 1969. RNA synthesis incells infected withherpessimplex virus. I. The pat-terns ofRNAsynthesis in productively infected cells. J. Virol.4:36-46.
31.Walboomers,J. M.M.,and J. TerSchegget. 1976. A newmethod for theisolation of herpes simplexvirus type 2DNA.Virology79:256-258.
on November 10, 2019 by guest
http://jvi.asm.org/