Ontogeny of the v-erbA oncoprotein from the thyroid hormone receptor: an alteration in the DNA binding domain plays a role crucial for v-erbA function.
Full text
Figure

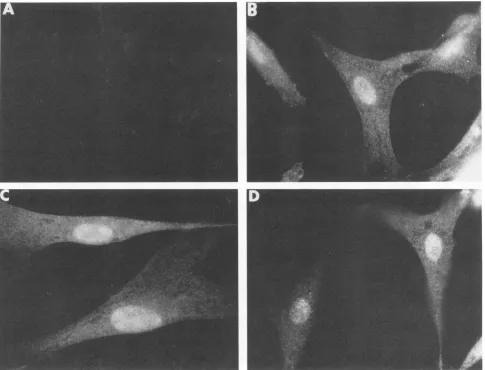
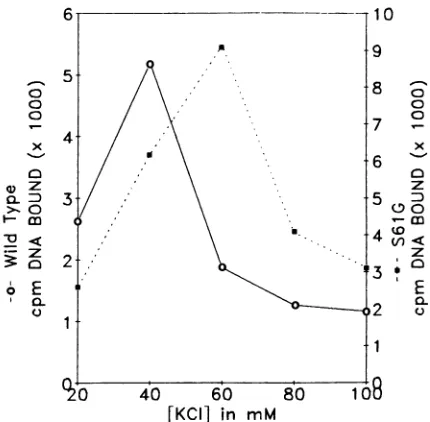

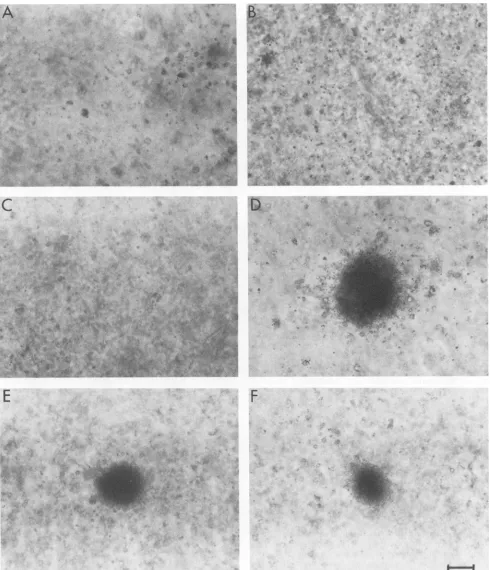
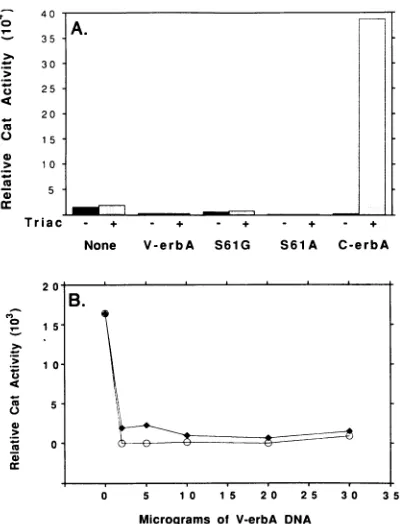
Related documents
To investigate directly whether individual differences in mea- sures of cortical motor excitability and/or physiological inhibition predicted individual variability in the
In general, this approach uses a process known as the Service Provision Model (SPM), (SRARA, 1985), to translate current and future service targets into financial terms in
We have used the polymerase chain reaction technique to clone the small multiply spliced mRNA species produced after infection of human cells by a molecular clone of
In this paper, we used a penalty approach to modify the node element based acoustic wave variational formulation and successfully applied to eliminate the spurious acoustic modes
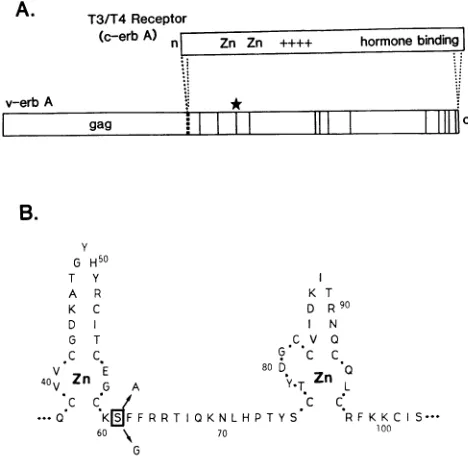
Symbols: , silent mutations; _ , amino acid substitutions; *, differences not present in all three IP-3-Ca clones; $, IP-3-Ca mutation not present in the virus 3-1 M gene sequence;
It is observed that the Build Index is highly effected by the change in the number of keywords and documents. This also means that with the change in the size of the index table
Total gray matter volume appears to be unaffected at this stage (28), although several studies have described subtle volumetric abnormalities (29-34).. In FEM patients fMRI
interpreted as two zones of local decrease of SP potential after the 2013 flood. Both
