0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
Identification of the Major Membrane and Core Proteins of
Vaccinia Virus by Two-Dimensional Electrophoresis
OLE N. JENSEN,1TONY HOUTHAEVE,1ANDREJ SHEVCHENKO,1SALLY CUDMORE,2TONY ASHFORD,2
MATTHIAS MANN,1GARETH GRIFFITHS,2ANDJACOMINE KRIJNSE LOCKER2*
Protein and Peptide Group1and Cell Biology Programme,2European Molecular Biology Laboratory, D-69117
Heidelberg, Germany
Received 17 May 1996/Accepted 23 July 1996
Vaccinia virus assembly has been well studied at the ultrastructural level, but little is known about the molecular events that occur during that process. Towards this goal, we have identified the major membrane and core proteins of the intracellular mature virus (IMV). Pure IMV preparations were subjected to Nonidet P-40 (NP-40) and dithiothreitol (DTT) treatment to separate the core proteins from the membrane proteins. These proteins were subsequently separated by two-dimensional (2D) gel electrophoresis, and the major polypeptide spots, as detected by silver staining and35
S labeling, were identified by either matrix-assisted laser desorption/ionization mass spectrometry, N-terminal amino acid sequencing, or immunoprecipitation with defined antibodies. Sixteen major spots that partitioned into the NP-40–DTT-soluble fraction were identified; 11 of these were previously described virally encoded proteins and 5 were cellular proteins, mostly of mito-chondrial origin. The core fraction revealed four major spots of previously described core proteins, two of which were also detected in the membrane fraction. Subsequently, the NP-40–DTT-soluble and -insoluble fractions from purified virus preparations, separated by 2D gels, were compared with postnuclear supernatants of infected cells that had been metabolically labeled at late times (6 to 8 h) postinfection. This relatively short labeling period as well as the apparent shutoff of host protein synthesis allowed the selective detection in such postnuclear supernatants of virus-encoded proteins. These postnuclear supernatants were subsequently treated with Triton X-114 or with sodium carbonate to distinguish the membrane proteins from the soluble proteins. We have identified the major late membrane and nonmembrane proteins of the IMV as they occur in the virus as well as in infected cells. This 2D gel map should provide an important reference for future molecular studies of vaccinia virus morphogenesis.
Vaccinia virus (VV), the best-characterized member of the
Poxviridae, has a 190-kb double-stranded DNA genome that
has been completely sequenced (18, 31). It has the capacity to encode over 200 proteins, of which about 100 are in the virion (14). These proteins are traditionally divided into three tem-poral classes. The early proteins are those that are synthesized prior to replication of the DNA, while the intermediate and late proteins are made sequentially after the onset of DNA replication. The late proteins, which are synthesized from 5 to 6 h postinfection onwards, are generally assumed to encode all of the viral structural proteins (41).
The first morphological evidence of VV infection is the appearance, at approximately 2 to 3 h after infection, of struc-tures enriched in DNA, the so-called viral factories (5). These structures are the sites of DNA replication and transcription. The first morphological evidence for membrane assembly, co-inciding with the onset of late protein synthesis, is the forma-tion of rigid, curved membrane structures, the so-called cres-cents. Although they were once thought to be the only example of de novo membrane synthesis, it now seems clear that these viral membranes are derived from the intermediate compart-ment between the endoplasmic reticulum and the Golgi com-plex (34, 61, 73). Although these crescent-shaped membranes usually give the appearance of a single bilayer, we have pro-posed that they are in fact compro-posed of two, tightly appro-posed, cisternal membranes (61). The crescents mature into the
spherical immature virus (IV), which forms the first infectious form of VV, the intracellular mature virus (IMV). This latter process coincides with the cleavage of at least three of the major core proteins (4a, 4b, and p25, the gene products of A10L, A3L, and L4R, respectively) (65, 66). Subsequently, a variable fraction of the IMVs becomes engulfed by a cisterna derived from the trans-Golgi network, forming the precursor of the second infectious form of the virus, the extracellular enveloped virus (EEV) (23, 48, 57).
VV morphogenesis has been well studied by electron mi-croscopy (9, 61), but little is known about the molecular events occurring during the assembly. With this in mind, we decided to make a comprehensive two-dimensional (2D) gel analysis, the goal of which was the identification of all of the major membrane and core proteins of the IMV. In previous studies the major proteins of VV, as well as fractions of the virus prepared by extraction of specific proteins, have been analyzed by 2D gel electrophoresis, but these previous analyses were not combined with an identification of the proteins (6, 14, 45). A recent attempt to identify the major IMV proteins relied on separation by 1D gels followed by Edman degradation of some of the major bands (63). However, because of the relatively large number of proteins in VV, upon separation by 1D gels one band may in fact correspond to several different proteins. Thus, no estimation of the abundances of some of the individ-ual proteins could be made. Also, the sequence of several bands was not revealed, simply because the N terminus was blocked, a serious limitation of the Edman degradation ap-proach when it is used for sequencing of intact proteins.
Peptide mixture analysis by mass spectrometry (MS) is a fast and sensitive method for protein identification by database * Corresponding author. Mailing address: European Molecular
Biology Laboratory, Meyerhofstr. 1, D-69117 Heidelberg, Germany. Phone: 49 6221 387 508. Fax: 49 6221 387 306. Electronic mail address: krijnse@embl-heidelberg.de.
7485
on November 9, 2019 by guest
http://jvi.asm.org/
searches (see reference 47 for a review). Peptide mass maps obtained by matrix-assisted laser desorption/ionization (MALDI) (22) time-of-flight MS and peptide sequence information ob-tained by electrospray ionization (15) tandem MS (2, 25) are specific probes for such protein sequence database searches (12, 21, 27, 37, 39, 46, 75). Both techniques are applicable to unseparated peptide mixtures at the low- to subpicomole level. Advantages of MALDI MS are fast and simple sample prep-aration, tolerance to sample impurities, and high sensitivity (see reference 38 for a recent review).
In this study we have separated the membrane and core proteins of pure IMV preparations by 2D gel electrophoresis. The major proteins of the IMV were identified by using either MALDI MS (i.e., peptide mass mapping), conventional Ed-man degradation, or defined antibodies. In addition, we have compared the major IMV proteins with the proteins made late in infected cells and have used Triton X-114 (TX-114) and sodium carbonate extraction to define the membrane proteins.
MATERIALS AND METHODS
Cells, virus, and antibodies.HeLa cells were grown as described previously (61). The deletion mutant vRB12, a mutant of strain Western Reserve lacking the 37-kDa EEV membrane-associated protein (3), was grown and purified as described previously (53). Briefly, four plates (24 by 24 cm) (Nunc) of 70% confluent HeLa cells were infected at a multiplicity of infection of 3 with vRB12 for 60 min at 378C, the inoculum was removed, and the infection was continued for 48 h. The cells were scraped from the dish and gently pelleted for 10 min at 1,500 rpm, and the pelleted cells were resuspended in 10 mM Tris-Cl, pH 9.0. The cells were broken by 10 to 12 strokes of a tightly fitting Dounce homogenizer (pestle B), and the nuclei were pelleted at 2,500 rpm in a Minifuge GL (Heraeus, Christ) for 5 min. Care was taken to remove all nuclei, and sometimes the supernatant was subjected to another round of centrifugation. The supernatant was layered on top of a 36% (wt/vol) sucrose cushion in 10 mM Tris-Cl (pH 9.0) and spun for 30 min at 24 krpm in an SW40 rotor. The pellet was resuspended in 2 ml of 10 mM Tris-Cl (pH 9.0), sonicated for 1 min in a water bath sonicator, layered on top of a continuous 25 to 40% (wt/vol) sucrose gradient in 10 mM Tris-Cl (pH 9.0), and spun for 30 min at 15 krpm in an SW27 rotor. At about one-third of the way from the bottom, a light-scattering virion-containing band could be detected, and this was collected. The virus was subsequently concen-trated by pelleting for 30 min at 24 krpm in an SW40 rotor, and the resulting pellet was resuspended in 200 to 400ml of 10 mM Tris-Cl, pH 9.0. The purity of each virus preparation was evaluated by negative-staining electron microscopy (EM) (data not shown). In one experiment some of the postnuclear supernatant (PNS) before and after the first pelleting step was retained. Equal amounts of total protein (as determined by a bicinchoninic acid protein assay [see below]) of this material and of the final purified virus were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the purification steps were monitored by silver staining of the total protein pattern.
The following antibodies were used: anti-p32 (D8L) (44), a kind gift of E. G. Niles; anti-M25 (L1R) (16), a kind gift of D. E. Hruby; anti-A14L (A14L) (51a), a kind gift of J. F. Rodriguez; and anti-p65 (D13L, against the C terminus of p65), a kind gift of R. Doms (62). The peptide serum recognizing p11 (F18R) was prepared as described previously (8) by using a peptide matching amino acids 80 to 100 (extreme C terminus).
Quantitation of virus preparations.Purified and concentrated vRB12 was quantitated as described by Joklik (32). Virus preparations were diluted in 10 mM Tris-Cl (pH 9.0), and the optical density at 260 nm (OD260) was measured.
On one occasion the protein concentrations of several virus preparations were measured by using a bicinchoninic acid protein assay (Pierce) according to the instructions of the manufacturer, and these results were compared with the OD260values. Since the measured protein concentrations corresponded well with
the OD measurements (that is, 64mg of protein per ml equals one OD260unit
[32]), for the subsequent preparations only the OD was measured. Concentrated virus preparations contained on average 1.5 mg of protein per ml (corresponding to 300 to 500mg of total protein for four plates [24 by 24 cm]) or 23.5 OD260units
per ml.
Preparation of core and membrane fractions and TX-114 and carbonate extraction of PNSs of infected cells.For an average experiment 100 to 150ml of purified vRB12 was sonicated for 1 min in a water bath sonicator and incubated for 30 min at 378C with 20 mM freshly prepared dithiothreitol (DTT) in 10 mM Tris-Cl (pH 9.0) and subsequently for another 30 min at the same temperature with an equal volume of 2% Nonidet P-40 (NP-40) and 40 mM DTT. The sample was sonicated for 30 s, layered on top of a 36% sucrose cushion in 10 mM Tris (pH 9.0), and spun for 30 min at 100,0003g. The supernatant (membrane protein-containing fraction) was concentrated by acetone precipitation at2208C and dissolved in first-dimension lysis buffer (9.8 M urea, 2% ampholines [Phar-macia] [pH 7 to 9], 4% NP-40, and 100 mM DTT), while the pellet (core fraction)
was directly dissolved in this buffer. The samples were incubated for 30 min at 378C prior to their separation by either isoelectric focusing (IEF) or nonequi-librium pH gradient gel electrophoresis (NEPHGE) in the first dimension. The TX-114 and sodium carbonate extraction of PNSs of infected cells was as de-scribed by Cudmore et al. (8).
IEF, NEPHGE, and silver staining.For the separation of the proteins with a pI between 3 and 7, IEF was performed with the Bio-Rad mini-2D-gel system. One-millimeter-diameter tubes containing 4% acrylamide-bisacrylamide, 2% NP-40, 5.2% ampholines of pH 5 to 7 (2.6% from Pharmacia, Uppsala, Sweden, and 2.6% from Serva, Heidelberg, Germany), and 1.6% ampholines of pH 3.5 to 10 (Pharmacia) were used, and the samples were run for 2.5 h at 1,000 V. For the basic proteins, NEPHGE was used. The tubes were prepared with the same amounts of acrylamide-bisacrylamide and NP-40 as for the IEF and 4% am-pholytes of pH 8 to 10.5, 2.5% amam-pholytes of pH 5 to 8, and 1.6% amam-pholytes of pH 3 to 10 (all from Pharmacia). The core fractions were run for 3 h at 450 V, while the membrane fractions were run at the same voltage for 4 h. After electrofocusing, the tubes were incubated for 10 min at room temperature in Laemmli sample buffer containing freshly prepared 100 mM DTT. The second-dimension separation was done on SDS–15% PAGE. The proteins were subse-quently detected by silver staining as described by Shevchenko et al. (60) or, in the case of35S-labeled samples, by autoradiography. When IEF standards were
run, Carbamylyte (Pharmacia) was used; for the IEF the pI was measured with both creatinine phosphokinase (pI range, 4.9 to 7.1) and carbonic anhydrase (pI range, 4.8 to 6.7) as standards. The pIs of the viral proteins were determined as described by the manufacturer by comparing their migrations with that of each standard individually and averaging both values (which never differed more than 0.2 pI unit). For the NEPHGE only glyceraldehyde-3-phosphate dehydrogenase (pI range, 4.7 to 8.3) was used as a standard, which allowed the determination of the pIs of most of the spots, except for p11 (see Table 1).
Identification of the proteins by MS. (i) In-gel enzymatic digestion of protein. Tryptic digestion of proteins was performed in the gel matrix (in-gel digestion) (11, 55) by a procedure optimized for subsequent MS analysis (60, 71). Briefly, stained protein bands or spots were excised from gels, cut into pieces (1 by 1 mm), and transferred to a 1.5-ml microcentrifuge tube. The gel pieces were washed in 50 mM ammonium bicarbonate–50% acetonitrile, and subsequently the protein was reduced (10 mM DTT in 50 mM ammonium bicarbonate, 568C, 45 min) and S alkylated (25 mM iodoacetamide in 50 mM ammonium bicarbon-ate, 20 min, room temperature) in the gel matrix. Excess reagent was removed, and the gel pieces were washed as described above and subsequently dried in a vacuum centrifuge. For enzymatic digestion, 1mg of bovine trypsin (Boehringer-Mannheim; sequencing grade) or porcine trypsin (Promega; modified, sequenc-ing grade) was dissolved in 70ml of 50 mM ammonium bicarbonate–2 mM CaCl2–10% acetonitrile and gradually added to the dry gel pieces at 48C. After
swelling of the gel pieces, a small volume of 25 mM ammonium bicarbonate was added to completely cover the gel pieces. Samples were incubated overnight at 378C. Excess liquid was then transferred to a microcentrifuge tube and pooled with subsequent extracts produced by incubations of gel pieces with 30 to 100ml of 5% formic acid–50% acetonitrile for 20 min. The peptide solution was dried in a vacuum centrifuge and redissolved in 20 to 50ml of 5% formic acid–10% acetonitrile. Aliquots (0.5ml), corresponding to 1 to 5% of the sample, were analyzed by MALDI MS.
(ii) MALDI MS.Fast-evaporation matrix films were produced as described previously (67, 69) by using a saturated solution of 4-hydroxy-a-cyanocinnamic acid (Sigma) in acetone. The peptide solution was deposited on the matrix film and allowed to dry at the ambient temperature. Sample deposits were rinsed once or twice with pure water and subsequently inserted in the MALDI mass spectrometer.
Mass analysis was performed on a Bruker REFLEX MALDI time-of-flight mass spectrometer (Bruker-Franzen Analytik, Bremen, Germany) equipped with the SCOUT multiple-sample inlet. The acceleration voltage was set to 30 kV, and the reflector (ion mirror) voltage was set to 32 kV. Spectra were acquired as the sum of ion signals generated by irradiation of the peptide-matrix deposit by 100 to 250 pulses from a 337-nm N2laser (model VSL-337ND; Laser Science,
Inc., Newton, Mass.). Internal mass calibration of all spectra was accomplished by using the known masses of 5 to 10 matrix-related ion signals (68). Data acquisition and analysis were performed with LaserOne software (P. Mortensen and M. Mann, European Molecular Biology Laboratory) running on an Apple Macintosh PowerPC 7100/80 connected to a LeCroy 9350AM digital oscilloscope (LeCroy, Chestnut Ridge, N.Y.) via a GPIB board (National Instruments, Aus-tin, Tex.).
(iii) Sequence database searches with peptide mass data.PeptideSearch soft-ware (36) running on the above-mentioned Apple Macintosh computer was employed for protein sequence database searches with peptide mass data. A nonredundant protein database (generated at the European Molecular Biology Laboratory) was used for initial searches, whereas a VV protein database was used in cases in which no high-ranking match was found in the nonredundant protein database. Initial database search parameters were as follows: cleavage at Lys or Arg residues (trypsin) allowing one or two uncleaved tryptic sites per peptide, at least five peptides required to map to a protein sequence with 0.1-Da accuracy for a positive match, protein molecular mass range of 0 to 300 kDa, and cysteine as anS-carbamidomethyl orS-acrylamide derivative.
on November 9, 2019 by guest
http://jvi.asm.org/
Identification of the proteins by NH2-terminal sequencing.The gene products
of A17L and A13L were identified by Edman degradation. Since these two proteins could not be stained with any conventional staining method used to detect proteins on blots, membrane fractions of the IMV were mixed with
35
S-labeled virus preparations. This mixture was separated by IEF or NEPHGE followed by SDS-PAGE. The gel was blotted onto a polyvinylidene difluoride membrane (Immobilon-P; Millipore Corp., Bedford, Mass.) with CAPS [10 mM 3-(cyclohexylamino)-1-propanesulfonic acid-NaOH (pH 11), 10% methanol]. The labeled proteins were visualized by autoradiography, and the spot of interest was cut from the membrane by using the exposed X-ray film to locate its position on the blot.
The blotted protein samples were loaded into a blot cartridge and intensively washed. They were then subjected to Edman degradation with an ABI 494A Procise sequencer running with user-optimized cycles.
Identification of the proteins by immunoprecipitation.35
S-labeled IMV was prepared as described previously (8). Either labeled virus was directly dissolved in first-dimension lysis buffer or proteins from the virus were immunoprecipi-tated by using the above-mentioned antibodies as described previously (13) with the modification that after the final wash in radioimmunoprecipitation assay buffer, the proteins were released from the staphylococcal protein A pellet by a 30-min incubation at 378C in first-dimension lysis buffer.
RESULTS
Preparation of pure IMV samples and separation of the core and membrane proteins. Toward the goal of elucidating the molecular events that occur during the morphogenesis of the first infectious form of VV, the IMV, we initiated the identi-fication of its major membrane- and core-associated proteins. To facilitate the isolation of IMV only, we have made use throughout this study of a deletion mutant, vRB12, of the strain Western Reserve which lacks the 37-kDa (F13R) EEV protein and therefore is deficient in EEV formation (3). IMV was isolated from infected HeLa cells and purified by a three-step procedure (see Materials and Methods). PNSs of infected cells were first layered on top of a 36% sucrose cushion, and the virus was pelleted. This pelleted material, as assessed by negative-staining EM (data not shown), contained virus and some mitochondria as well as undefined membranes. After sonication to remove virus aggregates, this pelleted material was further purified by sedimentation over a 25 to 40% sucrose gradient; the light-scattering (virus) band was collected and subsequently concentrated by pelleting. By negative-staining EM this final pellet was at least 95% pure, containing exclu-sively IMVs (data not shown).
From Fig. 1 the purification steps can be assessed. We sub-jected equal amounts (in terms of protein concentration) of the PNS, the pelleted material after the first centrifugation step, and the final purified virus to SDS-PAGE and silver staining. As can be seen, the first pelleted material already contained almost exclusively viral proteins.
The purified IMV preparations were subjected to treatment with 1% NP-40 and 20 mM DTT, a variation of a classical method (10, 24, 26, 56) to separate the VV membrane from core proteins. The membrane and core fractions were subse-quently separated with 2D gels. The first dimension was either an IEF covering a pI range from 7 to 3 or an NEPHGE that, depending on the running time, separated proteins with pIs of 7 to 11. The second dimension in both cases was an SDS–15% PAGE, after which the proteins were detected by silver stain-ing. The protein spots were identified either by MALDI MS of peptide mixtures after in-gel digestion, by N-terminal Edman degradation after electroblotting of the protein onto polyvi-nylidene difluoride membranes, or by immunoprecipitation with specific antibodies. In the last case, the proteins were immunoprecipitated from 35S-labeled vRB12 and separated
with 2D gels, and their positions were compared with those for the silver stained gels.
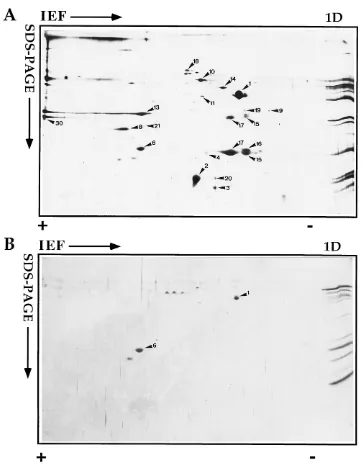
IEF gels.When the membrane fraction was analyzed by IEF, up to 20 polypeptide spots could be detected by silver staining
(Fig. 2A). Most of these spots could also be detected by IEF with 35S-labeled IMV (compare Fig. 2A and 7E). Since the
radioactive label was added at 6 h after infection (at the onset of the late protein synthesis), it can be expected that these proteins represent late (structural) proteins (see below). Table 1 shows a summary of the proteins we have identified in the membrane and core fractions of NP-40–DTT-treated virus preparations. By using IEF standards (Carbamylyte; see Ma-terials and Methods), we also determined the pIs of the pro-teins as they appear in our gel system. In general these corre-sponded well to the predicted pIs, except for the most acidic proteins (A4L and A17L products). We believe that this discrep-ancy is due to the first-dimension running conditions, which most likely do not separate acidic proteins (pI below 4.5) very well.
Compared with the membrane fraction, the core fraction appeared to contain relatively few abundant proteins, only two of which were readily detected by IEF. Moreover, upon iden-tification of these spots by MALDI MS, they were found to correspond to two protein spots in the membrane fraction (A4L product or p39 [35] [spot 1] and A10L product [referred to in this paper as p23] [66] [spot 6]). We have previously shown that the product of the gene A4L, p39, partitions par-tially in the membrane and parpar-tially in the core fraction upon NP-40–DTT treatment (8). We have rationalized this finding by suggesting that p39 may be located in the space between the inner membrane and the core, an idea that was supported by immunocytochemical data (8). Since the C terminus of A10L behaves similarly, it can be expected to be located in the same space (see Discussion).
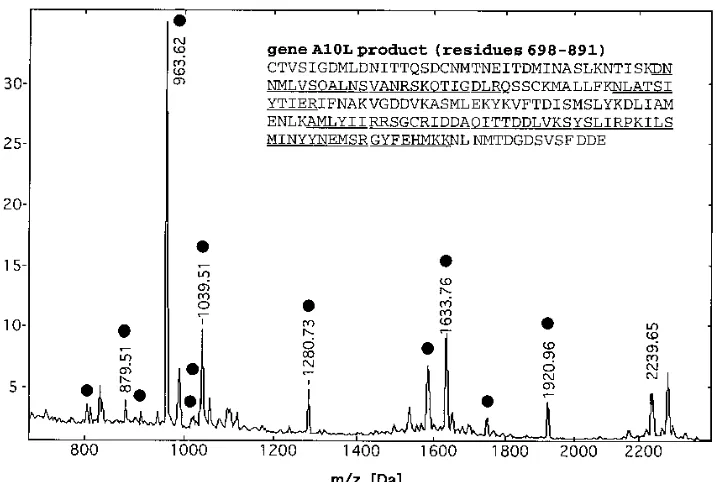
[image:3.612.338.530.70.287.2]Figure 3 shows a MALDI mass spectrum of a tryptic peptide mixture produced from protein spot 6 (or p23) from the 2D gel (Fig. 2A). Protein sequence database searching with the list of tryptic peptide masses identified the protein as the product of gene A10L. Fourteen experimentally obtained tryptic peptide
FIG. 1. Silver-stained SDS-PAGE of different fractions from the virus puri-fication. Equal amounts (in terms of protein concentration) of a PNS of infected cells (lane 1), pelleted material from this PNS after centrifugation through 36% sucrose (lane 2), and sucrose-gradient purified virus (lane 3) were run on SDS– 15% PAGE, and the proteins were detected by silver staining. On the left the positions of several marker proteins are indicated (numbers are molecular masses in kilodaltons), while on the right four abundant viral proteins (4a, 4b, p35, and p11) are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
masses were found to match predicted peptide masses to within 0.1 Da, covering 12% of the amino acid sequence. Fur-thermore, the presence of two C-terminal sequence tags (29) was used to confirm this identification (see Discussion). The A10L gene encodes a 103-kDa polypeptide, which is inconsis-tent with the apparent molecular mass of the protein, 23 kDa, determined from the 2D gel. However, alignment of the matching peptide masses to the polypeptide sequence revealed that 12 peptides mapped exclusively to the C-terminal domain of the protein, covering 42% of this 23-kDa domain (Table 2).
This observation is in agreement with previous studies showing that proteolytic cleavage between residues 697 and 698 of the gene A10L product releases the C-terminal 193-residue 23-kDa protein, a process that most likely coincides with a critical step in virus maturation (66).
[image:4.612.125.487.64.530.2]We have recently shown that gene product of A17L, p21, migrated as several spots with similar molecular weights but differing in pI (34). In this study the N-terminal amino acid sequence of only spot 15 was determined by Edman degrada-tion, but immunoblotting also identified spots 16 and 17 as
FIG. 2. 2D electrophoresis of the NP-40–DTT-soluble (A and C) and -insoluble (B and D) fractions. Purified IMV preparations were treated with NP-40 and DTT, and the membrane proteins were separated from the core proteins by centrifugation. The protein samples were separated in the first dimension by either IEF (A and B) or NEPHGE (C and D). The second dimension in all cases was SDS–15% PAGE, after which the proteins were detected by silver staining. The basic (1) side is on the left, and the acidic (2) side is on the right. On the far right in each panel are the same samples separated only by SDS-PAGE (1D). Up to 30 protein spots can be detected in the membrane fraction separated by IEF. The corresponding core fraction shows only two clear spots that also occur in the membrane fraction (spots 1 and 6). By NEPHGE of the membrane fraction (C), three clear spots can be detected. For spot 24 only the dimer form is well resolved after silver staining. The corresponding core fraction (D) shows two well-resolved abundant spots (spots 28 and 29), as well as a streaking of the two major core proteins 4a and 4b (double arrowheads); a group of less-abundant proteins in the middle of the gel is indicated by small arrows.
on November 9, 2019 by guest
http://jvi.asm.org/
products of the gene A17L (reference 34 and data not shown). It is not known what might lead to these different pI forms of the protein. Attempts to show co- or posttranslational modifi-cations added to the protein have been unsuccessful (33a). The same three protein species were also detected by Western blotting (immunoblotting) at a molecular mass of 35 kDa. These forms may correspond to dimers of p21 that have been described previously (51). We have not yet been able to obtain any MS data from these samples. As previously described, the N-terminal sequencing showed that in the virus the A17L gene product is processed, since it was missing the N-terminal 17 amino acids (references 51 and 70 and data not shown).
We also detected a relatively abundant protein migrating at 35 kDa (spot 13) that was identified by its peptide mass map as the gene product of H3L, thus confirming previous data show-ing a 35-kDa membrane protein on the surface of the IMV (7).
Spot 8 of the membrane fraction did not generate tryptic peptides that could be detected by MALDI MS but was iden-tified by immunoprecipitation as the L1R gene product, a myristylated protein of 27 kDa and with predicted pI of 7 which has been referred to previously as M25 (Fig. 4) (16, 50).
Besides VV-encoded proteins, the membrane fraction also contained several cellular proteins (HSP70, HSP60, actin, ATP synthase, and annexin 5), which were readily identified by MS peptide mapping and database searching (Table 1). We believe these proteins to be cellular contaminants that adhere to our virus preparations (see Discussion).
[image:5.612.122.481.69.539.2]Finally, MS detected a relatively minor protein in the mem-brane fraction (spot 3), the gene product of G4L, which from its sequence does not appear to have a transmembrane do-main.
FIG. 2—Continued.
on November 9, 2019 by guest
http://jvi.asm.org/
NEPHGE. NEPHGE of the NP-40–DTT-soluble (mem-brane) proteins yielded three major spots (Fig. 2C). The gene product of A14L (referred to in this paper as p16) was iden-tified by immunoprecipitation, showing that this protein, like p21 (A17L product) migrated as several spots (spot 24 in Fig. 5B). Like p21, besides migrating at its predicted molecular mass of 16 kDa, it also resolved as a 25 kDa protein (Fig. 5B). This latter form most likely corresponds to a dimer of p16 (45, 51a). On silver-stained gels, the monomer of p16 did not stain very well, and only the dimer form could be detected. Besides the abundant membrane protein p16, the NEPHGE also re-solved p32 (D8L; pI 8 [44]), the gene product of A13L (re-ferred to in this paper as p8), a protein that has been described before by Takahashi et al. (63), as well as two minor spots whose identities were not further investigated. It is worth not-ing that the spot for p8, which from its sequence is predicted to have a pI of 10, ran at pI 8 (Table 1). This suggests that p8 acquires a modification (e.g., phosphorylation) that makes it run at a less basic position.
NEPHGE of the core fraction (Fig. 2D) showed a discrete spot of one of the major core proteins, p25 (L4R product, spot 28 [74]), as well as a smear identified as p11 (F18R product, spot 29 [72]), both of which were identified by MALDI. The latter protein appeared as four or five spots of 11 kDa but with different pIs when immunoprecipitated from35S-labeled IMV
(Fig. 6B). These forms may correspond to different phosphor-ylation states (known to make the pIs of proteins more acidic) of p11 (33, 49, 54). Consistent with earlier data, the two other major core proteins 4a (A10L product) and 4b (A3L product) did not resolve into discrete spots (double arrowheads in Fig. 2D) but rather showed a broad smear over the entire length of
the gel. In the middle of the NEPHGE gel (at an estimated pI of approximately 8), several minor spots were consistently detected (small arrows in Fig. 2D and 8F); these were not identified but may encode nonstructural proteins of the IMV that can be expected to be associated with the viral core.
The detection of the IMV proteins by both silver staining and 35S labeling allowed some preliminary conclusions about
the abundances of some of the proteins. Of the proteins par-titioning in the membrane fraction, the most abundant ap-peared to be the gene products of A17L (p21), A14L (p16), A4L (p39), A27L (p14 [52]), and A13L (p8).
Comparison between the membrane and nonmembrane pro-teins of the IMV and viral propro-teins in infected cells. The results described above show that after NP-40–DTT treatment, several typical membrane proteins (such as the gene products of A17L, H3L, D8L, A14L, A13L, and L1R) as well as some proteins that do not possess a hydrophobic sequence expected for membrane proteins (products of A27L, A4L, and A10L) can be found in the soluble (membrane) fraction. We therefore wanted to establish an independent method to classify the typical membrane versus nonmembrane proteins of the IMV. For this, cells were infected in the presence of rifampin, a drug that blocks viral assembly prior to IV formation (19, 43). The cells were radioactively labeled from 6 to 8 h postinfection, a time point where host protein synthesis can be expected to be switched off and only viral late proteins are synthesized. Com-parison of the 2D gel patterns of 35S-labeled virus and such
[image:6.612.58.556.82.332.2]crude PNSs indeed showed that they were very similar (see below); the main proteins we detected in such radiolabeled PNSs appeared to be viral proteins. Two criteria were used to TABLE 1. VV membrane and core proteins identified in this study
Spot no. 2D gel data
(mol mass, pI)a Gene Proteinb
SwissProt accession
no.
Method(s)c
MALDI mass mapping
Reference(s) No. of
peptides
Sequence coverage (%)
1 40, 4 (5.4) A4L p39, protein A4 (C and M) P29191 MS 10d
48 8, 33
2 14, 5.2 (5.7) A27L p14, 14-kDa fusion protein (M) P20535 MS 9d 58 50
3 12, 5 (5.7) G4L Protein G4 (M) P21025 MSe
4 45 30
6 20, 6 (6.4) A10L C terminus of 4a (p23, amino acids 698–891) (C and M)
P20642 MS 14 (12)d,f 12 (46) 61
8 27, 7 (6.7) L1R M25, protein L1 (M) P20540 Ab 16
10 65, 6 (5.7) HSP60 Mitochondrial matrix protein P1 (human) (M)
P10809 MS 14d
19
11 45, 6 (6) ACTB b-Actin, cytoplasmic (human) (M) P02570 MS 9 26
13 37, 6.4 (6.4) H3L p35, immunodominant envelope protein (M)
P07240 MS 9 28 7
14 42, 5.5 ATB5B ATP synthaseb-chain (human) (M) P06576 MS 13 35
15, 16, 17 19, 4.3 (5.2–5.5) A17L p21, 23-kDa late protein (M) P16711 Ab, Edman 49
18 65, 6 (5.9) GRP75 Mitochondrial stress-70 protein precursor (human) (M)
P38646 MS 12 22
19 35, 4 ANX5 Annexin V (human) (M) P08758 MS 11d 33
20 12, 5 (5.8) A27L p14, 14-kDa fusion protein (M) P20535 MS 4d
17 50
24 16, 8.7 (7.8–8.3) A14L p16 (M) P20991 Ab 60
27 10, 10 (8.0) A13L p8, protein A13 (M) P20990 Edman 60
28 25, 9 (8.6) L4R p25 (C) P03295 MS 8d 27 70
29 11, 10 F18R p11 (C) P07396 MS, Ab 5 34 69
30 32, 9.3 (8.0) D8L p32 (M) P20508 Ab 42
aMolecular masses (in kilodaltons) are as determined by SDS–15% PAGE; pIs are as predicted from the sequence. Numbers in parentheses are the calculated pIs.
bM or C, protein found in the membrane or core fraction, respectively.
cMethod used for protein identification. MS, MALDI MS; Ab, antibody (immunoprecipitation); Edman, N-terminal Edman sequencing.
dOne or two terminal sequence tags (see text and reference 47) confirming the protein identity were found in the MALDI mass spectrum.
eThis protein identification was confirmed by nano-electrospray tandem MS sequencing (data not shown).
fSpot 6; precursor is a 103-kDa polypeptide (p4a). The C-terminal domain (residues 698 to 891) constitutes p23. Numbers in parentheses indicate the peptide
matches in the p23 polypeptide.
on November 9, 2019 by guest
http://jvi.asm.org/
identify the membrane and soluble proteins in PNSs of such infected and labeled cells. First, PNSs were subjected to treat-ment with Na2CO3, a method that separates soluble from
in-tegral proteins (17). In a second instance, PNSs were also treated with TX-114, which allows the separation of membrane proteins and soluble proteins (4). Under these two conditions, the typical membrane proteins (containing at least one hydro-phobic domain long enough to span a membrane) behaved as expected, pelleting with the membranes or partitioning in the detergent phase (Fig. 7 and 8). These included the proteins p21, p35, p32, p16, and p8 (gene products of A17L, H3L, D8L,
A14L, and A13L, respectively). This result is particularly im-portant for p32 and p35, two proteins that apparently associate with membranes in infected cells but failed to associate with rough microsomal membranes following in vitro translation (unpublished results). The spot for the M25 protein (L1R product) could not be clearly identified in the 2D gels of infected cells, and no unequivocal conclusion can be made about its behavior.
[image:7.612.127.486.68.309.2]Consistent with the fact that p14 (A27L product) does not contain a hydrophobic stretch long enough to span a mem-brane (52), the protein behaved in infected cells mostly like a
FIG. 3. MALDI MS of peptides produced by in-gel tryptic digestion of protein spot 6, the COOH terminus of protein 4a. The peptide masses were used for protein database searches and retrieved the gene product of A10L. Tryptic peptides that map to the polypeptide sequence to within a mass accuracy of 0.1 Da are indicated with a solid circle and are underlined in the amino acid sequence (inset). The measured and calculated peptide masses and the sequence assignments are given in Table 2.
TABLE 2. Measured and calculated molecular masses for tryptic peptides which identify the 23-kDa protein (spot 6 in Fig. 2) as the C-terminal part (residues 698 to 891) of the 103-kDa p4a polypeptidea
Mass (Da)b
Position Sequencec
Measured Calculated Difference
801.51 801.44 0.07 57–63 (K)QTIGDLR(Q)
878.50 878.50 0.00 125–131 (K)AMLYIIR(R)
910.43 910.40 0.03 171–177 (R)GYFEHMK(K)
962.61 962.55 0.06 150–157 (K)SYSLIRPK(I)
1,016.53 1,016.56 20.03 55–63 (R)SKQTIGDLR(Q)
1,034.63 1,034.61 0.02 125–132 (K)AMLYIIRR(S)
1,038.50 1,038.49 0.01 171–178 (R)GYFEHMKK(N)
1,279.72 1,279.68 0.04 75–85 (K)NLATSIYTIER(I)
1,597.75 1,597.80 20.05 91–104 (K)VGDDVKASMdLEKYK(V)
1,632.75 1,632.76 20.01 158–170 (K)ILSMINYYNEMSR(G)
1,744.90 1,744.85 0.05 39–54 (K)DNNMLVSQALNSVANR(S)
1,919.95 1,919.93 0.02 133–149 (R)SGCeRIDDAQITTDDLVK(S)
aThe peptides cover 90 of 193 residues, corresponding to 46% sequence coverage. The peptide mass accuracy (better than60.07 Da) and the presence of several
C-terminal sequence tags (see text for details) uniquely identify the protein in the nonredundant protein sequence database. bMonoisotopic, neutral masses.
cTerminal sequence tags are underlined.
dMet is oxidized.
eCys asS-acrylamide derivative.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.61.556.533.677.2]soluble protein (Fig. 7B and D). This indicates that in the absence of viral assembly, this protein does not associate with membranes. The p39 protein (A4L product), which always partitioned partially with the IMV membranes and partially with the core fraction, also showed a dual behavior in infected cells. While the bulk of the protein partitioned into the detergent phase after TX-114 extraction, it also partially pelleted with mem-branes after carbonate treatment (Fig. 7) (8).
The precursor forms of the core proteins 4a, 4b, and p25 (A10L, A3L, and L4R products, respectively [65, 66]) and p11 (F18R product) (again migrating as several spots) behaved mostly as expected, partitioning in the aqueous phase and in the soluble fraction after carbonate extraction (Fig. 8). A small amount of p4b (the precursor of the protein 4b), however, was consistently detected in the detergent phase after phase sepa-ration with TX-114 (Fig. 7C). This observation is surprising considering that its sequence lacks significant stretches of hy-drophobic amino acids. In contrast to the situation in the virus, the precursor forms of 4b (A3L product) and 4a (A10L prod-uct) (identified by immunoprecipitation [not shown]) were well resolved when isolated from infected cells (compare Fig. 2D with Fig. 7B and D). The reason for their inability to be resolved as discrete spots when extracted from the IMV is not known, but it has been speculated that it is due to the insolu-bility of these proteins (45). One factor to consider is that for both proteins, upon cleavage, an obligatory step in the forma-tion of the IMV from the IV (19, 42), the predicted pI becomes more basic (from 6 to 8 for 4a and from 6 to 7.3 for 4b).
On IEF of infected cells, the precursor of p25, p28 (L4R product), was well resolved (Fig. 7B and D), while in the virus this protein could be detected only by NEPHGE (Fig. 2D and 8F). Upon cleavage of this core protein (65), its predicted pI shifts from 6 to 10. This drastic pI shift is interesting, since this protein has been described as binding both single- and double-stranded DNA in vitro (74) and such DNA-binding proteins are generally basic. According to our view on VV assembly (see, e.g., reference 13), the IVs initially mature without con-taining the genome. If this model is correct, then after the entry
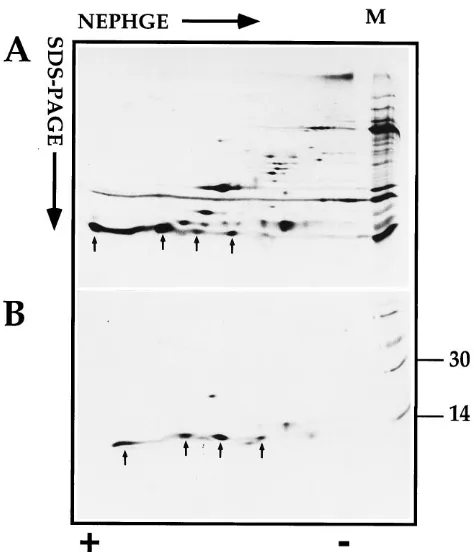
FIG. 4. Identification of the gene product of L1R by immunoprecipitation followed by 2D gel analysis. (A) 2D gel pattern of35
S-labeled IMV separated by IEF as described for Fig. 2A and B. Some of the major spots, as detected by autoradiography, that are also seen in Fig. 2A are indicated (arrowheads). The spot for L1R (spot 8) is indicated with a small arrow. On the right (lane M)
14
C-labeled marker proteins that were only run in the second dimension are shown; of these, the 30- and 14-kDa marker proteins are indicated. (B) A
35
S-labeled IMV preparation was subjected to immunoprecipitation with an antibody to the gene product of L1R and shows one clear spot (as well as a minor spot [not indicated]) coinciding with spot 8 in panel A.
FIG. 5. Identification of spots 24 and 30 by immunoprecipitation with specific antibodies. (A) NP-40–DTT-soluble proteins of35
S-labeled IMV separated by NEPHGE, run as described for Fig. 2C and D. The positions of spots 24 and 30 are indicated. (B) Immunoprecipitation of35
S-labeled cell lysate with antibodies to p16 (the gene product of A14L) is evident. Note that the monomer of this 16-kDa protein migrates as several spots with the same molecular mass but with different pIs, while the major (central) spot is also resolved as a dimer. In cell lysates p16 also resolves with a molecular mass intermediate between those of the monomer and dimer forms. The origin of this spot will be described else-where (33a). (C) Immunoprecipitation of a35
S-labeled cell lysate by using anti-bodies to D8L, identifying spot 30 as product of this gene. In panel A the marker (lane M) is the same sample as used for the NEPHGE run only in the second dimension, while in panels B and C14
C-labeled marker proteins were used, of which the 30- and 14-kDa marker proteins are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
of the DNA into the IV and its subsequent maturation to form the IMV, the drastic change in pI that accompanies the cleavage of p25 could thus be a factor that regulates its binding to DNA. On NEPHGE a basic protein with a molecular mass of about 25 kDa could be detected in all fractions of cell lysates (Fig. 8A to D). Since this protein was absent in the virus, its identity was not further investigated.
Finally, in infected cells an abundant soluble protein (as assessed by both criteria) with a pI of 5 and a molecular mass of about 65 kDa could be detected (Fig. 7B and D). By immu-noprecipitation, this protein was identified as the product of D13L, the target of rifampin (Fig. 9) (1, 64). Consistent with earlier data, this protein was not detected in the IMV (40). As expected from its lack of a hydrophobic domain long enough to span a membrane, p65 behaved entirely as a soluble protein; it partitioned in the aqueous phase and did not pellet along with the membrane proteins (Fig. 7).
DISCUSSION
VV assembly is a complicated process that has been exten-sively described at the ultrastructural level but is poorly under-stood at the molecular level. Crucial to the understanding of the molecular events is the need to identify the many mole-cules involved. In this study we have made an extensive 2D gel map of the major membrane and core proteins of the IMV. By comparing the silver staining pattern with that for35S-labeled
late abundant proteins, we could be reasonably sure that we have identified most of the late (structural) proteins of the IMV. This comparison has proven to be especially important since some of the (mostly highly hydrophobic) proteins stained only with silver and could not be detected by using other
commonly used protein stains, such as Coomassie brilliant blue, amido black, or Ponceau S (unpublished observations).
We have made extensive use of MALDI MS peptide map-ping for protein identification for a completely sequenced virus genome. This work further demonstrates the feasibility, as well as some of the current limitations, of protein identification by this technique. MALDI MS identified 14 of the 30 excised protein spots from the 2D gels and thereby complemented the more established methods, i.e., the use of monoclonal antibod-ies and N-terminal Edman degradation (Table 1). The success rate of approximately 50% is typical for protein identification at the 0.5- to 5-pmol level by the MALDI reflector time-of-flight MS technique used in our laboratory (66a). Considering the nature of many of the proteins identified in this study, i.e., relatively hydrophobic membrane-associated polypeptides which are difficult to analyze by any analytical methods, this is an encouraging result.
The protein spots that were not identified by MALDI mass mapping were of low abundance, or they consisted of small basic proteins (A13L product) or small hydrophobic proteins (L1R, A17L, and A14L products). The addition of a nonionic detergent (e.g., N-octyl-glycopyranoside) during in-gel enzy-matic digestion and peptide extraction did not improve the tryptic peptide yield for the latter proteins as judged by MALDI MS analysis. Better in-gel digestion and peptide ex-traction methods are necessary to improve the performance of MALDI MS for identification of integral membrane proteins, because the approach currently used requires at least four tryptic peptides from a protein for its detection. The recent introduction of delayed-extraction MALDI (reviewed in refer-ence 38) allows the accurate mass determination of peptides, which facilitates the certain identification of proteins when only a few peptides are detected (28).
Since the completion of this work, we have implemented a strategy that uses MS only for protein identification. It com-bines MALDI MS peptide mapping with nano-electrospray tandem MS sequencing (71) for the certain identification of proteins from polyacrylamide gels at the subpicomole level (59). For those proteins which are not identified by their pep-tide mass maps, data provided by tandem MS sequencing of one or two peptides suffice to uniquely retrieve a protein in a database. Therefore, this approach does not rely so much on the enzymatic cleavage and extraction efficiency achievable with the current sample preparation method.
[image:9.612.59.296.67.343.2]Proteins identified by MALDI peptide mass mapping were either VV-encoded proteins (10) or human-encoded proteins (5), the latter being mainly mitochondrial membrane-associ-ated proteins. The protein database searches were performed with a 0.1- or 0.2-Da mass accuracy requirement for the pep-tide masses in order to increase the discrimination between the correct protein match and false-positive matches. This high peptide mass accuracy is routinely achieved and allows us to confidently assign an identity to a protein without limiting the database search space or obtaining complementary informa-tion. The protein sequence coverage of the MALDI peptide maps (summarized in Table 1) is well above 15%, a require-ment for a certain protein identification by MALDI MS in our laboratory. Note that some small proteins in the 11- to 14-kDa range (A27L, G4L, and F18R products) produce relatively few detectable tryptic peptides that, however, constitute a large percentage of the polypeptide sequences. To further improve the confidence level for protein identification by MALDI mass mapping and database searching, we have devised a simple approach that takes advantage of the fact that most proteins contain tandem tryptic cleavage sites, e.g., Lys-Lys or Arg-Lys (29). If a cleavage occurs C terminal to either of these residues,
FIG. 6. p11 occurs in several forms with different pIs. (A) Autoradiogram corresponding to Fig. 7F, representing the core proteins of the IMV separated by NEPHGE. (B) p11 was immunoprecipitated from35S-labeled IMV and
sepa-rated by NEPHGE. The different forms of p11 are indicated by arrows. The positions of the14C-labeled 14- and 30-kDa marker proteins are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
two peptides which differ in mass by a Lys (128.1-Da) or by an Arg (156.1-Da) residue are produced. These peptides may give rise to a diagnostic pattern in the MALDI mass spectrum which can be used as an N- or C-terminal peptide sequence tag for database searching. We have shown that such a tag im-proves the search specificity significantly when used in combi-nation with the complete peptide mass map. Seven of the peptide mass maps contained one or more terminal sequence tags which supported the protein identifications (Table 1).
When only poor-quality peptide mass data are obtained, it may be necessary, as a second step, to search a species-specific or an otherwise filtered subset of the nonredundant protein sequence database and to use complementary methods to con-firm a protein identification. Care should be taken when searching species-specific databases, as this excludes the pos-sibility of identifying contaminating polypeptides from a sec-ond species, as is the case with human mitochsec-ondrial proteins present in VV preparations or human keratins introduced dur-ing sample preparation. Another possible pitfall in database
searching with peptide mass data is illustrated by the identifi-cation of the p23 protein (spot 6) as being part of the 103-kDa precursor polypeptide (gene A10L product). The detected tryptic peptides map to the C-terminal part of the precursor polypeptide, which is in agreement with the fact that this 23-kDa protein covers residues 698 to 891 of the precursor polypeptide. If the protein mass range had been limited during database searching, this protein would not have been retrieved on the basis of the peptide mass map.
A general observation we made was that the membrane proteins of the IMV are all relatively small in size; the largest is 35 kDa (H3L product), and the smallest is 8 kDa (A13L product), and the two most abundant (as assessed by silver staining and35S-labeling) ones are 21 kDa (A17L product) and
16 kDa (A14L product). This observation can possibly be ra-tionalized by the fact that small proteins are translated more rapidly and efficiently and may need shorter folding times. Although most of the membrane proteins resolved with a pI of between 3 and 7, three proteins (A14L, A13L, and D8L
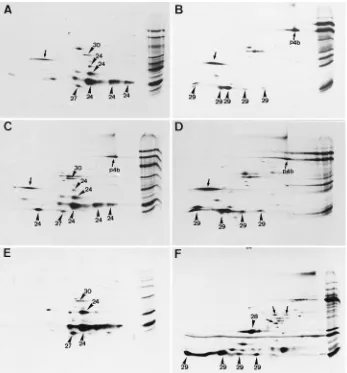
prod-FIG. 7. Comparison by IEF of the membrane and nonmembrane proteins of the IMV with those in rifampin-blocked35
S-labeled cell lysates. The membrane (E) and core (F) proteins of35
S-labeled IMV treated with NP-40 and DTT were separated by IEF as described for Fig. 2. (A to D) Rifampin-blocked cell lysates metabolically labeled from 6 to 8 h postinfection. (A and B) PNSs were subjected to sodium carbonate extraction followed by separation of the membrane-associated (A) from the soluble (B) proteins by centrifugation. (C and D) PNSs were treated with TX-114 to separate the membrane (C) from the soluble (D) proteins. Some of the major membrane and nonmembrane proteins are indicated with arrowheads and numbers, corresponding to those in Fig. 2A and B. The positions of the precursors of 4b and p25 (p4b and p28, respectively) and of p65 (gene product of D13L [see text]) are indicated with small arrows. In all cases the basic side is on the left and the acidic side is on the right of the autoradiogram. On the far right is the same sample run only in the second dimension (A to D) or14
C-labeled markers run in the second dimension only (E and F).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:10.612.135.481.68.445.2]ucts) could not be resolved by this approach, thereby empha-sizing the need for separating protein mixtures also by NEPHGE, which facilitated the separation of basic proteins. Consistent with the general observation that RNA- or DNA-binding proteins generally tend to be basic, the two major DNA-binding proteins of the IMV, p25 (L4R product) and p11 (F18R product) could be resolved only by NEPHGE.
In addition to proteins encoded by the VV genome, we also consistently detected several cellular proteins. Moreover, some of these proteins were also present in the 35S-labeled gels,
indicating that their synthesis is not shut off during the VV infection. Among these cellular proteins we mainly found mi-tochondrial proteins. During routine virus purification, small amounts of mitochondria can be found after the first pelleting step, as assessed by negative-staining EM (unpublished obser-vations). The final sucrose-purified virus preparations were, however, always devoid of (intact) mitochondria and contained IMVs only. We offer, therefore, three possible explanations for the occurrence of these mitochondrial proteins in our virus preparations. First, they might be derived from broken
mito-chondria and copurify with the virus because they somehow adhere to it. In this respect, it is interesting that these proteins are fully accessible when IMV preparations are treated with proteases, indicating an external location (not shown). In the case of HSP60 and HSP70, whose synthesis is evidently up-regulated rather than switched off during VV infection (30, 58), another possibility could be that they copurify as abundant cytosolic proteins, just like actin, a cytosolic protein that we also routinely detected in our virus preparations. Finally, this apparent upregulation could have a real function; these pro-teins could play an active role in VV assembly and possibly even in the VV structure. In this respect, it is worth noting that we also found these proteins copurifying when we used cesium chloride instead of sucrose gradients (not shown).
[image:11.612.134.481.69.442.2]In general, by using NP-40 and DTT, the membrane proteins were well separated from the core proteins, with two excep-tions. Both p39 (A4L product) and the C-terminal cleavage product of A10L, p23, partitioned consistently in both the membrane and core fractions. We have recently provided ev-idence that p39 may be part of the spike exposed on the
FIG. 8. Comparison by NEPHGE of the membrane and core proteins of the IMV with those in rifampin-blocked35S-labeled cell lysates. The membrane (E) and
core (F) proteins of35S-labeled IMV are shown. Some of the major spots are indicated with arrowheads. (A to D) Rifampin-blocked cell lysates metabolically labeled
from 6 to 8 h postinfection. (A and B) PNSs were subjected to sodium carbonate extraction followed by separation of the membrane-associated (A) from the soluble (B) proteins by centrifugation. (C and D) PNSs were treated with TX-114 to separate the membrane (C) from the soluble (D) proteins. Some of the major membrane and nonmembrane proteins are indicated by arrowheads and numbers, corresponding to those in Fig. 2C and D. The positions of p4b (the precursor of 4b) and of an unknown protein (see text) are indicated with small arrows. In panel F the positions of several small spots are also indicated with small arrows. In all cases the basic side is on the left and the acidic side is on the right of the autoradiogram. On the far right is the same sample run only in the second dimension.
on November 9, 2019 by guest
http://jvi.asm.org/
outside of the core (8, 53), and we have speculated that it might provide the structural link between the core and the inner of the two IMV membranes. We have further reasoned that its location in the IMV might explain why p39 can be extracted upon NP-40–DTT treatment. Since p23 (C terminus of A10L) behaves similarly, it is tempting to speculate that it is also located on the outside of the core, possibly as part of the spike. Another possibility is that, in contrast to the proteins 4a/4b, p25, and p11, p39 and p23 are more loosely bound to the core and therefore partition partially into the membrane fraction. We consider this possibility unlikely, however, since reextrac-tion of cores with NP-40 and DTT does not extract the remain-ing p39 and p23 from them (data not shown).
The C terminus of the A10L product (p23) appeared as a well-resolved spot in IEF, in contrast to both 4a and 4b, which, consistent with earlier observations (45), did not resolve into discrete spots. This observation has been interpreted as reflect-ing insolubility of both proteins in first-dimension lysis buffer (45). We have shown, however, that the precursors of both 4a and 4b are well resolved and that only the processed forms show this aberrant behavior. If indeed an insolubility is respon-sible for this failure of the two major core proteins of the IMV to be resolved as discrete spots, then this feature of 4a and 4b, both highly abundant core proteins, could possibly contribute to the extreme stability of the viral core.
Consistent with earlier data, we found that the spot corre-sponding to the gene product of D13L, the target of rifampin, was absent in the virus (40). This protein has been shown to extensively label the concave side of the crescents and the
inner membrane aspect of the IVs (61), as well as of a VVts
mutant whose morphogenesis, at the restrictive temperature, was blocked at the stage beyond the IV but prior to IMV formation (13). Since our data presented here as well as pre-vious data (38) show that p65 is not present in the IMV, it follows that upon the transition of the IV to the IMV, this protein is somehow lost (e.g., by selective degradation). We have speculated before that p65 may play a role as scaffold protein in the IV. Its absence from the IMV thus resembles the situation in some phages, in which a scaffold protein present in the prohead (which, we believe like the IV, lacks the genome) is lost in the mature head (which now, like the IMV, contains the DNA [20]).
In conclusion, we have now identified the major IMV mem-brane and core proteins. These 2D gel patterns can now be used diagnostically, without the need for antibodies. They could also serve as a reference to map the EEV membrane proteins, by comparing 2D gel maps of the IMV with prepa-rations of EEV.
ACKNOWLEDGMENTS
We thank Anna Shevchenko for technical assistance with in-gel protein digestion.
O.N.J. is the recipient of a postdoctoral fellowship from the Euro-pean Union Biotechnology programme, and J.K.L. was supported by a fellowship from the Human Frontier Science Program Organization.
REFERENCES
1.Baldick, C. J., and B. Moss.1987. Resistance of vaccinia virus to rifampicin conferred by a single nucleotide substitution near the predicted NH2 termi-nus of a gene encoding a Mr 62,000 polypeptide. Virology156:138–145. 2.Biemann, K., and H. Scoble.1987. Characterization by tandem mass
spec-trometry of structural modifications in proteins. Science237:992–998. 3.Blasco, R., and B. Moss.1991. Extracellular vaccinia virus formation and
cell-to-cell virus transmission are prevented by a deletion of the gene en-coding the 37,000-dalton outer envelope protein. J. Virol.65:5910–5920. 4.Bordier, C.1981. Phase separation of integral membrane proteins in Triton
X 114 solution. J. Biol. Chem.256:1604–1607.
5.Cairns, H. J. F.1960. The initiation of vaccinia infection. Virology11:603–623. 6.Carrasco, L., and R. Bravo.1986. Specific proteins synthesized during the viral lytic cycle in vaccinia virus-infected HeLa cells: analysis by high-reso-lution, two-dimensional gel electrophoresis. J. Virol.58:569–577. 7.Chertov, O. Y., I. N. Telezhinskaya, E. V. Zaitseva, T. B. Golubeva, V. V.
Zinov’ev, L. G. Ovechkina, L. B. Mazkova, and E. G. Malygin.1991. Amino acid sequence determination of vaccinia virus immunodominant protein p35 and identification of the gene. Biomed. Sci.2:151–154.
8.Cudmore, S., R. Blasco, R. Vincentelli, M. Esteban, B. Sodeik, G. Griffiths, and J. Krijnse Locker.1996. A vaccinia virus core protein, p39, is membrane associated. J. Virol.70:6909–6921.
9.Dales, S., and B. G. T. Pogo.1981. Biology of poxviruses. Virology mono-graphs. Springer Verlag, Vienna.
10. Easterbrook, K. B.1966. Controlled degradation of vaccinia virions in vitro: an electron microscopic study. J. Ultrastruct. Res.14:484–496.
11. Eckerskorn, C., and F. Lottspeich.1989. Internal amino acid sequence analysis of proteins separated by gel electrophoresis after tryptic digestion in polyacrylamide matrix. Chromatographia28:92–94.
12. Eng, J. K., A. L. McCormack, and J. R. Yates.1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom.5:976–989.
13. Ericsson, M., S. Cudmore, S. Shumna, R. C. Condit, G. Griffiths, and J. Krijnse Locker.1995. Characterization ofts16, a temperature-sensitive mu-tant of vaccinia virus. J. Virol.69:7072–7086.
14. Essani, K., and S. Dales.1979. Biogenesis of vaccinia: evidence for more than 100 polypeptides in the virion. Virology95:385–394.
15. Fenn, J. F., M. Mann, C. K. Meng, S. F. Wong, and C. M. Whitehouse.1989. Electrospray ionization for mass spectrometry of large biomolecules. Science 246:64–71.
16. Franke, F., E. M. Wilson, and D. E. Hruby.1990. The use of cell-free system to identify the vaccinia virus L1R gene product as a major late myristylated virion protein M25. J. Virol.64:5988–5996.
17. Fuijki, Y., A. Hubbard, L. S. Fowler, and P. B. Lazarov.1982. Isolation of intracellular membranes by means of sodium carbonate treatment. Applica-tion to endoplasmic reticulum. J. Cell Biol.93:97–102.
[image:12.612.58.296.66.343.2]18. Goebel, S. J., G. P. Johnson, M. E. Perkus, S. W. Davis, J. P. Winslow, and E. Paoletti.1990. The complete DNA sequence of vaccinia virus. Virology 179:247–266.
FIG. 9. Identification of p65 in rifampin-blocked cell lysates. (A) Same au-toradiogram as in Fig. 7D, corresponding to the aqueous phase obtained after subjecting PNSs of infected rifampin-blocked cells to extraction with TX-114. The position of p65 is indicated with double arrowheads. (B) p65 (double ar-rowheads) was immunoprecipitated from35
S-labeled rifampin-blocked cell ly-sates and separated by IEF. On the far right (lane M) are14
C markers run only in the second dimension, of which the 68-, 45-, and 14-kDa marker proteins are
indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
19. Grimley, P. M., E. N. Rosenblum, S. J. Mims, and B. Moss.1970. Interruption by rifampicin of an early stage in vaccinia virus morphogenesis: accumulation of membranes which are precursors of virus envelopes. J. Virol.6:651–659. 20. Hendrix, R. W., and R. L. Garcea.1994. Capsid assembly of dsDNA viruses.
Semin. Virol.5:15–26.
21. Henzel, W. J., T. M. Billeci, J. T. Stults, S. C. Wong, C. Grimley, and C. Watanabe.1993. Identifying proteins from two-dimensional gels by molec-ular mass searching of peptide fragments in protein sequence databases. Proc. Natl. Acad. Sci. USA90:5011–5015.
22. Hillenkamp, F., M. Karas, R. C. Beavis, and B. T. Chait.1991. Matrix-assisted laser desorption/ionization mass spectrometry of biopolymers. Anal. Chem.63:1193A–1202A.
23. Hiller, G., and K. Weber.1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 55:651–659.
24. Holowczak, J. A., and W. K. Joklik.1967. Studies of the structural proteins of virions and cores. Virology33:717–725.
25. Hunt, D. F., J. R. Yates, J. Shabanowitz, S. Winston, and C. R. Hauer.1986. Protein sequencing by tandem mass spectrometry. Proc. Natl. Acad. Sci. USA83:6233–6237.
26. Ichihashi, Y., M. Oie, and T. Tsuruhara.1984. Location of DNA-binding proteins and disulfide-linked proteins in vaccinia virus structural elements. J. Virol.50:929–938.
27. James, P., M. Quadroni, E. Carafoli, and G. Gonnet.1993. Protein identi-fication by mass profile fingerprinting. Biochem. Biophys. Res. Commun. 195:58–64.
28. Jensen, O. N., A. Podtelezhnikov, and M. Mann.Delayed extraction im-proves specificity in database searches by MALDI peptide maps. Rapid Commun. Mass Spectrom., in press.
29. Jensen, O. N., O. Vorm, and M. Mann.1996. Sequence patterns produced by incomplete enzymatic digestion or one-step Edman degradation of peptide mixtures as probes for protein database searches. Electrophoresis17:938–944. 30. Jindal, S., and R. A. Young.1992. Vaccinia virus infection induces stress
response that leads to association of Hsp 70 with viral proteins. J. Virol. 66:5357–5362.
31. Johnson, G. P., S. J. Goebel, and E. Paoletti.1993. An update on the vaccinia virus sequence. Virology196:381–401.
32. Joklik, W. K.1962. The purification of four strains of poxvirus. Virology 18:9–18.
33. Kao, S. Y., and W. R. Bauer.1987. Biosynthesis and phosphorylation of vaccinia virus structural protein VP11. Virology159:399–407.
33a.Krijnse Locker, J.Unpublished observations.
34. Krijnse Locker, J., S. Schleich, D. Rodriguez, B. Goud, E. J. Snijder, and G. Griffiths.1996. The role of a 21kDa membrane protein in the assembly of vaccinia virus from the intermediate compartment. J. Biol. Chem. 271: 14950–14958.
35. Maa, J.-S., and M. Esteban.1987. Structural and functional studies of a 39,000-Mrimmunodominant protein of vaccinia virus. J. Virol.61:3910–3919.
36. Mann, M.1996. PeptideSearch software for Apple Macintosh computers (available via anonymous FTP at FTP://mac-mann6.embl-heidelberg.de/Sat-urn/Pub/Software).
37. Mann, M., P. Højrup, and P. Roepstorff.1993. Use of mass spectrometric molecular weight information to identify proteins in sequence databases. Biol. Mass Spectrom.22:338–345.
38. Mann, M., and G. Talbo.1996. Developments in matrix-assisted laser de-sorption/ionization peptide mass spectrometry. Curr. Opin. Biotechnol. 7:11–19.
39. Mann, M., and M. Wilm.1994. Error tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem.66:4390–4399. 40. Mohondas, A., and S. Dales.1995. Function of spicules in the formation of
vaccinia virus envelopes elucidated by a conditional lethal mutant. Virology 214:494–502.
41. Moss, B.1990. Poxviridae and their replication, p. 2079–2111. InB. N. Fields, D. M. Knipe, R. M. Chanock, M. S. Hirsch, J. L. Melnick, T. P. Monath, and B. Roizman (ed.), Virology. Raven Press, New York. 42. Moss, B., and E. N. Rosenblum.1973. Protein cleavage and poxvirus
mor-phogenesis: tryptic peptide analysis of core precursors accumulated by block-ing assembly with rifampicin. J. Mol. Biol.81:267–269.
43. Moss, B., E. N. Rosenblum, E. Katz, and P. M. Grimley.1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature (London)224:1280–1284. 44. Niles, E. G., and J. Seto.1988. Vaccinia virus gene D8 codes for a virion
transmembrane protein. J. Virol.62:3772–3778.
45. Oie, M., and Y. Ichihashi.1981. Characterization of vaccinia polypeptides. Virology113:263–276.
46. Pappin, D. J. C., P. Højrup, and A. J. Bleasby.1993. Rapid identification of proteins by particle-mass fingerprinting. Curr. Biol.3:327–332.
47. Patterson, S. D., and R. Aebersold.1995. Mass spectrometric approaches for the identification of gel-separated proteins. Electrophoresis16:1791–1814. 48. Payne, L. G.1978. Polypeptide composition of extracellular enveloped
vac-cinia virus. J. Virol.27:28–37.
49. Pogo, B. G. T., J. R. Katz, and S. Dales.1975. Biogenesis of poxviruses:
synthesis and phosphorylation of a basic protein associated with the DNA. Virology64:531–543.
50. Ravanello, M. P., and D. E. Hruby.1994. Conditional lethal expression of the vaccinia virus L1R myristylated protein reveals a role in virion assembly. J. Virol.68:6401–6410.
51. Rodriguez, D., J. F. Rodriguez, and M. Esteban.1993. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L. J. Virol.67:3435–3440. 51a.Rodriguez, J. F., et al.Unpublished data.
52. Rodriguez, J. F., and M. Esteban.1987. Mapping and nucleotide sequence of the vaccinia virus gene that encodes a 14-kilodalton fusion protein. J. Virol. 61:3550–3554.
53. Roos, N., M. Cyrklaff, S. Cudmore, R. Balsco, J. Krijnse Locker, and G. Griffiths.1996. A novel immunogold cryo EM method to investigate the structure of the IMV/EEV. EMBO J.15:2343–2355.
54. Rosemond, H., and B. Moss.1973. Phosphoprotein component of vaccinia virions. J. Virol.11:961–970.
55. Rosenfeld, J., J. Capdevielle, J. C. Guillemot, and P. Ferrara.1992. In-gel digestion of proteins for internal sequence analysis after one- or two-dimen-sional gel electrophoresis. Anal. Biochem.203:173–179.
56. Sarov, I., and W. K. Joklik.1972. Studies on the nature and location of capsid polypeptides of vaccinia virions. Virology50:579–592.
57. Schmelz, M., B. Sodeik, M. Ericsson, E. Wolffe, H. Shida, G. Hiller, and G. Griffiths.1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans Golgi network. J. Virol.68:130–147.
58. Sedger, L., and J. Ruby.1994. Heat shock response to vaccinia virus infec-tion. J. Virol.68:4685–4689.
59. Shevchenko, A., M. Wilm, O. Vorm, O. N. Jensen, A. V. Podtelejnikov, G. Neubauer, A. Shevchenko, P. Mortensen, and M. Mann.1996. A strategy for identifying gel-separated proteins in sequence databases by MS alone. Bio-chem. Soc. Trans.28:893–896.
60. Shevchenko, A., M. Wilm, O. Vorm, and M. Mann.1996. Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal. Chem. 68:850–858.
61. Sodeik, B., R. W. Doms, M. Ericsson, G. Hiller, C. E. Machamer, W. van’t Hof, G. van Meer, B. Moss, and G. Griffiths.1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic retic-ulum and the Golgi stacks. J. Cell Biol.121:521–541.
62. Sodeik, B., G. Griffiths, M. Ericsson, B. Moss, and R. W. Doms.1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribu-tion of viral protein p65. J. Virol.68:1103–1114.
63. Takahashi, T., M. Oie, and Y. Ichihashi.1994. N-terminal amino acid se-quences of vaccinia virus structural proteins. Virology202:844–852. 64. Tartaglia, J., A. Piccini, and E. Paoletti.1986. Vaccinia virus
rifampicin-resistance locus specifies a late 63,000Da gene product. Virology150:45–54. 65. Van Slyke, J. K., C. A. Franke, and D. E. Hruby.1991. Proteolytic maturation of vaccinia virus core proteins: identification of a consensus motif at the N termini of the 4b and 25K virion proteins. J. Gen. Virol.72:411–416. 66. Van Slyke, J. K., S. S. Whitehead, E. M. Wilson, and D. E. Hruby.1991. The
multistep proteolytic maturation pathway utilized by vaccinia virus P4a pro-tein: a degenerate conserved cleavage motif within core proteins. Virology 183:467–478.
66a.Vorm, O., O. N. Jensen, A. Shevchenko, and M. Mann.Unpublished data. 67. Vorm, O., and M. Mann.1994. Improved mass accuracy in matrix-assisted laser desorption/ionization time-of-flight mass spectrometry of peptides. J. Am. Soc. Mass Spectrom.5:955–958.
68. Vorm, O., and P. Roepstorff.1996. Detector bias gating for improved detec-tor response and calibration in matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom.31:351–356.
69. Vorm, O., P. Roepstorff, and M. Mann.1994. Improved resolution and very high sensitivity in MALDI TOF of matrix surfaces made by fast evaporation. Anal. Chem.66:3281–3287.
70. Whitehead, S. S., and D. E. Hruby.1994. Differential utilization of a con-sensus motif for the proteolytic maturation of vaccinia virus proteins. Virol-ogy200:154–161.
71. Wilm, M., A. Shevchenko, T. Houthaeve, T. Fotsis, and M. Mann.1996. Femtomole sequencing of proteins from polyacrylamide gels by nanoelec-trospray mass spectrometry. Nature (London)379:466–469.
72. Wittek, R., M. Haenggi, and G. Hiller.1984. Mapping of a gene coding for a major late structural polypeptide on the vaccinia virus genome. J. Virol. 49:371–378.
73. Wolffe, E. J., D. M. Moore, P. J. Peters, and B. Moss.1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol.70:2797– 2808.
74. Yang, W. P., and W. R. Bauer.1988. Purification and characterization of vaccinia virus structural protein VP8. Virology167:578–584.
75. Yates, J. R., S. Speicher, P. R. Griffin, and T. Hunkapiller.1993. Peptide mass maps: a highly informative approach to protein identification. Anal. Biochem.214:397–408.