Vol.63,No. 12 JOURNALOFVIROLOGY, Dec. 1989, p. 5258-5267
0022-538X/89/125258-10$02.00/0
Copyright© 1989, American Society for Microbiology
Genetic Evidence for Multiple Nuclear Functions of the Herpes
Simplex Virus ICP8 DNA-Binding Protein
MIN GAOAND DAVID M. KNIPE*
Departmentof Microbiology and Molecular Genetics, HarvardMedical School, 200 LongwoodAvenue,Boston,
Massachusetts 02115
Received 26 June 1989/Accepted 6 September 1989
We have isolated severalmutantherpes simplex viruses, specifically mutated in the infected cellprotein 8
(ICP8)gene,todefine the functional domains of ICP8, the major viralDNA-binding protein.Tofacilitatethe
isolation of thesemutants,wefirst isolatedamutantvirus, HD-2,with the lacZgenefused tothe ICP8geneso
thatan
ICP8-40-galactosidase
fusion proteinwasexpressed.Thisvirus formedblueplaquesonICP8-expressing cell lines in the presenceof5-bromo-4-chloro-3-indolyl-,1-D-galactopyranoside. Mutated ICP8geneplasmidscontransfectedwith HD-2 DNAyieldedrecombinant viruses with themutantICP8geneincorporatedinto the viralgenome. These recombinantswereidentifiedbyformation of whiteplaques.Four classes ofmutantswere
defined: (i)someexpressed ICP8that could bindtoDNA but could not localizetothe cellnucleus; (ii) some
expressed ICP8 that didnotbind toDNAbut localizedtothenucleus; (iii)someexpressedICP8 that neither
boundtoDNAnorlocalizedtothenucleus; and(iv)oneexpressedICP8 that localized to the cell nucleus and
bound to DNA invitro,but the mutant virus did notreplicateits DNA. These classes ofmutantsprovide genetic
evidence that DNAbindingand nuclear localizationaredistinct functions of ICP8 and that ICP8 hasnuclear
functions other than binding to DNA. Furthermore, the portionof ICP8 needed for a nuclear function(s)
distinct from DNAbindingis the part of ICP8showingsequencesimilaritytothat of thecellularprotein cyclin
or proliferatingcell nuclearantigen.
The majorDNA-binding protein, or infected cell protein 8 (ICP8), is expressed by herpes simplex virus type 1as aX or delayedearly gene product.Thisgeneproductisrequired for viral DNA synthesis (7, 10, 29, 49, 51) and normal regulation of viral geneexpression (16, 17). Thefunctionsandactivities specifiedby ICP8 have not been completely established. The known properties of ICP8 include (i) the ability to bind to DNAin vitroand invivo, (ii) the ability to localizetothe cell nucleus, and (iii) the ability to promote assembly of nuclear structures involving viral and cellular DNA replication pro-teins.
ICP8 binds to single-stranded (ss) or double-stranded DNAinvitro(2, 21, 37, 42) and can be isolated in deoxynu-cleoprotein complexesfrominfectedcells (25, 26). The latter observationhasbeeninterpreted tomeanthatICP8 interacts directly with DNA in the infected cells. Because DNA binding by ICP8 molecules encoded by temperature-sensi-tive (ts) mutants is thermolabile in vivo (25, 26) or in vitro (42), thisproperty maybe an essentialfunction of ICP8. The portion of ICP8 involved in DNA binding has not been preciselymapped. Someofthealterationsin ICP8 molecules that are thermolabile for DNA binding are at residues 119, 348, and 450 (15). Because these sequence alterations render the protein conditionally defective, these changes may di-rectly or indidi-rectly affect the DNA binding site on ICP8. Leinbach and Heath (27) have reported that the carboxyl-terminal 69 kilodaltons of ICP8 can bind to DNA in vitro. Thus, the DNA-binding domain of ICP8 may map within this portionof themolecule, but a more precise mapping of this propertyisneeded.
ICP8 contains the necessary signals for nuclear localiza-tion in the absence of other viral proteins (39), but these signalshave notbeen mapped. One mutant protein, lacking
*Correspondingauthor.
residues 326to584, failstolocalize tothe nucleus (34),but it is not known whether the defect is due to lack of an
essential localization signalor analteredconformation pre-venting nuclear uptake.
Cell biological studies of ICP8 nuclear localization have shown that ICP8 localizes to infected cell structures called prereplicative sites in the absence ofviral DNAreplication (12, 39). In these structures, ICP8 behaves as if it were
bound tothe nuclear matrix. Viruses expressing analtered ICP8 molecule failtoassembleprereplicative sites,andfrom theseresults, weconcluded that ICP8 promotes the
assem-blyof these structurescontainingDNA replication proteins (12). Theportion(s)of ICP8 neededtopromoteassemblyof prereplicative sites has notbeenidentified.
Toidentifytheportionof ICP8 neededforthese functions andtoattempttoseparatethevarious functions ofICP8,we
have devised a genetic system for the efficient transfer of mutations totheICP8 gene in the viralgenome. Thisreport outlines the initial characterization ofa set ofviruses
con-tainingmutationsspecifically introduced intotheICP8gene. Thephenotypes ofthese virusesindicatethat DNAbinding and nuclear localization are genetically independent func-tions ofICP8 and that ICP8 specifies nuclearfunctions in addition toDNAbinding.
MATERIALS ANDMETHODS
Cellsandviruses.Verocellsweregrownandmaintainedas describedpreviously (23). The growth medium for the Neor cell lines S2 and B10 (see below) included 200 ,ug of the antibiotic G418 per mlduringthe first passage of cells after thawingor500
pg
of G418 per ml every five passages.The herpes simplexvirus type1 wild-type strain KOS1.1
was propagated and assayed as described previously (23, 25). Mutant viruses were grownin ICP8-expressingS2 and 5258
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 1. Complementationof ICP8 mutants byB10and S2 cellsa
Titer (109PFU/ml)b
Virus B10 S2 Neor
33.50C 390C 33.50C 390C 33.50C 390C
KOS1.1 1.7 1.7 0.7 2.0 1.3 1.8
tsl3 4.3 3.1 2.3 4.0 1.0 <0.001
tsl8 3.0 3.2 2.3 2.7 1.7 <0.001
tsHAl 2.3 3.7 3.7 1.7 2.4 <0.001
aCultures of B10, S2, andNeorcells were infected with each virus and incubated at 33.5°C or 39°C.
bPlaquenumberswerecountedfor2to3days.
B10 celllines. Forallexperiments, monolayer cultures were infectedwith KOS1.1or mutantvirus at amultiplicityof 20 PFUpercell.
Isolation of ICP8-expressing cell lines. Vero cells were transformed withtheplasmidpSG18-SacI (25,40) or p8B-S (15) and pSVneo (45)essentially asdescribed by Deluca et al. (13). After growth in medium containing the antibiotic G418 (a neomycin analog), 21 drug-resistant colonies were picked,grownintocultures, andscreenedfor theirability to complement the growth oftheICP8mutants
tsl3,
tsl8,and tsHAl (10, 20). At thenonpermissive temperature, these ts mutants formed plaques in 7 of21 cell lines derived from culturesreceiving theICP8 gene but notefficiently inNeor cells which were derived from cultures transfected with plasmidpSV2neoalone. The cell cloneB10, derivedfroma culture transfectedwithplasmid p8B-S, and S2cells,derived from a culture transfected with pSG18-SacI, yielded the highest levels of complementation and were chosen for further use (Table 1). KOS1.1 formed plaques inNeor cells as well as in B10 and S2 cells at both temperatures. The mutant viruses tsl3, tsl8, and tsHAl formed plaqueseffi-ciently
onlyat33.5°Cin Neor cells butformedplaquesatthe wild-type level at both temperatures in B10 and S2 cells. Southern blot hybridizationwasperformedtodeterminethe copy number of the ICP8gene in these cell lines, and B10 and S2 cells contained approximately 1 and 10 copies per haploidgenome, respectively (datanot shown).Plasmids.Plasmids p8B-S, pSV8,andpml,aswell as the nucleotide numbering system, were described previously (15, 47). The plasmid p8B-S was constructed by cloninga 5.9-kilobase-pair (kbp) BamHI-SacI fragment (map units 0.374 to 0.411), including the ICP8 gene promoter, into pUC18. Theplasmid pSV8 was constructed by inserting a
5.5-kbp SmaI-Sacl fragment (map units 0.374 to 0.409) downstream ofthesimian virus 40 early promoter, and the plasmidpml was derived from plasmid pSV8 by changing codons499and 502oftheICP8genefrom
cysteine
codonstoglycine codons. Mutant ICP8 gene plasmids used in this studywerederived frompICP8or pSPICP8,inwhicha5.5 kbp SmaI-Sacl fragment (map units 0.374 to 0.409) was inserted into pUC19orpSP64, respectively. Plasmids pnlO and pn2 were generated by linearization of the plasmid spICP8 (whichwasachievedby partial
digestion
withSmaI) andsubsequent insertion ofa 14-nucleotide (nt)XbaIlinker(5'-CTAGTCTAGACTAG-3';
NewEngland
BioLabs, Inc.,
Beverly, Mass.) containing stop codons in all three reading framesat nts 4084 and 3695, respectively. Therefore, pnlO encodes thefirst1,160aminoacidresidues,andpn2encodes the first 1,029 amino acid residues of ICP8 as well as 4additional amino acids,
Pro-Ser-Leu-Asp,
encodedby
the XbaI linker sequence. Plasmid pd301 wasgenerated by aninternal in-frame deletion of a 2,001-base-pair (bp) NotI fragment(nts 1395to3396). Plasmids pdlOl andpdlO2were
constructed in thefollowingways:theplasmid pSPICP8was
linearized by partialdigestion with SmaI, and a 12-ntBglII linker, 5'-GGAAGATCTTCC-3', was ligated in. A1,642-bp deletion was generated by digestion with BglII (converted from a SmaI site at nt652) and BamHI (nt 2294) to yield plasmid pdlO1. Thus, pdlOl lacks codonsforresidues 17to
563 but has aninsertion ofone Arg codon encoded by the BglII linker sequence. An 1,188-bp deletion was generated by digestionwithBglII (convertedfromSmaI at nts 652and 1840) to yield plasmid pdlO2. Thus, pdlO2lacks codons for residues 17 to 411oftheICP8coding sequence butencodes threeadditional amino acids,Arg-Ser-Ser, in the BgIII linker sequence. Both plasmids pdlOl and pdlO2 also contain a
14-ntXbaI linker at nt 4419, downstream of theICP8 poly(A) signal. Because there are other SmaI sites around nts 4084 and 1840,bothpnlO andpdlO2weresequenced to determine the exactmutation sites.
Isolation ofmutant viruses. Themutant virus HD-2, con-taining a lacZinsertion in the ICP8 gene, wasisolated by the following steps. After deletion of a 780-bp XhoI fragment from plasmid pICP8, the plasmid was briefly digested with BAL 31, a BglII linker was added, and the lacZ gene (Pharmacia, Inc.,Piscataway, N.J.) was inserted. The lacZ gene ofpMC1871 contains no transcription promoter and lacksthe firsteightnonessential amino-terminal codons. The mixture of ICP8:lacZ plasmids wastransfected with KOS1.1 DNA into B10 cells (22). After infection with the progeny virus, B10 or S2 cells were overlaid with medium 199: 1% calf serumcontaining0.1% human immune serum for 1 to 2 days at37°C. To detect
P-galactosidase
activity, the medium was then changed to 199 medium: 1.0% agarose containing 400,ug of5-bromo-4-chloro-3-indolyl-(3-D-galactopyranoside (X-Gal) per ml for 8 to 16 h. Recombinant viruses were identified as blue plaques at a frequency of about 0.1 to 0.5%. One mutant virus was plaque purified and designated as HD-2.HD-2 served as therecipient virus to generate all mutant viruses in this study except d301. After cotransfection of infectious HD-2 DNA with mutated ICP8 plasmids, the potential recombinant viruses were first isolated as white plaques from the population of parental blue plaques in the presence of X-Gal and then selected for furtheranalysis.
The mutantvirus d301 was constructedby cotransfection of B10 cells with infectious KOS1.1 DNA and plasmid pd301, inwhich a2,001-bpNotIfragmentwas deleted from theICP8codingsequence(Fig. 1). Theprogeny viruses from the marker transfer were tested for their ability to grow in B10 cells but not in Vero cells. One ofthe more then 300 viruses tested couldgrow in B10butnotin Vero cells.
Analysis of viral proteins. Cell monolayer cultures were infected with KOS1.1 or mutant virusand thenlabeled with [35S]methionine and harvested as indicated in the text. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of infected-cell lysates was performed as described previ-ously (23). Afterelectrophoresis, gels werefixed,dried, and exposed to Kodak SB5film,or theproteinsweretransferred by electrophoresis (30 V, 0.2 A overnight) to nitrocellulose filters for Western blotanalysis. Detection of immune com-plexes on blots by a Western blot procedure involving a
color reaction for the alkaline phosphatase activity was
conducted as specified bythe manufacturers(Promega Bio-tec,Madison,Wis.). Therabbitpolyclonalserum3-83(24)or
the mouse monoclonal antibody lOE-3 (41) (diluted 1:400) wasused to detect ICP8. We used adensitometerto scanthe
on November 10, 2019 by guest
http://jvi.asm.org/
5260 GAO AND KNIPE
gated goat anti-rabbit
immunoglobulin
were used for the detection ofn2ICP8.KOSlJ
nlO2I
n2
-dlOl
dlC2
d3 l1
pml 11
499 5M
FIG. 1. Locations of the ICP8 gene nonsense
and point (pm) mutations used in this study. Th ICP8 codingregionontheHSV-1genomeis showi figure. Restriction sitesshownare BamHI,(B),N
(S).
negatives of the color reactions of Western tate the total amount of ICP8 present in a
Variousamountsof ICP8wereusedtoensur was performed within the linearresponse ra
shown).
ssDNA cellulose chromatography. ssDNA matography of infected-cell extracts was pe scribed previously (21), except that flasks
fected Vero cells were labeled from 4 to 6
with [35S]methionine.
Analysis of viral DNA. (i) Preparation of DNAs were prepared as described previou
tiousviral DNA formarkertransfer andmai
purified from infected cells by centrifugatic equilibrium density gradients as described j
Viral DNA used for Southern blot analysis
the following steps. Cells at a late stage of
frozen and thawed two orthree times and i cated for 30 s at 0 to 4°C. Cell debris we
centrifugationat480x g.Theresultingsuper
subjected to centrifugation at23,500 x g. T
extractedwith phenol-chloroform-isoamyl a
three times. After ethanol precipitation, vi] dissolved inTE buffer.
(ii) Southern blot analysis. Viral DNAs we] appropriate restriction enzymes, separated
electrophoresis, and transferred to nitrocel]
themethod of Southern(44). PlasmidDNAs forhybridizations werelabeledasdescribed
(iii) Measurement of viral DNA synthesis. tureswere infected withappropriate virusa
6to10 hwith20,uCi of[3H]thymidineperml was isolated by the method of Challberg ( were digested with the appropriate restrictic
separated by agarose gel electrophoresis. A
resis, the gel was treated with 1.0 M sodiui
fluorography (8).
Indirect immunofluorescence. Indirect cence was performed as described previou
1:10 dilution of 793 anti-ICP8 monoclona Pereira, unpublished results) and a 1:100 4
damine-conjugated goatanti-mouse antibod viruses except n2. A 1:30 dilution of anti-IX
clonal serum (36) and a 1:200 dilution offlu
1196aa RESULTS
Eiwaa Constructionofmutantviruses.(i)Strategies.Toattemptto 1033aa define the functional domains of ICP8, we constructed several differenttypesofmutations in the cloned ICP8gene
1650aa (Fig. 1): (i) nonsensemutations(pnlOand pn2), (ii)internal
804aa deletions (pd301, pdlOl, andpdlO2), and(iii) a site-specific mutation (pml) (15). Wewanted tointroduce the mutations 529aa into the ICP8genein the viralgenomeby markertransfer.
196aa However, the ICP8 gene is closed to orL, and orL se-quences are always spontaneously deleted from plasmid
clones (46, 49, 50). Thus, marker transfer of the ICP8 (n), deletion(d), mutation fromplasmidssuchaspSG18-SacIposestheriskof te location of the also transferring an altered oriL. To avoid this potential nat thetopofthe problem, all of the mutated ICP8 plasmidsused formarker
lot (N),and Sal transfer in this study were constructed from a plasmid in
which the viralDNAsequencesdid notextendto oriL.The
viral sequenceintheplasmidusedstarts at nt437, only170
bp upstreamfromthe ICP8codingsequences (Fig. 1 [15]). blots to quanti- Recently, the Escherichia coli
P-galactosidase
gene hasl given extract. been used to generate insertion mutant viruses that form
ethat theassay blueplaquesinthepresenceof X-Gal (6, 18, 19, 28, 35).To nge(resultsnot facilitate screening of recombinant viruses after marker
transfer,amutantvirus whichcontainsalacZgeneinsertion cellulose chro- in the ICP8 coding regionwas constructed. This recombi--rformed as de- nantvirus, designated asHD-2,formed blue plaquesin the
(75 cm2) of in- ICP8-expressingcell lines in thepresenceofX-Gal, but did h postinfection notformplaquesin Verocells. Toverify that the lacZgene
was inserted in the ICP8 gene of HD-2, a marker rescue DNA. Plasmid experimentwas performedwith aclonedfragment
contain-isly (11). Infec- ing only the ICP8 gene (nts 437 to 3995) from wild-type
rkerrescuewas KOS1.1 DNA. The percent rescue ([titer ofvirus in Vero :n through NaI cells/titer of virus in B10 cells] x 100) for HD-2 was 19%
previously (22). (resultsnotshown). Becausethe lacZgenecontains a stop
waspurified by codon,onlytheamino-terminal regionofICP8wasexpected
Finfection were tobe synthesizedfrom HD-2. TomaptheICP8:lacZ fusion
were then soni- regionmoreprecisely, thejunction region wascloned from
re removed by HD-2 DNA, and the DNA sequence indicated that the .natantwasthen junction sitewasafternt 1450(K. Baradaran,M. Gao,and 'he pelletswere D. M. Knipe, unpublished results). Thus, HD-2 encodeda
Llcohol(24:24:1) fusionprotein containingtheamino-terminal 281 amino acid ral DNAs were residues of ICP8 and the P-galactosidase protein and
ex-pressedunder thecontrol of the ICP8genepromoter.When
redigestedwith Verocellswereinfected withHD-2, nowild-typeICP8was
by agarose gel detected,butanovel band of 145kilodaltons,approximately
lulose filters by thesizepredicted for the fusionprotein,wasobserved(Fig.
usedasprobes 2, lane3). The
ICP8-p-galactosidase
fusionproteinreactedpreviously (16). with both the 3-83 rabbit polyclonal anti-ICP8serumanda
Monolayer cul- mousemonoclonal anti-,B-galactosidase antibody (Promega ,ndlabeled from Biotec)in Westernblot analysis and inindirect
immunoflu-,and total DNA orescence(datanotshown).
7). Viral DNAs (ii) Isolation of ICP8 mutant viruses. The HD-2 virus
rnenzymesand formed blueplaques in B10cells in the presenceof X-Gal. ,fter electropho- Mutated ICP8geneplasmidscotransfected with HD-2 DNA
m salicylate for yielded recombinant viruses withmutantICP8genes
incor-poratedintotheviralgenome.Putativerecombinantviruses immunofluores- were identified as white plaques in the presence of the
isly (38) with a parental blue virusplaques. With this approach, ICP8gene
l antibody (L. mutations in the plasmids were introduced into the ICP8
dilution of rho- gene in the viral genome. White plaques appeared at fre-y for allmutant quencies ranging from 2 to 39%. The frequency of white
CSP 11/12 poly- plaques in cells transfected with HD-2 DNA and pdlOl or
iorescein-conju- pd102wasnotabovethebackground.Thiswasprobablydue
0i1 0O40
N B
0.39
i S
J.VIROL.
-
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.62.301.68.217.2]MULTIPLE NUCLEAR FUNCTIONS OF ICP8 5261
1 2 3 4 5 6 7
kbp
W.
g~~-ICP8-Ba
FIG. 2. Polypeptide profilesof HD-2- and KOS 1. 1-infected Vero
cells. Cell monolayercultures were infected atamultiplicity of 20 PFUpercellin the presence of 400Rgofphosphonoacetateperml,
labeled with [35S]methionine from 5.5 to 6 h postinfection, and harvestedat6 hpostinfection. Sampleswere separated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis.Lanes: 1,
mock-infectedcells; 2,KOS1.i-infectedcells; 3,HD-2-infected cells. The marks to the right indicate the positions of the fusion protein
(ICP8-ogal;approximately145kilodaltons)andwild-typeICP8(128 kilodaltons).
tothelimitedamountof viral sequencesavailable for
recoin-bination between pdlOl orpdlO2 and HD-2 DNA.
Thefollowingtypesofanalyses wereperformedtoverify that the recombinant viruses contained the appropriate
mu-tation as a result of marker transfer from the ICP8 gene
plasmid constructs. The virus from each white plaque was
plaque purified three times. Viral DNA was isolated, di-gested with appropriate restriction enzymes, and initially analyzedbyagarosegelelectrophoresis. The DNAfragment
pattern was usually sufficient to discriminate the mutant
DNA from HD-2 DNA. Southern blot analysis was
per-formed to confirm the presence of mutations in viral DNA and thepurityof themutantviruspopulations. Forexample, the8.2-kbpBamHIGfragmentof KOS 1. 1 DNA(Fig.3,lane 1)wasdividedinto 6.8- and1.4-kbp fragmentsin n2 DNAby digestionwith BamHI and XbaI because of the XbaI linker
(lane 3). Thejunction region of BamHIG and V (2.3 kbp)
wasreplaced bythelacZgeneinHD-2;therefore, digestion of HD-2 DNA withBamHl andXbal generated a 12.6-kbp fragment (Fig. 3, lane 2). Comparisons ofKpnl digests of KOS1.1(Fig. 3,lane4),HD-2(lane 6),and n2(lane 5)DNAs revealed that the patterns of KOS1.1 and n2 DNAs were
identical but differed from that of HD-2 DNA because of the lacZinsertion (lane 6). In addition, because ofourplasmid
constructs, theoriL-containingKpnIfragments (1.85kbp)of HD-2and n2 DNAs(Fig. 3,lane 6 and5,respectively)were
identicalto KOS1.1 (lane 4) andapproximately55 bp larger than the fragment in plasmid pSG18-Sacl (lane 7), demon-stratingtheintegrity of oriL in the mutantviruses.
94-
6.6-
4.4- 2.3-
2.0-
l-FIG. 3. Southern blotanalysis of KOS1.1, HD-2, and n2 DNAs. KOS1.1, HD-2, and n2 DNAs were digested with BamHI-XbaI
(lanes 1, 2, and 3) and KpnI (lanes 4, 6, and 5), and subjected to electrophoresis inparallel with KpnI-digested pSG18-SacI (lane 7). Digested DNAswereseparatedon a0.8%agarosegel, blottedonto
anitrocellulosefilter,andhybridizedto32P-labeledplasmid pICP8. Thelocations of molecularsize markersareshownattheleft.
Growthproperties. Allmutant virusescontaining the
mu-tations described in Fig. 1 failed to grow inVero cells and
required B10orS2 cells forpropagation. All of the mutant viruses isolated in this study grew to titers nearwild-type
levelsin theseICP8-expressing cell lines.Because the ICP8 geneistheonlyknown viralgeneintheB10andS2celllines,
it is likely that the viral defects were due to lesions in the ICP8 gene. The plaque sizes produced by these mutant viruses were slightly smaller than those ofKOS1.1. In all
cases,themutantvirusesmaintained theirmutantphenotype
afterpropagation inB10and S2 celllines in that the titers of all mutantviruses werebetween 108 and 109PFU/mlin the cell lines but <2.0 x 103inVero cells. This suggestedthat recombinationbetweenthemutantviruses and thewild-type
ICP8 gene in the cell lines occurred at an insignificant frequency.
ICP8 expressed by mutant viruses. To confirm that the mutantsexpressedthepredictedICP8polypeptides,extracts of 35S-labeledmutant- andKOS1.1-infectedVerocellswere
analyzed by polyacrylamide gel electrophoresis (data not shown)and Westernblotting (Fig. 4). Thesizesofthe ICP8 polypeptides specified bythemutantviruseswereconsistent with theexpected mutationalalterations. The lOE-3 mouse monoclonal antibody reacted with ICP8 polypeptides of mutantspml, dlOl, d102, and d301 (Table 2) but not with those of nlOand n2(datanotshown). These results indicated thatthe lOE-3 monoclonal antibody reacts withan epitope
contained, at least in part, within the carboxyl-terminal 36 amino acids of ICP8. These results also show that dlOl, d102, andd301 contain in-frame deletions.
Viral DNAreplication. Toassaytheabilityofeach mutant
1 2 3
VOL.63, 1989
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.123.228.74.326.2] [image:4.612.359.510.78.330.2]5262 GAO AND KNIPE
.m.IS
to
1 2 3 4 5 6 7 8 9 10 11
Kbp
9,4- "a*
4
6,6- ....
92-
44-
69-2 .3-
2.0-FIG. 4. Western blot analysis ofmutantvirus ICP8 polypeptides. Vero cells were infected with viruses in the presence of phosphonoacetate and harvested at 6hpostinfection. Proteins in the cellular extracts were separated by sodium dodecyl sulfate-poly-acrylamide gel electrophoresis and electroblotted onto nitrocellu-lose. Thefilter was then probed with polyclonal rabbit antiserumto ICP8 (3-83). Detection of the immune complexon thefilter utilized acolor reaction for akaline phosphatase activity. The locations of molecular size markersareshownatthe left.kd, Kilodaltons.
virus to replicate its DNA, we infected Vero cells with the
mutant viruses individually and labeled the cultures with [3H]thymidine from 6 to 10 hpostinfection. The cellswere
harvested, and DNA was isolated. Each DNA sample was digestedwith BamHI and XhoI and subjectedtoagarosegel electrophoresis. For all ICP8 gene mutants, undetectable
amountsof[3H]thymidinewereincorporated into viralDNA (Fig. 5, lanes 6 through 11) similar toDNA prepared from cellsinfected with wild-type virus inthe presenceofsodium phosphonoacetate(Fig. 5, lane 5), a compound that prefer-entially inhibitsthe herpes simplex virus type 1 DNA
poly-merase. Incorporation of
[3H]thymidine
into wild-type viral DNA was detected when 1/10 the normal amount of totalTABLE 2. Solubility of ICP8 encoded by viralmutants %ofICP8
Virus Antibodyb
Supernatanta Pellet
KOS1.1 81 19 10E-3
pml 12 88 10E-3
dlOl 22 78 10E-3
d102 33 77 10E-3
d301 42 58 10E-3
KOS1.1 77 23 3-83
nlO 92 8 3-83
n2 10 90 3-83
aThesupernatantandpelletfractionsweredefinedas thesamplesobtained
by centrifugation after DNaseItreatment(21).
[image:5.612.350.525.76.288.2]bTheantibodyusedfor the Westernblots tovisualize ICP8was rabbit polyclonal3-83(24)ormousemonoclonal 1OE-3(41).Thenegatives of the color reactionsof Westemblotswerescannedby densitometer.
FIG. 5. Measurementof viral DNAreplication. Vero cellswere mock infected(lane 1) orinfected with KOS1.1 in the absenceof
phosphonoacetate (lanes 2 through 4), or in the presence of phosphonoacetate (lane5), pml (lane 6), nlO(lane 7), n2(lane8), d102(lane 9), dlO1 (lane 10), or d301 (lane 11) and labeled with [3H]thymidine from 6to 10 h postinfection, and total DNA was
isolated. Ten microgramsof eachDNA sample(exceptin lane 3[5 ,ug] and lane 4 [1 pLg]) was digested with BamHI and XhoI and separatedbyagarosegel electrophoresis. Afterelectrophoresis, the
gelwastreated with 1.0 M sodiumsalicylate forfluorography.
DNA was loaded (Fig. 5, lane 4). Therefore, we conclude that the viralmutants synthesized levelsofviral DNAless
than 10% of the wild-type level of DNA. In the mock-infectedsamples (Fig. 5, lane 1), aband of host DNAwas
seen. Thus, in mutant-infected cells, viruses could not
promoteviral DNAsynthesis,buttheystill had theabilityto inhibitincorporationof[3H]thymidineinto cellular DNAby
6hpostinfection.
DNA-binding properties of ICP8. (i) Solublity. Prior to
studyingtheDNA-binding propertiesofmutantICP8
mole-cules, the solubilities of the ICP8polypeptides under stan-dard extraction conditions were examined (Table 2). The
solubility of the altered ICP8 polypeptides varied signifi-cantly. For example, nlO ICP8 wasjust as soluble as was
wild-typeICP8,butfor n2 andpml, only approximately10%
wassoluble.
(ii)Abilitytobind to DNA. TheDNA-binding propertiesof the soluble fraction of ICP8 molecules encoded by the mutant viruses were examined by chromatography on
ss-DNA-cellulosecolumns.Becauseof theinsolubility ofsome mutant ICP8 polypeptides, all extracts were clarified by centrifugationbeforeapplicationtothe columns. The soluble fractionof theextractswasapplied tothecolumns,and the
proteins were eluted with increasing NaCl concentrations.
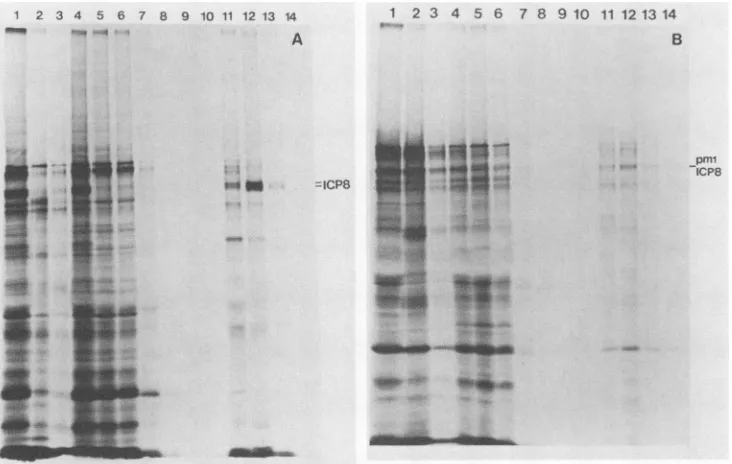
Figure 6 shows the results for KOS1.1 and pml ICP8. Almost allof KOS1.1 ICP8 bound tothe column (compare
lanes 4 and 5 inFig.6A),and themajority of boundICP8was
elutedat0.5 MNaCl(lane 12). In contrast, themajority of
pmlICP8cameoutin theflowthrough (comparelanes 4 and 5 in Fig. 6B). The amount of each form of ICP8 elutedat each of the different NaCl concentrations from ssDNA cellulose columns was determined (Table 3). Mutant nlO
-.
5 4
kd
200
-J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.98.271.77.302.2] [image:5.612.65.301.567.681.2]1 2 3 4 5 6 7 8 9 10 11 12 13 14
_ - A
Wl
1 2 3 4 5 6 7 8 9 10 11 121314
IF ..
B
M_U7
pmt-1 , ICP8
FIG. 6. ICP8 binding to ssDNA-cellulose. Vero cellswereinfected with eitherKOS1.1 (A) orpml(B) in the presence ofphosphonoacetate
andlabeled with[35S]methioninefrom4to6hpostinfection. Various protein fractions resolvedonssDNAcellulose columnsweresubjected
to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Lanes: 1, totalcellularlysate; 2, pellet fromhigh-saltDNaseextraction;3, pellet after dialysis; 4, extract putonssDNAcolumn; 5 to10,flowthrough and wash; 11, 0.3MNaCleluate;12, 0.5 M NaCleluate; 13, 1.0 MNaCl eluate; 14, 4.0 M NaCl eluate. Position of ICP8 (either KOS1.1 ICP8orpmlICP8) is indicatedontherightof eithergel.
ICP8 boundto ssDNA-cellulose asefficientlyand tightlyas did wild-type ICP8. The lowest level of binding was ob-served with d301 ICP8 (21%) in which 56% of ICP8 was missing. The majority of the ICP8 encoded by amino, terminal deletion mutants dlOl and d102 bound to DNA cellulose(72 and75%, respectively). In contrast, the major-ity of n2 ICP8 was found in the flowthrough and wash fractions (54%). This result is consistent with the results of Leinbach and Heath (27) in which the carboxyl-terminal region, but notthe amino-terminal region, of ICP8 synthe-sized in vitro bound to ssDNAcellulosecolumns.Therefore, we conclude that the portion ofICP8 from residues 564 to 1160contains aregion required forDNAbinding.
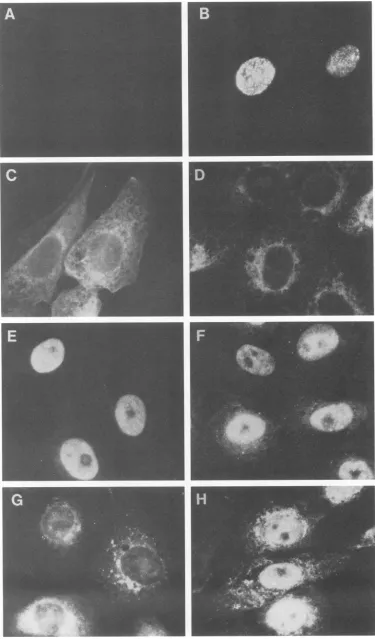
Nuclear localization of ICP8 molecules encoded by viral mutants.Wild-typeICP8 waspreviously shownby immuno-fluorescence microscopy to localize into the nucleus in infected cells (14, 23, 38, 39). In the presence ofa DNA synthesis inhibitor, ICP8 was found at the prereplicative
sites(Fig. 7B; [39])andduringviral DNAreplication, ICP8 was distributed in the nucleusas replication compartments (39). The cellulardistribution of wild-typeandmutantICP8 molecules is shown in Fig. 7. The nlO ICP8
polypeptide,
which lacks the last 36 amino acids from thecarboxyl
terminus and boundto an ssDNA cellulose columnjust astightly as wild-type ICP8, remained within the
cytoplasm
(Fig. 7C). In contrast, the pml ICP8polypeptide,
which bound poorly to an ssDNA-cellulose column, was found predominantlyin the nucleus(Fig.
7E).Theseresultsclearly demonstrate that the nucleus localizationsignal(s)of ICP8isseparatefromthe
DNA-binding
function.ThedlOl ICP8 localizedprimarilywithin the nucleus(Fig. 7H)and bound toanssDNAcellulose column(Table 3),but this virus didnot
replicate
itsDNA. The phenotypeof this mutant providesgenetic
evidence that ICP8 has nuclear functions other thanbinding
toDNA. [image:6.612.125.491.76.308.2]DISCUSSION
TABLE 3. Ability ofmutantICP8tobindtossDNAcellulose % of ICP8:
Virus In flowthrough Eluted at indicatedNaCIconcn
Bound and wash 0.3 M 0.5M 1.0M 4.0 M
KOS1.1 2 23 67 8 <1 98
pml 69 7 20 4 <1 31
nlO 2 13 77 8 <1 98
d102 25 26 22 20 6 75
d10la 28 47 22 5 <1 72
d301a 79 6 13 2 <1 21
n2a 54 1 39 3 <1 46
aData obtained from densitometry of the negatives prepared from the
Westernblots.
Phenotypic groups ofICP8 mutant viruses. On thebasis of their phenotypic
properties,
we have classified the ICP8 mutantviruses into fourgroups(Table
4). ThemutantICP8polypeptides
ofgroup A (nlO andd102)
bound to ssDNA columns but failed to localize to the nucleus. The mutantICP8polypeptides ofgroup B (pml and
d301)
localized tothe nucleus but did not bind to DNA. The mutant ICP8
polypeptide
of group C(n2)
neither bound to DNA norlocalizedtothe
nucleus,
and themutantICP8polypeptide
of group D (d0l1) not only localized to the nucleus but also boundto DNA.Itisnotsurprisingthatnoneof ICP8mutantviruses could grow in the Vero cells because noneofthem
promoted
viral DNAreplication.
Failuretoinduce viral DNAsynthesis by
=lICP8
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.58.296.601.713.2]FIG. 7. Subcellular localization of ICP8 encoded by mutant viruses. Vero cells wereinfected with KOS1.1orICP8mutantviruses.At 4 hpostinfection,the cells were fixed, permeabilized, and incubated with either 793 anti-ICP8 monoclonalantibodyandrhodamine-conjugated goatanti-mouse immunoglobulin(panels A through F and H) or anti-ICSP11/12 polyclonalserumandfluorescein-conjugatedgoatanti-rabbit immunoglobulin (panel G). Immunofluorescence micrographs: A, mock-infected cells; B, KOS1.1-infected cells in the presence of phosphonoacetate; C, nlO-infected cells; D, d102-infected cells; E,pml-infected cells; F, d301-infected cells; G, n2-infected cells; H,
dlOl-infectedcells.
5264
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.121.496.38.677.2]TABLE 4. Phenotypic classes of ICP8 mutant viruses
Group Mutants
Growth
on bindinga LocalizationbA nlO, d102 - + C
B pml, - - N
d301
C n2 - - C
D dlOl - + N
a+,
>50%
binding.bMutant ICP8molecules predominantly localized in the cytoplasm (C) or
nucleus (N), as definedby indirect immunofluorescence.
thegroup A mutantscould beattributedtothe fact that ICP8 was excluded from the nucleus. However, we do not know whether restoration of nuclear localization would be suffi-cientfor normal function oftheprotein.Thegroup D mutant dlOl exhibits anovel phenotype because it localized pre-dominantly to the nucleus (Fig. 7H), and themajority of it canbindto ssDNAinvitro,but the mutant virus stillfails to replicateviralDNA. This indicates that the role of ICP8 in viral DNA replication must be more than just simply binding to ssDNA. Matsumoto et al. (32) observed that the amino-terminal one-fifth of ICP8 shares some sequence similarity with rat proliferating cell nuclear antigen (PCNA), also knownascyclin (3) ortheDNApolymerase-deltaauxiliary protein (48). PCNA is a highly conserved protein whose synthesis is tightly associatedwith the cellcycle,occurring immediately before DNA synthesis (5, 30). PCNA, like ICP8, isanuclearprotein, and the intranucleardistribution of both PCNA and ICP8 is controlled by DNA synthesis itselfor events triggered byDNAreplication (5, 39). More-over, the sites of nuclear localization ofboth PCNA and ICP8 have been found to correspond to sites of ongoing DNAsynthesis (12, 31). Inaddition, both PCNA andICP8 are required for DNA synthesis and can stimulate the activity of DNA polymerase (4, 33, 43, 48). Because the group D mutantvirus dlOllacks the aminoterminus of ICP8, it isconceivable that the amino terminus of ICP8or aportion of
it,
like PCNA, has some nuclearfunction(s).
This func-tion(s) may involve interactions with different structural or functional elements in theinfected cell nucleus, e.g., inter-actionwithand stimulation ofDNApolymerase (9).Theregion required for DNA binding. In thisstudy,mutant nlOICP8 molecules bound to ssDNA invitroastightly and efficiently as wild-type ICP8 molecules did but failed to localize to the nucleus, suggesting that the absence of 36 carboxyl-terminal amino acids of ICP8 only affected nuclear localization,not DNAbinding. This isinagreementwith our previous finding that the abilities of ICP8 to localize tothe nucleus and bind to the nuclear matrix are distinct from DNA-bindingability (25). Significant quantitative differences in DNA binding were observed when ICP8 was further truncatedto residue1029 (n2,
46%,
comparedwith98%
for nlO polypeptide [Table 3]). This indicates that theregion
between residues 1029 and 1260 of ICP8 is required for efficient DNA binding. In addition, approximately 72 and 75% ofdlOl andd102 ICP8polypeptides
bound tossDNA cellulosecolumns,respectively (Table 3).This demonstrated that theregion fromresidues 564 to 1160 containsanssDNA binding site. It should be noted that themajorityofmutantICP8 molecules which bound to ssDNA-cellulose eluted fromthe columns at a lower salt concentration(0.3MNaCl) than thatofwild-type ICP8 (0.5M
NaCl), suggesting
that the amino terminus of ICP8 may be required for tight and efficientbinding.The mappingof the ssDNAbinding domain of ICP8must take into account not onlythe phenotypes ofthe deletion mutants but also the phenotypes of the ts mutants (15). Several interpretations of these results are possible. First, theamino-terminalportionof ICP8 is notabsolutely required for ssDNA binding but may be involved in stabilizing the binding activityof thecarboxylterminus. This couldexplain the failure of the ICP8 molecules oftsl3,tsl8,andtsHAl at thenonpermissivetemperature to bind to DNA.Second,the amino terminusof ICP8 may be involved in the interactions with the carboxyl terminus of another molecule ofICP8, thus providing cooperative ssDNA binding. This is sup-ported bythe fact thatmutantICP8polypeptidesofdlOl and d102 didnotbind ssDNA-cellulose columnsas
efficiently
aswild-type ICP8. It is also possible that ICP8 contains not
only multiple binding sites but also an inhibitory region which could modulate the DNA-binding activities of this polypeptide.
It is interesting to note that thepml ICP8 polypeptide boundpoorlyto the ssDNA-cellulose column(31% binding [Table 3]),while the dlOl polypeptide, whichismissingthe zinc finger structure motif
(Cys-X2-4-Cys-X2-15-His-X2-4-His), bound relativelywell
(72% binding).
A similar obser-vationwasmade for the DNA-binding domainof the simian virus40 Tantigen(1).Thus, thisputative zincfingercannot be an essential determinant of DNAbinding
forICP8;
however, it may still be involved inmodulating
the DNA-binding activity. It will beinteresting
to examine ssDNA-binding activitiesof ICP8 when either of thesecysteines
oradjacent cysteines are changed. Complete deletion of this region may lead the truncated dlOl polypeptide to fold differently, allowingthebinding regionaccess to ssDNA.
The results of this study demonstrate that the nuclear localization signal(s) andssDNA-bindingdomain(s) ofICP8 areseparable. Analysisof these andadditionalICP8mutants
willprovidea moredetailed definitionofthe
ssDNA-binding
domain(s)
and its role in viral DNAreplication
and the natureof the other nuclear functions of ICP8.ACKNOWLEDGMENTS
Wethank L. Pereira and K. L.Powell for793anti-ICP8 mono-clonalantibodyandanti-ICSP 11/12polyclonalserum,respectively. Wealso thank members ofourlaboratoryfortheircomments onthe manuscript.
This workwassupportedbyPublic HealthServicegrantCA26345 fromtheNational Cancer Institute. D.M.K. was supported byan American CancerSociety FacultyResearch Award.
LITERATURECITED
1. Arthur, A. K., A. Hoss, and E. Fanning. 1988. Expression of simian virus T antigen in Escherichia coli: localization of
T-antigen origin DNA-binding domain to within 129 amino acids.J. Virol. 62:1999-2006.
2. Bayliss,G.J.,H. S.Marsden,andJ. Hay. 1975.Herpessimplex
virusprotein:DNA-bindingproteinsininfectedcellsand in the virusstructure. Virology68:124-134.
3. Bravo, R., and J. E. Celis. 1980. A search for differential polypeptide synthesisthroughoutthecellcycleofHelacell. J. Cell Biol. 84:795-802.
4. Bravo, R.,R.Frank,P. A.Blundeil, and H. MacDonald-Bravo. 1987.Cyclin/PCNA istheauxiliaryproteinofDNA
polymerase-delta. Nature(London)326:515-517.
5. Bravo, R., and H. MacDonald-Bravo. 1985. Changes in the nuclear distribution of cyclin (PCNA) but not its synthesis dependonDNAreplication. EMBOJ.4:655-661.
6. Chakrabarti,S.,K.Brechling,and B. Moss.1985.Vacciniavirus
on November 10, 2019 by guest
http://jvi.asm.org/
5266 GAO AND KNIPE
expression vector: coexpression of ,-galactosidase provides visual screening of recombinant virus plaques. Mol. Cell. Biol. 5:3403-3409.
7. Challberg, M. D. 1986. A method for identifying the viral genes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA83:9094-9098.
8. Chamberlain, Y. J. 1979. Fluorographic detection of radioactiv-ity in polyacrylamide gel with the water-soluble fluor, sodium salicylate. Anal. Biochem. 98:132-135.
9. Chiou, H. C., S. K.Weller, and D. M. Cohen. 1985. Mutations in the herpes simplex virus major DNA-binding protein gene leading to altered sensitivity to DNA polymerase inhibitors. Virology 145:213-226.
10. Conley, A. J., D. M. Knipe, P. C. Jones, and B. Roizman. 1981. Molecular genetics of herpes simplex virus. VII. Characteriza-tion of a temperature-sensitive mutant produced by in vitro mutagenesis and defective in DNA synthesis and accumulation of polypeptides. J. Virol. 37:413-428.
11. Davis, R., J. Roth, and D. Botstein. 1981. Advanced bacterial genetics. Cold Spring HarborLaboratory,Cold Spring Harbor, N.Y.
12. de Bruyn Kops, A., and D. M. Knipe. 1988.Formation of DNA replication structures in herpes virus-induced cells requires a viral DNA binding protein. Cell 55:857-868.
13. DeLuca, N. A., A. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of HSV-1 in the gene encoding the immediate-early regulatory proteinICP4. J. Virol. 56:558-570.
14. Fenwick, M. L., M. J. Walker, and J. M.Petkevich. 1978. On the association of virus proteins with the nucleus ofcells infected with herpes simplex virus. J. Gen. Virol. 39:519-529.
15. Gao, M., J. Bouchey, K. Curtin, and D. M.Knipe. 1988. Genetic identification of a portion of the herpes simplex virus ICP8 protein required for DNA binding. Virology 163:319-329. 16. Godowski, P., and D. M.Knipe.1985. Identification of a herpes
simplex virus function thatrepresses late geneexpression from parental virus genomes. J. Virol. 55:357-365.
17. Godowski, P. J., and D. M. Knipe. 1986.Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc. Natl. Acad. Sci. USA 83:256-260.
18. Goldstein, D. J., and S. K. Weller. 1988. Herpes simplex virus
type-1inducedribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characteri-zation of an ICP6lacZ insertion mutation. J. Virol. 62:196-205. 19. Ho, D. Y., and E. S. Mocarski. 1988. P-Galactosidase as a marker in the peripheral andneuraltissues of the herpes simplex virus-infected mouse. Virology 167:279-283.
20. Holland, L. E., R. M. Sandri-Goldin, A. L. Goldin, J. C. Glorioso, and M. Levine. 1984. Transcriptional and genetic analyses of the herpessimplex virustype 1genome: coordinates 0.29 to 0.45. J. Virol. 49:947-959.
21. Knipe, D. M., M. P.Quinlan, and A. E. Spang. 1982. Charac-terization of two conformational forms of the major DNA-binding protein encoded by herpes simplex virus 1. J. Virol. 44:736-741.
22. Knipe, D. M., W. T. Ruyechan,and B. Roizman. 1979. Molec-ulargenetics of herpes simplex virus. III. Fine mapping of a genetic locus determining resistance to phosphonoacetate by two methods ofmarker transfer. J. Virol. 29:698-704. 23. Knipe, D. M., and A. E. Spang. 1982. Definition of a series of
stages in the association of two herpesvirus proteins with the cellnucleus. J. Virol.43:314-324.
24. Knipe, D. M., D.Senecheck, S. A. Rice, and J. L. Smith. 1987. Stages in the nuclear association of the herpes simplex virus transcriptional activator proteinICP4. J. Virol. 61:276-284. 25. Lee, C. K., and D. M. Knipe. 1983. Thermolabile in vivo
DNA-binding activity associated with a protein encoded by mutants ofherpes simplex virus type 1. J. Virol.46:909-919. 26. Leinbach, S. S., and J. F. Casto. 1983. Identification and
characterization ofdeoxyribonucleoprotein complexes contain-ing the majorDNA-binding protein of herpes simplex virus type 1.Virology 131:274-286.
27. Leinbach, S. S., and L. S. Heath. 1988. Acarboxyl-terminal peptide of the DNA-binding protein ICP8 of herpes simplex
virus contains a single-stranded DNA-binding site. Virology 166:10-16.
28. Ligas,M.W.,and D.C.Johnson. 1988.Aherpes simplexvirus
mutant in which glycoprotein D sequences are replaced by ,B-galactosidase sequences binds to butis unable to penetrate into cells. J. Virol. 62:1486-1494.
29. Littler, E., D. Purifoy, A. Minson, and K. L. Powell. 1983. Herpes simplex virus nonstructural proteins. III. Functionof the major DNA-binding protein. J. Gen. Virol. 64:983-995. 30. MacDonald-Bravo, H.,and R. Bravo.1985. Induction of nuclear
protein cyclin in serum-stimulated quiescent 3T3cellsis inde-pendent ofDNA synthesis. Exp. Cell Res. 156:455-461. 31. Madsen, P., andJ. E. CelHs. 1985. S-phase patterns of cyclin
(PCNA) antigen staining resemble topographical patterns of DNAsynthesis. FEBS Lett. 193:5-11.
32. Matsumoto, K., T. Moriuchi, T. Koji,and P. K. Nakane. 1987. Molecular cloning of cDNA coding for rat proliferating cell nuclearantigen(PCNA)/cyclin. EMBO J. 6:637-642.
33. O'Donnell, M. E., P. Elias, B. E. Funnell, and I. R. Lehman. 1987. Interaction between the DNA polymerase and
single-strandedDNA-bindingprotein (infectedcellprotein 8)ofherpes simplex virus 1.J. Biol. Chem. 262:4260-4266.
34. Orberg, P. K., and P. A. Schaffer. 1987. Expression ofherpes
simplex virus type 1 major DNA-binding protein, ICP8, in transformedcelllines: complementation of deletion mutantsand inhibition ofwild-type virus. J. Virol. 61:1136-1146.
35. Panicali, D., A. Grzelecki, and C. Huang. 1986. Vaccinia virus vectors utilizing the,-galactosidaseassay for rapid selectionof recombinant viruses and measurement of gene expression.
Gene 47:193-199.
36. Powell, K. L., E.Littler, andD.J. M.Purifoy. 1981. Nonstruc-tural proteins of herpes simplex virus. II. Majorvirus-specific DNA-binding protein. J. Virol. 39:894-902.
37. Powell,K.L.,and D.J.M.Purifoy. 1976.DNA-bindingproteins of cells infected by herpes simplex virus type 1 and type 2. Intervirology 7:225-239.
38. Quinlan, M.P., and D. M.Knipe. 1983. Nuclearlocalizationof herpesvirus protein: potential role for the cellular framework. Mol. Cell. Biol. 3:315-324.
39. Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:657-868.
40. Quinlan,M.P., and D. M.Knipe. 1985. Stimulation of expres-sion of a herpes simplex virusDNA-binding proteinby twoviral functions. Mol.Cell. Biol. 5:957-963.
41. Rose, D. S. C., K. Shriver, D. S. Latchman, and N. B. LaThangue. 1986. A filamentous distribution for the herpes simplex virus type 2-encoded major DNA binding protein. J. Gen. Virol. 67:1315-1325.
42. Ruyechan, W. T., A. Chytil, and C. M. Fisher. 1986. In vitro characterization ofa thermolabile herpes simplex virus DNA-binding protein. J. Virol. 59:31-36.
43. Ruyechan, W. T., and A. C. Weir. 1984. Interaction with nucleic acids and stimulation of the viral DNA polymerase by the herpes simplex virus type 1 major DNA-binding protein. J. Virol. 52:727-733.
44. Southern, E. M. 1975. Detection of specific sequences among DNA fragments separated by gelelectrophoresis. J.Mol. Biol. 98:503-517.
45. Southern, P. J., and P. Berg. 1982.Transformationof mamma-lian cells to antibiotic resistance with a bacterial gene under control of theSV40 early region promoter. J. Mol. Appl. Genet. 1:327-341.
46. Spaete, R. R., and N. Frenkel. 1982. The herpes simplex virus amplicon: a new eukaryotic defective-virus cloning-amplifying vector. Cell 30:295-304.
47. Su, L., and D. M. Knipe. 1987. Mappingof the transcriptional initiation site of the herpes simplex virus type 1 ICP8 gene in infected and transfected cells. J. Virol. 61:615-620.
48. Tan, C. K., C. Castillo, A. G. So,and K. W.Downey. 1986. An J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
auxiliary protein for DNA-polymerase-delta from fetal calf thymus. J. Biol. Chem. 261:12310-12316.
49. Weller, S. K., J. Lee, D. J.Sabourin, and P. A. Schaffer. 1983. Genetic analysis of temperature-sensitivemutantswhich define thegeneforthemajor herpessimplex virustype1DNA-binding protein.J. Virol. 45:354-366.
50. Weller, S.K., A.Spadaro,J.E.Schaffer,A. W.Murray, A. M.
Maxam, and P. A. Schaffer. 1985. Cloning, sequencing, and functionalanalysisoforiL, anHSV-1originof DNAsynthesis.
Mol.Cell. Biol.5:930-942.
51. Wu,C.A.,N.J.Nelson,D.J.McGeoch, and M. D.Chaflberg.
1988. Identification ofherpes simplex virus type 1 genes
re-quiredfor origin-dependentDNA synthesis. J. Virol. 62:435-443.

![FIG.2.cells.PFUlabeleddodecylharvestedinfected(ICP8-ogal;marksPolypeptide profiles of HD-2- and KOS1.1-infected Vero Cell monolayer cultures were infected at a multiplicity of 20 per cell in the presence of 400 Rg of phosphonoacetate per ml,with [35S]methi](https://thumb-us.123doks.com/thumbv2/123dok_us/1323230.86084/4.612.359.510.78.330/pfulabeleddodecylharvestedinfected-markspolypeptide-profiles-monolayer-infected-multiplicity-presence-phosphonoacetate.webp)
![FIG. 5.geld102mockisolated.phosphonoacetatephosphonoacetateseparated[3H]thymidine,ug] Measurement of viral DNA replication](https://thumb-us.123doks.com/thumbv2/123dok_us/1323230.86084/5.612.350.525.76.288/fig-geld-mockisolated-phosphonoacetatephosphonoacetateseparated-thymidine-measurement-viral-replication.webp)