0022-538X/85/080357-09$02.00/0
CopyrightC)1985, American SocietyforMicrobiology
Identification of
a
Herpes Simplex
Virus Function That
Represses
Late
Gene Expression
from Parental Viral Genomes
PAUL J. GODOWSKI AND DAVID M. KNIPE*
DepartmentofMicrobiology and Molecular Genetics, HarvardMedicalSchool, Boston, Massachusetts02115 Received5February 1985/Accepted 6 April 1985
The expressionof herpes simplex virus Y2(late)genesis inhibited before the onset of viral DNAreplication. Wereportthat the block in theexpression of certain 'Y2genesis relieved,atleast in part, bydefects in the i ICP8 protein. Wehaveexamined theexpression ofthe X2geneencodingglycoprotein C (gC) incellsinfected withatemperature-sensitive ICP8mutant. Under conditionsinwhich viralDNAreplicationisinhibited, cells
infected with the ICP8 mutant overproduce the gC family of mRNAs relative to the level observed in cells infected with a wild-type virus. The gC mRNA synthesized incells infected with the ICP8 mutant virus is correctly initiated and spliced and is translated with the same relativeefficiency as in cells infected with a replicating wild-type virus. These resultssuggest thatICP8 isinvolvedin the negative regulationof Y2genes
expressed from parental viral genomes. The level ofgC expression was greatest in cells infected with a replicating wild-type virus. These datasuggestthat DNAreplicationandgenomeamplificationarenotabsolute requirementsfor "2geneexpression but mayfacilitate full-levelexpression ofthese genes.
Acommon theme illustrated in the productive infections ofeucaryotic cells by DNA viruses is the tightly controlled, developmentally regulated expression of viralgenes.Certain viral gene regulatory circuits provide appropriate model systems for thestudy of cellulargene expression. Although several viral- or cellular-positive regulatory factors have beenidentified, relatively few well-defined negative regula-toryfunctions have beencharacterized. In general, viral late genes are expressed at very low levels before viral DNA replication. By an as yet undetermined mechanism, DNA replicationorgenomeamplification results inaburst of late
geneactivity. The factors that restrict lategene expression before DNAreplication and the mechanism by which viral DNAreplication increases late gene expression are poorly
understood. Elucidation of the mechanism by which these viralgenesareregulated may proveuseful in understanding
theregulation of cellulargeneswhoseexpression is linkedto DNAreplication.
Herpes simplex virus (HSV) is a large DNA virus that
replicates in the cell nucleus (40, 44, 47). The infection of permissive cells results in the induction of more than 50
polypeptides, many of which have been shown to be
en-codedby the virus. Thegeneshave been dividedinto three general classes, a, 1, and -y, based on differences in their
orderofexpression and the viralgeneproductsnecessaryfor theirsynthesis. The -yclass hasbeen furthersubdivided into
the
lYl
and Y2classes. In theinitialstagesofalytic infection,the viralagenesareexpressed from the parentalgenomes.a
geneproducts activate the expression of viral ,Bgenesfrom unreplicated genomes. The aand P genes are required for
theefficientexpression of viral
lyl
andY2 genes.Unlikeaor genes, after DNAreplication,theexpression ofyl and Y2genesgreatly increases. ZYi andY2genes aredistinguished by
theobservation that _Y2gene expression is barelydetectable in a normal lytic infection before viral DNA replication,
whereas-Ylgene expression is readily detected (40).
We have shown that HSVtemperature-sensitive mutants encoding a defective p gene product, ICP8, overproduce certaina, ,B, and-Ylproteins and mRNAsatthe
nonpermis-*Correspondingauthor.
sive temperature (14). These results suggested an involve-ment of ICP8 in the regulation of HSV gene expression. ICP8 bindssingle- and double-stranded DNA (24, 29, 36) and isrequired for viral DNA replication(8, 49). These mutants aredefective for viral DNAreplicationatthenonpermissive temperature (8, 18, 49). It wasof interest todetermine the effect of these defectsontheexpression ofY2genes. Inthis study,we show that thetight blockto Y2geneexpression in theabsence of DNA replication isrelieved, at least inpart,
by defects in ICP8. The expression ofY2genesisgreatestin cellsinfectedbyareplicating wild-type virus. These studies suggest that, before DNA replication, Y2 gene expression may be regulated,directlyorindirectly, by ICP8.
MATERIALS AND METHODS
Cells, viruses, and inhibitors. The procedures used for propagating and titrating viral stocks have been previously described(14, 26). HSV-1 strains KOS1.1and KOS1.1 tsl8 (18, 21)wereprovided by R. Sandri-Goldin and M. Levine, University of Michigan, Ann Arbor. Sodium phos-phonoacetate (PAA)wasagift of Abbott Laboratories, North
Chicago,Ill. PAAwasusedataconcentration of 400 ,ug/ml.
PAA was added 2 h before infection or at the time of infection and maintained throughout infection. The expres-sion ofY2genes wassimilar whether PAAwasaddedbefore
or at the time ofinfection (data not shown). Thymine-1-3-D-arabinofuranoside(AraT; Sigma Chemical Co., St. Louis, Mo.)wasusedataconcentration of 3.8x
10-4
M.Cellswere pretreated for 1 h with 1.9 x10-4
M AraT in Dulbecco modified Eagle medium-10% serum before infection. To construct KOS1.1 18Rsl, Vero cells were transfected (25)with0.5 ,ug each of intact tsl8 DNA and plasmidpSG18-B51 (18) which had beendigested with BamHItorelease theviral DNA insert. After4 daysat33°C, the cells wereharvested, andviruswastitratedat33 and39.5°C. The ts+ recombinants
were plaque purified four times at39.5°C.
Northern blotanalysis of viralmRNAs. Confluent cultures ofVero cellswere infected ata multiplicity of20 PFU per
cell. At 8 h postinfection, the cells were harvested, and
cytoplasmic nucleic acids were isolated after Nonidet P-40 lysis as previously described (14). Ten-microgram portions 357
on November 10, 2019 by guest
http://jvi.asm.org/
of total cytoplasmic RNA were denatured, fractionated in 1% neutral agarose gels containing formaldehyde, blotted
onto nitrocellulose, and hybridized with nick-translated probes as previously described (14). Autoradiography was
performed with intensifying screens at -70°C.
Sl analysis of viral mRNAs. To identify the 5' end of the glycoprotein C(gC) mRNA, totalcellular RNAwas isolated after the lysis of cells with guanidine isothiocyanate as previously described (32). Fifty-microgam samples of RNA were precipitated with ethanol. The precipitate was col-lected by centrifugation at 15,000 x g for 15 min and then
washedwith 75% ethanol. Theprecipitate was dissolved in 20,ulof a solutioncontaining 100to 200 ng of5'-end-labeled DNA probe, 80% formamide, 400 mM sodium chloride, 40 mMPIPES (piperazine-N,N'-bis(2-ethanesulfonic acid) [pH 6.4]), and 1 mM EDTA (4), heated at 76°C for 10 min, and then incubated at64°C for 8 h. The samples were diluted into 200,ul of buffer (0°C) containing 280 mM sodium chloride, 50 mMsodiumacetate, 4.5 mM zinc acetate, 25 ,ug of denatured salmon sperm DNA per ml, 5% glycerol, and 500 U ofSi
nuclease (Boehringer Mannheim, Mannheim, Federal Re-public of Germany) per ml. After incubation at 35°C for 1 h, the reaction was terminated by the addition of ammonium acetate to 0.5 M and EDTA to 12.5 mM. The reaction was extracted with equal volumes of phenol and chloroform, and the nucleic acids were then precipitated with 2 volumes of ethanol. The products of the S1 nuclease reaction were resolved by electrophoresis in 8%polyacrylamide gels
con-taining8 M urea(33). Thegelswerefixed in10% acetic acid
for 20 min and then dried. Autoradiography wasperformed
with Kodak XRP-5film at -70°C with an intensifying screen. To identify the gC mRNA splice acceptor sites, 2.5 ,ug of
polyadenylated RNA (isolated from totalcytoplasmic RNA by oligodeoxythymidylic acid-cellulose [Collaborative Re-search, Inc., Waltham, Mass.] chromatography) was mixed
with 100 ,ug ofEscherichia coli tRNA (Boehringer Mann-heim) and precipitated with 2 volumes of ethanol. The precipitate was treated as described above, and theproducts ofthe S1 nuclease reaction wereseparated by
electrophore-sis in 4% polyacrylamide gels containing 8 M urea (33).
Preparation of nick-translated and end-labeled probes. Plasmid DNAs were isolated by CsCl equilibrium density
gradient centrifugation of alkaline lysates from plasmid-containing cells (32). The HSV-1 or a-tubulin DNA inserts were cleaved from the vector DNA by digestion with the appropriate restriction enzyme. For nick translation, the fragments were purified by electrophoresis through neutral agarose gels and then isolated by electroelution (34). They
were labeled by nick translation (39)with
[a-32P]dCTP
(1,600Ci/mmol;New England NuclearCorp., Boston, Mass.)to a
specific activity of 1 x
108
to 3 x108
cpm/,ug. Before endlabeling, restrictionendonuclease-generatedDNAfragments
were treated with calf intestinealkaline phosphatase
(Boeh-ringer Mannheim). DNA probes labeled at the 5' end were prepared with polynucleotide kinase and
[-y-32P]ATP
(ICNChemicals, Inc.) (33).
Synthesis of viral DNA in infected cells. Confluent
monolayer cultures of Vero cells were infected as described above with 20 PFU per cell of the indicated HSV strain. As indicated, cells were incubated from the time of infection
until thetime ofharvesting in thepresence of 400 ,ugofPAA per ml of medium. At 1 h postinfection the inoculum was
removed, and the cells were overlaid with medium 199-1%
inactivated calf serum containing 20 ,uCi of [methyl-3H]thymidine (New England Nuclear) per ml. At 8 h post-infection, DNA was extracted from the cells as previously
described (22). The DNA was banded in Nal equilibrium densitygradientsin aBeckmanTiSO rotor at44,000rpmfor 48 h at 15°C. Fractions of equal volume (175
pAl)
werecollected with aMicrofractionator(Hoefer Scientific Instru-ments, Inc., San Francisco, Calif.). Portions (20
[lI)
of the fractionswerespottedontoDE-81filters(WhatmanLabora-tory Products, Inc.,
Clifton,
N.J.), andradioactivity
wasdeterminedas previously described (32).
Quantitationof viralDNAmolecules ininfected cells.Vero
cellswere infected with eithertsl8 or KOS1.1 in the pres-ence orabsenceofPAA asdescribed above. At8 h
postin-fection, the cellswereharvested and
lysed
with1%NonidetP-40 asdescribed fortheisolation of
cytoplasmic
RNA(14).Thenuclearpelletwaswashed witha
detergent
mixture(26)
and then suspended in 10 mM
Tris-hydrochloride-10
mM EDTA (pH8.0) with 0.6% sodiumdodecyl
sulfate. Pronasewasaddedto 200,ug/ml, and the nucleiwereincubated for12 h at37°C. NaClwasaddedto0.3M, andthe
suspension
wasextractedonce withan equal volume of
phenol
and chloro-formand then oncewithchloroform. Theaqueousphase
wastreated with 10 ,ugofRNase A per mlfor30minat
37°C,
and nucleic acids wereprecipitated
with 2 volumes of ethanol. Equal quantities of DNA weredigested
tocompletion
withBamHI.As an internal controlfor
complete
digestion,
1 ,ugof lambda DNA wasadded to each
sample,
anddigestion
was monitored by
electrophoresis
through
neutral agarosegels and ethidium bromide
staining.
The nuclear DNA molecules wereresolvedby
electrophoresis through
a0.8% neutralagarosegelandtransferredtonitrocellulose(32).
The conditions forhybridization
of theprobe
to DNAim-mobilized on the nitrocellulose
strips
wereexactly
as de-scribedpreviously
(14) forhybridization
toRNAforNorth-ernblot
analysis.
Immunoprecipitation and
polyacrylamide
gel
electrophore-sis.Infectedormock-infected Vero cellswere
pulse-labeled
for 15 min at 8 h
postinfection
with[35S]methionine
asdescribed
previously
(14,26).
The cells(6
x106)
wereharvested and
pelleted by
centrifugation
for35 s at15,000 xg. The cell pellet was
suspended
in 150,ul
ofRIPA buffer (150mMNaCl,
1% sodiumdeoxycholate,
1% TritonX-100,
0.1% sodium
dodecyl sulfate,
10 mMTris-hydrochloride [pH
7.2]) (13)
containing
1 mMphenylmethylsulfonyl
fluoride(PMSF)
(Sigma)
and 10jiM
N-a-p-tosyl-L-lysine
chloromethyl
ketonehydrochloride
(TLCK) (Sigma)
andthen
subjected
to 15 1-spulses
with a celldisruptor (Heat
Systems-Ultrasonics, Inc.,
Plainview,
N.Y.).
The extract was clarified bycentrifugation
at15,000
x g for 15 min at4°C.
Ascites fluidcontaining
C16antibody
(19;
provided
by
J. C.
Glorioso, University
ofMichigan,
AnnArbor)
wasaddedto 10%
(vol/vol)
and thenincubatedonice for45min. One-half volume of a 10% solution(vol/vol)
offormalde-hyde-treated
Staphylococcus
aureus(IgSorb;
Enzyme
Cen-ter Inc.) in RIPA buffer with PMSF and TLCKwas added and incubated at 0 to
4°C
for 30 to 60 min. Theim-munoprecipitate
wascollectedby
centrifugation
at15,000
xg for 30 s at
4°C.
Thepellet
was washed four times withRIPA buffer
containing
PMSF and TLCK. Theproteins
were dissolved in gel sample buffer and then resolved
by
electrophoresis
through
a 7.25%polyacrylamide
gel (26).
Labeled
proteins
were detectedby
fluorography (27).
RESULTS
Expressionof Y2mRNAsby HSV-1mutantviruses
encoding
adefective ICP8. Todeterminethe effectof ICP8 on Y2 viral geneexpression,
we have examined theexpression
of theHSV-1
gC
gene. Themajor
transcription
product
of the Y2gC
on November 10, 2019 by guest
http://jvi.asm.org/
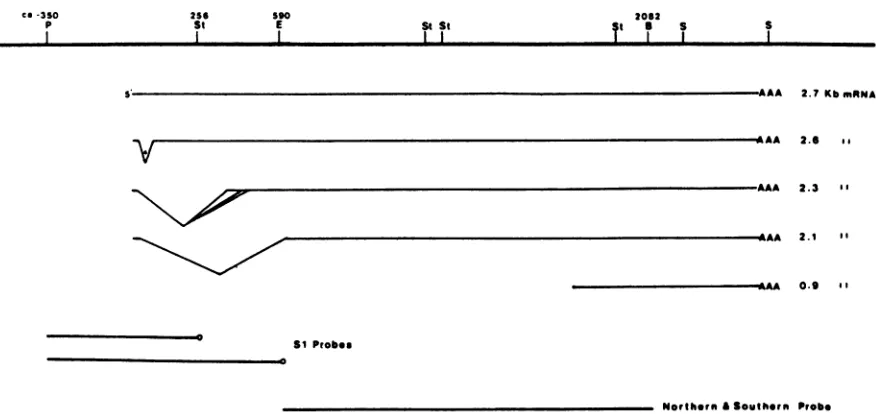
co-3SO
p 256St 590E St St I I
2062
St a S S
2.7KbmnNA
2.6 ..
V
2.3 B'
2.1 I'
0.9 I'
SI Probes
Northern &Southtrn Probe
FIG. 1. Mapof the HSV-1 gCgene. Thesitesfortherestrictionenzymes Pstl(P),SstII(St), EcoRI (E), BamHI(B),andSall(S) and their position relativetotheinitiation site of themajor2.7-kbgCmRNAtranscriptidentifiedbyFrinketal.(11)areindicated. The minorprocessed
forms ofthe2.7-kb mRNA, 2.6, 2.3, and 2.1kb,arealso shown.The 2.3-kb mRNAactuallyconsistsofatleast threespeciesthatdifferslightly
in the splice acceptorsites. The independently initiated 0.9-kb -Y2 mRNAcoterminal with the gC familyofmRNAsis also shown. The fragments used in the S1 mapping experiments (Pstl-SstII andPstl-EcoRI) and thefragment used as aprobe for thegC mRNA andto determine thegCgene copynumber(EcoRl-BamHlfragmentI-I)areindicated.
geneisanunspliced 2.7-kilobase (kb)mRNA. A numberof minortranscripts(2.6, 2.3, and 2.1 kb)arederivedfromthe
2.7-kb mRNAby splicing.These mRNAs arereferred to as
the "gC family" ofmRNAs(10, 11). The minortranscripts
shareashort leader sequence atthe 5' end that isspliced to
sequences ca. 100, 400, or625 bases downstream from the
startofthe 2.7-kb mRNA(Fig. 1).Nucleotide sequencedata
indicatethatboththe 2.7-andtheminor2.6-kb mRNAscan
encode thegCpolypeptide. Thefunctions ofthe 2.4- and the
2.1-kb mRNAs are notknown. Inaddition, a0.9-kb mRNA
overlaps the 3' end ofthe 2.7-kb mRNA. This unspliced
mRNAterminatesatthesamesiteasdoes the2.7-kb mRNA but appears to be initiated at its own promoter. A 4.3-kb
minortranscript also maps in this region but has not been
fully characterized (11).
WeusedNorthern blot analysis tocharacterizetheeffects ofDNA replication on the accumulation ofthese mRNAs. These mRNAs were very abundant in samples of RNA
isolated from cells infectedwith awild-type virus (Fig. 2A, lane 6). The inhibition of viral DNA replication in cells
infectedbythewild-type virusresulted in adrastic decrease in the accumulation ofthe gC family of mRNAs (Fig. 2A, lane 4). The 0.9-and 4.3-kb mRNAs were also reduced when DNA replication was blocked. In other experiments, we usedquantitativeslot blots to compare the amountsof these
transcriptsin cellsinfected with ts+ virus inthe presence or
absence ofAraT, anotherinhibitorof viral DNA replication (2, 17). The amount of stable RNA transcribed from this
region inthe presenceofAraTwasonly1 to2% ofthe level observed in the absence of AraT (data not shown). In
agreement with the observations of Frink et al. (10, 11), these mRNAs appear tobe regulated as Y2 transcripts.
We then examined the effects of a defect in ICP8 on the
expression of these mRNAs. ICP8 is an essential function
forviral DNA replication. Cells infected with the tempera-ture-sensitive ICP8 mutant KOS1.1 ts18 at the nonpermis-sive temperature (39.5°C) synthesize less than 0.1% of the
amount of DNA synthesized in cells infected by the
wild-typevirus (18).Themutantproteinis defective forbindingto
viral DNA in vivo(28). In cells infectedwith ts18at39.50C andblocked forDNAreplication by the
temperature-sensi-tive defectorby thetemperature-sensitive defect andPAA,
the inhibition of gC mRNA accumulation was partially
overcome(Fig. 2A,lanes5 and3, respectively).Under these latter conditions, thegC family ofmRNAs accumulated to ca. 10%of the levelfound in cells infected by a replicating
wild-type virus. It is noteworthy that the 0.9-kb mRNA
accumulated to a level nearly equal to that found in cells infectedby areplicating wild-type virus. At the
permissive
temperature (330C)therewasonlyaverylowamountof the gC familyandofthe 0.9- and 4.3-kb mRNAs in PAA-treated cellsinfected with either tsl8 orts+ virus (Fig. 2A, lanes 1 and2).Thus, theregulation ofthese mRNAs in cellsinfected
with the mutant appeared nearly normal at the permissive
temperature.
To control for the possibility that mRNA samples were
degraded or lost, the same blot was probed with a
nick-translated x-tubulin cDNA clone (30). The amount ofthe 1.6-kb a-tubulin mRNA varied somewhat but did not
cor-relatewith the amount of gC mRNAs(Fig. 3).
The conditional, lethal temperature-sensitive phenotype of tsl8 is the resultofamutation in theICP8gene (18, 28). However, it is formally possible that a second, nonlethal temperature-sensitive mutation is responsible forthe altered
regulation ofthese Y2 mRNAs. Weconstructedats+ deriva-tiveofts18bymarkerrescuewith thecloned HSV-1BamHI
V DNA fragment (HSV-1 map coordinates 0.397 to 0.411). Thisfragment is contained almost entirely within the gene boundaries of ICP8 (18,38;L. Su and D.Knipe, unpublished results). We then determined whether this ts+ derivative (KOS1.1 18Rsl) regulated Y2 gene expression normally. In cells infected with 18Rsl at 39.5°C, theaccumulation ofgC
mRNAswasstrongly inhibitedbythetreatmentof cells with PAA (Fig.2B, lanes 2 and 3). We also selecteda
spontane-ous ts+ revertant of tsl8. This revertant also regulated Y2 mRNAsnormally (E.Richards,P.Godowski,and D.Knipe,
I I I I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.90.528.67.274.2]A
330 39.S* B 39.50
Kb Kb
-s.2
4.3-
2.7-
0.9-_.
*-5.2
4.3- I
2.7- #:
-2---.0 -2.0
0.9-1 2 3 4 5 6 12 3
FIG. 2. gC mRNA levels in cells infected with wild-type or mutant virus. Ten-microgram samples of total cytoplasmic RNA from PAA-treatedoruntreated cellsweresubjectedto electropho-resisinformaldehyde-agarose gels andtransferredtonitrocellulose as described in the text. The blot was probed with the HSV-1 EcoRI-BamHI fragment I-I (see the legend to Fig. 1). (A) RNA sampleswerefrom cells infected with either tsl8orts+virus, inthe presence (+PAA) or absence (-PAA) of PAA, at the indicated temperature. Lane 1, tsl8, +PAA,33°C; lane2,ts+, +PAA,33°C; lane3,ts18, +PAA,39.5°C;lane 4,ts+, +PAA,39.5°C; lane5,tsl8, -PAA, 39.5°C; and lane 6,ts+, -PAA, 39.50C. (B)RNAisolated from cells infectedasfollows: lane 1, tsl8, +PAA, 39.50C; lane2, KOS1.1 18Rsl, +PAA,39.5°C; and lane3, KOS1.1 18Rsl, -PAA, 39.50C.The sizes of themajorRNAspecies detected with the probe areindicated in kilobasesonthe left ofeachpanel.These sizeswere determined relative to migration of 28S (5.2 kb) and 18S (2.0 kb) rRNAmarkers, indicatedontheright ofeachpanel. Autoradiogra-phywasperformedat -70°C withintensifyingscreens.
This might result in a difference in the number of
parental
viral genomes that enterthe nucleus and express
Y2
genes. Wecompared thegCgene copynumber inthenuclei of cells infected with the ts or ts+ virus. Total nuclear DNA was prepared frominfected cells, digested with BamHI,fraction-atedbyagarosegel electrophoresis, and then transferred to
nitrocellulose. The blotwasincubated with the same labeled DNAfragment usedasprobe to determine the level of thegC
mRNAs. ThegCgene copy numberwasvery similar incells treated with PAA and infected at 39.5°C with ts18 or thets+ virus(Fig. 5,lanes 1 and2).As expected, there was ahuge
increase in thegCgene copy number in thenucleiof cells in which viral DNA replication was occurring (Fig. 5, lane3).
Taking into consideration that 50-fold less DNA from this sample was loaded onto the gel, we calculate that there were, on the average, 200- to250-fold more copies of thegC
gene in the nuclei of cells in which viral DNAreplicationwas
occurring. Obviously, this analysis does not indicate the fraction of these DNAs that serve as templates for transcrip-tion. We conclude thatthedifference in theexpression of _Y2 genes from parental genomes in cells infected with tsl8 or
ts+ virusat 39.5°C was not due to differences in the number ofviral DNA molecules thatenterthenucleus.
Fine structureanalysis ofy mRNAs produced in the pres-ence orabsence of viral DNAreplication. Theprocessofviral DNA replicationinfluences the primary sites of initiation for the mRNAs transcribed from the simian virus 40 early transcription unit(5, 12, 16) and from thegeneencoding the adenovirus 72,000-molecular-weight protein (72K protein)
33° 39*50
Kb
unpublished data). These results indicated that the altered
regulation ofY2mRNAsincellsinfectedwithtsl8 at39.5°C
was related to the defectin ICP8.
Viral DNAreplication and DNA copy number in
mutant-infected cells. We determined theextentof theblock ofDNA
replicationin cells treated with PAA andinfectedwith either tsl8 or the ts+ virus. Infected or mock-infected cultures werelabeled with
[3H]thymidine
from Ito8 hpostinfection.The amount oflabeled DNA that sedimented inNal
gradi-ents withthedensity of viralDNAwas
determined,
and the results are shown in Fig. 4. The amount of labeled DNAsedimentingwith thedensity of viralDNAfrom PAA-treated cellsinfected with either tsl8orts+ virus wassimilartothe
amount found in the mock-infected culture. Thus, PAA
inhibited viral DNA replication very efficientlyunder these conditions. Theefficiencyofplaque formationat33°Cof the
mutanttsl8,thewild-typevirus,and the rescuedvirus 18Rsl in the presence of various concentrations of PAA was
identical(datanotshown).Therefore,differences in
sensitiv-itytoPAAdidnot accountfor the alteredregulationof these
Y2mRNAs.
Intheseexperiments, cellswereinfected with20PFU per cellofeitherthe ts orts+ virus. Conceivably, the tempera-ture-sensitivedefectcould affect the viralparticle/PFUratio.
-5.2
-2.0 1.6- I
f
0 * o 111 2 3 4 5 6
FIG. 3. Control for mRNA recovery from cells infected with
wild-typeor mutantvirus. The Northern blot shown inFig.2Awas rehybridized withana-tubulin cDNAprobe (30). The origins ofthe RNA samplesarethesame asdescribed in thelegendto Fig. 2A. Thesize of themajortranscriptdetectedwiththisprobeisindicated inkilobasesonthe left.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.85.282.66.304.2] [image:4.612.365.510.387.664.2]0
z
c 1.57D1D
0II
3
2
5 I0o s 20 5 10 Is 20
FRACTION
FIG. 4. DNA replication in PAA-treated (+PAA) or untreated (-PAA)cells. The distribution in Nal isopycnic density gradients of
[3H]thymidine-labeled DNA(0)isolated from infectedormock-infected cells incubatedat 39.5°Cwasdeterminedas described inthe text. Panels:(A)mock-infected, +PAA; (B) ts+,-PAA;(C)ts+,+PAA; and(D)tsl8,+PAA. Thedensity of gradient fractions(ingrams percubic centimeters)ascalculated from therefractive index(31) is also shown(A).
(6).
Therefore,
we analyzed the site ofinitiation ofthe gC transcriptional unit in cells infected with the mutant orwild-type vi'rus.Thesite ofinitiation oftheunspliced2.7-kb
gCmRNAwas analyzed in S1 mapping experimentswith a
DNA probe5' end labeledatthe SstII site (nucleotide 256) andextendingtothe PstI site (nucleotide -350)asshown in
Fig'. 1.RNAisolated fromcellsinfectedwith thereplicating
ts+ virusprotected DNAfragments migrating with asize of
ca. 255 and256 nucleotides (Fig. 6, lane5). This result is in agreement with that of Frinketal. (11). DNAfragmentsof
identical size were protected by RNA isolated from cells
infected' with tsl8 and treated with AraT. Reduced but detectable amounts of these bands were observed in cells treated with AraT and infected with ts+ virus (Fig. 6, cf.
lanes3 and 4). The amount of correctly initiated gC mRNA
incells infectedwithtsl8was ca.
10%
of thataccumulating in cells infectedwith the replicating wild-type virus. This is based on the observation that the amount of the 255- and 256-nucleotide band protected by 50 ,ug of RNA fromtsl8-infectedcells was very'similar to the amount protected
by the 5 ,ug of RNA isolated from cells infected with a
replicating ts+ virus (Fig. 6, cf. lanes 4 and 6). Multiple bands migrating with a size greater than 255 and 256
nucleotides were also observed in this experiment. The 278-nucleotide band may be the result of SI cutting at a sensitive site in reannealed probe, because the intensity of this band variedfrom'experimenttoexperimentand because
asimiliar-sized bandwasobservedin themock-infected cell RNA on longer exposures. The 310-nucleotide band was
reproducibly observed in multiple experiments with either total or polyadenylated RNA samples isolated from
virus-infected cells. The amount of this band in the various
samples closelyparalleledthat of the 255- and 256-nucleotide doublet. We suspect that the 310-nucleotide band identifies eitheraninitiation siteorspliceacceptorsiteforaY2 mRNA;
however,
additional experiments are required to provethispoint. Itis worthnotingthe presenceofarelativelyAT-rich
region ending 38 nucleotides upstream from the putative
initiation site identifiedby the310-nucleotide band (11). We conclude that thegCmRNAsynthesized incellsinfectedby tsl8 initiates at the same site(s) as that produced in cells infected bythereplicating ts+ virus.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.159.458.68.460.2]Kb
- ~*4U ---6.2
1 2 3 4
FIG. 5. Comparison of gC gene copy number incells infected with ts+ virus. Cells were infected at 39.5°C and incubated in the presence(+ PAA) or absence (-PAA)ofPAA. At 8h postinfection, total nuclear DNA was isolated. The DNA was digested with BamHI, fractionatedin a1%neutral agarose gel, andtransferredto nitrocellulose. The blot wasprobed withtheHSV-1EcoRI-BamHl fragmentI-I(Fig. 1). Lane 1,tsJ8, +PAA; lane 2, ts +PAA; lane 3, ts+, -PAA; and lane 4, purified HSV-1 DNA digested with BamHI. Lanes 1 and 2contained3.5 ,ugof total nuclearDNA, and lane 3 contained 0.07 ,ug of total nuclear DNA. The size of the HSV-1BamHI fragmentI(inkilobases) is indicatedontheright of the panel.
werepulse-labeled with [35S]methionine, extracts were pre-pared, and proteins were precipitated with the monoclonal antibody C16 (19). This monoclonal antibody im-munoprecipitates the fully glycosolated 130,000-Mr form of gC as well as the 110,000-Mr precursor, pgC (7, 43). The precipitated proteins were then analyzed by sodium dodecyl
sulfate-polyacrylamide gel electrophoresisandfluorography. In pulse-labeled cells infected with a replicating virus, the label was incorporated primarily into pgC (Fig. 8, lane 7). The amount of this protein that was immunoprecipitated from PAA-treated cells infected with the ts+ virus was greatly reduced (Fig. 8, lane 3). Under these latter condi-tions, small amounts of this protein were reproducibly observed. The amount ofpgC in PAA-treated or untreated cells infected with ts18was greaterthan 10% of the level of pgC observed inwild-type infections (Fig.8, cf.lanes 2 and 4with lane 5). Thus, the block in synthesis of this _Y2 protein waspartiallyovercomein cells infected with tsl8. Thelevels
600- 285-236-
--Probe
-310 -278
V
=255/256
217-
"-We determined whether the splice acceptor sites ofthe 2.6-and2.3-kbmRNAs wereutilized. Theprobe usedwas5' endlabeled attheEcoRI site(nucleotide560) and extended to the PstI site at nucleotide -350. RNA isolated from untreated cellsinfectedwith the ts+ virus protected amajor fragment of 590 nucleotides that corresponds to the
unspliced mRNA. Bands of605, 640(corresponding to the 278- and 310-nucleotide bands discussed above,
respec-tively), 205, 160, and 120nucleotides (correspondingto the
multiple splice acceptorsites ofthe 2.3-kbmRNA [10, 11])
werealso observed (Fig. 7, lanes 3 and 5). Toour surprise,
we did not detect a470-nucleotide fragment corresponding to the acceptor site ofthe minor 2.6-kb mRNA (11).
Con-ceivably, this reflects differences in splice acceptor
utiliza-tion due to the particular HSV strain used or to different
experimental conditions. These results indicate that the
same acceptorsites used during anormal
lytic
infection bythe ts+ viruswereusedininfectionsbytsl8. (Fig. 7, lanes 3 and4).
7Y2
mRNAs are translated in cells infected with ICP8 mu-tants. We also determined whether cells infected by ICP8mutants werecompetenttotranslate Y2 mRNAs. Cells were
infectedat 39.5°C with tsl8or ts+ virus in the presence or
absence of PAA. Afteran8-hincubationat39.5°C, the cells
[image:6.612.125.234.69.356.2]1 2 3 4 5 6
FIG. 6. Sianalysis of gCmRNAinitiationsites.At8 h postinfec-tion,total cellular RNAwasisolated frominfectedormock-infected cells incubated at 39.5°C in the presence (+AraT) or absence (-AraT)ofAraT. Theprobe usedwas5'endlabeledattheSstII site (nucleotide -256) and extended to the PstI site (ca. nucleotide -350) (seeFig. 1). Lane 1, DNAmarkers;lanes 2to5,S1 reactions thatcontained 50 ,ug of RNA isolated from cellsinfectedunder the following conditions: lane 2, mock-infected, -AraT; lane 3, ts+,
+AraT;lane4,tsl8,+AraT; and lane5,ts+, -AraT. Lane 6 shows theproducts ofanS1 reactionthatcontained51LgofRNAfromts+ cellsin theabsenceofAraTand45 ,ugofRNAfrommock-infected cells in the absence ofAraT. The products of the reactionswere separatedby electrophoresisinan8 Murea-8% polyacrylamidegel. Autoradiography was performed at -70°C with an intensifying
screen. The size of the HSV marker DNA(amixturegeneratedby digestionoftheprobewithTaqI [nucleotide 217],HhaII[nucleotide
236], SmaI [nucleotide 285], or PstI [ca. nucleotide 600]) in nucleotidesis shownonthe left.The size oftheprotectedfragments
alongwith thepositionof thereannelaledprobeareindicatedonthe right.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.355.517.268.538.2]-1353
-1078
-872
-603
640-
605-0590
._.& -1-. -pgC
S
l
.v>205-
:.160-ALz
-310
281
1
-271* -234
-194
118
[image:7.612.364.508.72.343.2]1 2 3 4 5 6 7
FIG. 7. S1 analysis of gC mRNA splice acceptor sites. Polyadenylated (pA+) RNAs prepared from PAA-treated cells in-fected at39.5°Cwith tsl8orfromuntreated cells infected withts+ viruswasanalyzed inS1mappingexperimentswithaprobe labeled at the EcoRI site (nucleotide 590) as shown in Fig. 1. Lane 1,
Undigested probe; lanes 2 to 4, S1 reactions that contained the following RNAs: 100 ,ug of tRNA, 2.5 jig of pA+ RNA from PAA-treatedcells infectedwithtsl8 and 100,ugoftRNA,and 2.5,ug ofpA+RNAfromuntreatedcellsinfected withts+virus and 100,ug oftRNA, respectively.Lane 5, DNA markers. Lanes6and 7 show
shorterexposuresof lanes4and 5, respectively. The productsof the
reaction were separated by electrophoresis in an 8 M urea-4%
polyacrylamide gel. The size of the markers (in nucleotides) is shown onthe right. The sizes of the bands corresponding to the positionsexpectedfor the spliceacceptorsites (>)areshownonthe
left.Also indicatedis the major initiation site (-)identifiedby Frink etal.(10, 11).
[image:7.612.101.257.92.551.2]1 2 3 4 5 6 7
FIG. 8. Synthesis of Y2 proteins in PAA-treated (+PAA) or untreated (-PAA) cells. At 8 h postinfection, infected or mock-infected cells were pulse-labeled with[35S]methionine, and pgC was precipitated with the monoclonal antibody C16 (18). Labeled pgC was analyzed by electrophoresis andfluorography asdescribed in the text. Lanes 1 to 7 contain proteins immunoprecipitated from extractsof cells infectedasfollows. Lane 1,Mock-infected, +PAA; lane 2, tsl8, + PAA; lane 3, ts+, +PAA; lane 4, ts18, -PAA; lane5,
ts+, -PAA, 1/10 of the immunoprecipitated sample shown in lane7; lane 6, mock-infected, -PAA; and lane 7, ts+, -PAA.
ofsynthesis of this protein, as measured by immunoprecipi-tation, were proportional to the amounts ofgC mRNAs in the cytoplasm determined by Northern hybridization (Fig. 2A).
DISCUSSION
We have examined the effect of a defect in the major
DNA-binding protein of HSV-1 on the expression of Y2
mRNAs. When DNA replication is blocked by drugs that
inhibit the viral DNA polymerase, the accumulation ofgC mRNAs is drastically reduced although not totally
elimi-nated. The major DNA-binding protein appears to be in-volved in the negative regulation Of Y2 gene expression because defects of this protein resulted in an increased accumulationofY2mRNAs. We did not detect aninfluence
of viral DNA replication or ICP8 defect on the site of
initiation or in the relative utilization of the multiple splice acceptorsites. Under all conditions examinedin thisstudy, thegC mRNAcan be translated in vivo.
It is very unlikely that the increase in synthesis of Y2
mRNAs in ICP8 mutant-infected cells results from an
in-crease in residual DNA synthesis compared with PAA- or
AraT-treated cellsinfected with thets+ virus. At39.5°C,the mutant is defective in ICP8, an essential function for viral DNAreplication (8, 18, 28, 49). Theviral DNApolymerase appears to be equally sensitive to PAA in these strains because theefficiencyofplatingoftsl8, ts+,or18Rsl virus
I
0 a, a
>1
on November 10, 2019 by guest
http://jvi.asm.org/
at33°Corts+or18Rsl virus at 39.5°C in various concentra-tions of PAA are similar (data not shown). In addition, the increased expression of this
Y2
mRNA is not the result of a difference in the number of DNA molecules that enter the nucleus at 39.5°C. Thus, theincrease in the accumulation of gC mRNAs appears to result from a defect in ICP8.Control of viral late geneexpression. It is well documented thattheaccumulation HSV
Y2
mRNAs is greatlyreduced in the absence ofDNA replication (10, 11, 17, 18). Preliminary experiments suggest that the rate of transcription of the gC gene is strongly inhibited in the absence of viral DNA replication (P. Godowski and D. Knipe, unpublished data). However, theresults reported here suggest that DNA repli-cation is not anabsolute requirement forY2
geneexpression. This is consistent with experiments reported by others. Several investigators have constructed cell lines containing viral or chimeric genes with HSVYl
(9, 41) orY2
(42) promoters stably integrated into the cellular genome. Al-though superinfection of these cell lines with HSV results in transactivation of these genes, amplification of the DNA sequences containing these genes does not appear to be necessary for promoter activity.One model toexplain theregulation of
Y2
geneexpression is asfollows. The transcription ofY2
genes is dependent on the synthesis of (x proteins. However,Y2
genes are tran-scribed only at very low levels from unreplicated genomes. The low level ofY2
transcription would be due to the intrinsically weak activity of these promoters and inhibition byviral and cellular trans-acting factors. Viral DNA replica-tion alters thetemplate structure, resulting in the activation ofY2
promoters. This, along with genome amplification, results in high-level expression ofY2
genes.In the context of this model, ICP8 is involved, either directly or indirectly, in the repression of transcription of these genes. ICP8 can bind to either single- or double-stranded DNA (24, 29, 36), but no sequence specificity has been observed. ICP8 could repress transcription bycoating
double-stranded viral DNA in a nonspecific manner. Al-ternatively, ICP8, perhaps together with additional proteins, recognizes specific features of viral DNA not reproduced in invitro DNA-binding assays, such as chromatin structure.
A second possibility is that ICP8 acts at a post-transcriptional level to control
Y2
mRNA accumulation. In this model,Y2
mRNAs are transcribed from parental genomes but are rapidly degraded. DNA replication would result in genome amplification, high-level transcriptionofY2
genes, and thus, accumulation of detectable levels of
Y2
transcripts. In the context of this model, ICP8 could act directly or indirectly to affect the processing or stability of
viral mRNAs. It has been reported that the functional half-life of y mRNAs is relatively short (20, 50). ICP8 could
affect the processing or stability of these transcripts, either atthe nuclear matrix (37) or in a transient association with cytoplasmic ribonucleoprotein particles (M. Quinlan, Ph.D. thesis, Harvard University, Cambridge, Mass., 1984). De-fects in ICP8 might result in a less rapid turnover ofthese
messages and their subsequent accumulation. During the infection of cells by wild-type virus, ICP8 could bind to progeny DNA molecules and prevent its binding to RNA, resulting in more stable -y mRNAs.
Multifunctional proteins and their role in viral replicative cycles. The simian virus 40 T antigen, the adenovirus 72K protein, and the HSV ICP8 play multifunctional roles intheir
respective viral replicative cycles. These proteins are es-sential for viral DNA replication and function in the
regula-tion of viral gene expression. Elegant studies have shown
that simian virus 40 T antigen
directly regulates
early
geneexpression by
binding
tohigh-affinity
sites near the viral originofreplication (1). Simian virus 40 Tantigen
increases late gene expression directly bybinding
to viral DNA andperhaps indirectly through genome
amplification (23).
Theadenovirus 72K
protein
is essential for adenovirus DNAreplication (46). This
protein
also appears toregulate
theexpressionofearlygenes transcribed from the
EIa, EIb, EII,
and EIII
regions
apparently by
affecting
thestability
ofthe mRNAs(3).Thisproteinspecificallyrepressesthetranscrip-tion of the E4transcriptionunitboth in vivo and in vitro(15, 35). LiketheICP8
protein,
it bindstosingle-stranded
DNA(45).Tothebestofour
knowledge,
sequence-specific
bind-ing
ofDNAby
the adenovirus 72Kprotein
has yet to be demonstrated.The mechanismby
which ICP8 affects HSV geneexpression
is still unclear. Currentdataareconsistentwith ICP8
affecting
thetranscription
of HSVearly
genes,but this has not been demonstrateddirectly (14).
Thisstudy
shows thatICP8lowers the
expression
of HSV Y2 genes fromparental
viral genomes. The results of thisstudy
are alsoconsistentwith the observations of Silver and Roizman
(42)
that ICP8 is not
required
directly
for the activation of Y2 promoters.However,
ICP8mayactindirectly
toincrease Y2 geneexpression by
promoting
viral DNAreplication.
Themechanism
by
which ICP8 affects viral geneexpression
and thepossible significance
of this function in thelytic
and latent life cycles of HSV areundergoing
furtherinvestiga-tion.
ACKNOWLEDGMENTS
Wethank EdWagner,LouHolland,andIgorLemishka for thegift
of recombinant DNA clones and Joe Glorioso for the giftof C16
antibody. We also thankLarry Cohen and David Kimmelman for their assistance withtheanalysisof thegCmRNAstructure,Rhonda
Bassel-Dubyforhelpwith the kinasereactions,andM. L.Levin and Janet Smithfor their assistance.
This workwassupported byPublic HealthServicegrant CA26345 from the National Cancer Institute. D.M.K. was
supported by
a FacultyResearch Awardfrom the American CancerSociety.
P.J.G. is a predoctoral trainee supported by the National Institutes of Healthtraininggrant5T32GM07196.LITERATURE CITED
1. Acheson,N. H.1980.Lytic cycleofSV40andpolyoma virus,p. 125-204. InJ. Tooze (ed.), DNA tumorviruses. Cold
Spring
HarborLaboratory, ColdSpring Harbor, N.Y.
2. Aswell,J.F.,G. P.Allen,A.T.Jamieson,D. E.Campbell,and G. A. Gentry. 1977. Antiviral activityofarabinosylthymine in herpesviral replication: mechanism of action in vivo and in vitro. Antimicrob. AgentsChemother. 12:243-254.
3. Babich, A., and J. R. Nevins. 1981. The stability of early
adenovirusmRNAiscontrolledbytheviral 72 kd
DNA-binding
protein. Cell 26:371-379.
4. Berk,A.J.,andP. A.Sharp.1977. Sizingand
mapping
ofearly
adenovirusmRNAsbygel electrophoresisof S1 endonuclease-digested hybrids. Cell 12:721-732.
5. Buchman, A. R., M. Fromm, and P. Berg. 1984.
Complex
regulation of simian virus 40 early-regiontranscription
from differentoverlapping promoters. Mol. Cell. Biol. 4:1900-1914. 6. Chow, L. T., T. R. Brocker, andJ. B. Lewis. 1979.Complex
splicing patterns ofRNAsfrom theearlyregionsofadenovirus 2. J. Mol. Biol. 134:265-303.
7. Cohen,G.H.,D.Long,and R.J.Eisenberg.1980.
Synthesis
and processingofglycoproteins gDandgC ofherpessimplex
virus type 1. J. Virol. 36:429-439.8. Conley,A.J.,D.M.Knipe,P.C.Jones,andB. Roizman. 1981. Moleculargeneticsofherpessimplex virus. VII. Characteriza-tion of a temperature-sensitive mutant
produced
by in vitromutagenesisand defective in DNA
synthesis
andaccumulationon November 10, 2019 by guest
http://jvi.asm.org/
of ypolypeptides. J. Virol. 37:191-206.
9. Dennis, D., and J. R. Smiley. 1984. Transactivation ofa late
herpes simplex virus promoter. Mol. Cell. Biol. 4:544-551. 10. Frink,R.J.,K. P. Anderson,and E. K. Wagner. 1981. Herpes
simplex virus type 1 HindIll fragment L encodes spliced and
complementary mRNAspecies. J. Virol.39:559-572.
11. Frink,R.J.,R.Eisenberg, G. Cohen, and E. K. Wagner. 1983. Detailedanalysisof theportionof theherpessimplexvirus type 1genome encoding glycoprotein C. J. Virol. 45:634-647. 12. Ghosh, P. K., and P. Lebowitz. 1981. Simian virus 40 early
mRNA's containmultiple5'termini upstream and downstream fromaHogness-Goldberg sequence;ashift in 5' termini during the lytic cycle is mediated by large T antigen. J. Virol. 40:224-240.
13. Gielkens, A. L. J., D. Van Zaane, H. P. Bloemers, and H. Bloemendal. 1976.Synthesisof Rauscher murine leukemia
virus-specific polypeptides in vitro. Proc. Natl. Acad. Sci. U.S.A. 73:356-360.
14. Godowski,P.J.,andD. M.Knipe. 1983. Mutations in the major
DNA-binding proteingene ofherpes simplexvirus type 1 result in increased levels of viral gene expression. J. Virol. 47: 478-486.
15. Handa, H.,R.E.Kingston,and P. A.Sharp. 1983. Inhibition of adenovirusearly region IV transcription in vitro bya purified
viralDNA binding protein. Nature (London) 302:545-547. 16. Hansen, U., D. G. Tenen, D. M. Livingston, and P. A. Sharp.
1981.TantigenrepressionofSV40earlytranscription fromtwo promoters. Cell 27:603-612.
17. Holland, L. E., K. P. Anderson, C. Shipman, Jr., and E. K.
Wagner.1980. ViralDNAsynthesis isrequired for the efficient
expression of specific herpes simplex virus type 1 mRNA
species.
Virology 101:10-24.18. Holland, L. E., R. M. Sandri-Goldin, A. L. Goldin, J. C.
Glorioso, and M. Levine. 1984. Transcriptional and genetic
analysesof theherpes simplexvirus type1genome:coordinates 0.29to0.45. J.Virol. 49:947-959.
19. Holland, T. C., F. L. Homa, S. D. Marlin, M. Levine, and J. Glorioso. 1984. Herpes simplex virus type 1 glycoprotein
C-negative mutants exhibitmultiplephenotypes, including secre-tionof truncatedglycoproteins. J. Virol. 52:566-574.
20. Honess,R.W.,and B.Roizman. 1974.Regulation of herpesvirus macromolecularsynthesis. I.Cascaderegulationof the synthe-sis of three groups of viralproteins.J. Virol. 14:8-19. 21. Hughes, R. G., Jr., and W. H. Munyon. 1975.
Temperature-sensitivemutants ofherpes simplex virus type 1 defective in
lysis butnottransformation.J. Virol. 16:275-283.
22. Jacob,R.J.,and B. Roizman.1977.Anatomy of herpes simplex virus DNA. VIII. Propertiesof thereplicating DNA. J. Virol. 23:394-411.
23. Keller,J. M., andJ. C. Alwine. 1984. Activation of the SV40 late promoter:direct effects of T antigen in the absence of viral DNAreplication. Cell 36:381-389.
24. Knipe, D. M., M. P.Quinlan, and A. E. Spang. 1982. Charac-terization oftwoconformational forms of the major
DNA-bind-ing
protein encoded by herpes simplex virus 1. J. Virol.44:736-741.
25. Knipe,D.M.,W. T.Ruyechan,andB.Roizman. 1979. Molecu-lar genetics of herpes simplex virus. III. Fine mapping of a
genetic locus determining resistance to phosphonoacetate by
twomethodsofmarker transfer. J. Virol.29:698-704. 26. Knipe, D. M.,and A. E. Spang. 1982. Definition of a series of
stages in theassociationof twoherpesviral proteins with the cell nucleus. J. Virol.43:314-324.
27. Laskey,R.A., and A. D. Mills. 1975. Quantitative film detection of3H and 14C inpolyacrylamide gels by fluorography. Eur. J.
Biochem. 56:335-341.
28. Lee, C. K., and D. M. Knipe. 1983. Thermolabile in vivo
DNA-binding activity associated with a protein encoded by mutantsofherpes simplexvirustype 1.J. Virol. 46:909-919. 29. Lee, C. K., and D. M. Knipe. 1985. An immunoassay for the
study of DNA-binding activities ofthe herpes simplex virus
proteinICP8. J.Virol. 54:731-738.
30. Lemischka, I. R., S. Farmer, V. R. Racaniello, and P. A.
Sharp.1981. Nucleotide sequence and evolutionofamammalian cx-tubulin messenger RNA. J. Mol. Biol. 151:101-120. 31. Lohman,P. H. M.,M. L. Sluyter, and I. A. A. Matthijis. 1973.
Repair replication in human cells studied by sodium iodide isopycnic centrifugation ofDNA in a fixed-angle rotor. Anal. Biochem. 54:178-187.
32. Maniatis, T., E. F. Fritsch, andJ. Sambrook. 1982. Molecular cloning: alaboratory manual. Cold Spring HarborLaboratory,
ColdSpring Harbor, N.Y.
33. Maxam, A., andW.Gilbert. 1980.
Sequencing
end-labeledDNA with base specific chemical cleavages. Methods Enzymol. 65:499-559.34. McDonell, M. W., M. N. Simon, and F. W. Studier. 1977. Analysisofrestriction fragments ofT7DNAanddetermination of molecularweights by electrophoresis in neutral and alkaline gels. J. Mol. Biol. 110:119-146.
35. Nevins, J. R., and J. J. Winkler. 1980. Regulation of early
adenovirus transcription: a protein product of early region 2 specifically represses region4 transcription. Proc. Natl. Acad. Sci. U.S.A. 77:1893-1897.
36. Powell, K.L., D. J. M.Purifoy, and R. J. Courtney. 1975. The synthesis of herpes simplex virus proteins in the absence of virus DNA synthesis. Biochem. Biophys. Res. Commun. 66:262-271.
37. Quinlan, M. P., and D. M. Knipe. 1983. Nuclearlocalizationof herpesvirus proteins: potential role for the cellular framework. Mol. Cell. Biol. 3:315-324.
38. Rafield, L. F., andD. M. Knipe. 1984. Characterization ofthe major mRNAstranscribedfrom the genes of glycoprotein B and DNA-binding protein ICP8 of herpes simplex virus type 1. J. Virol. 49:960-969.
39. Rigby, P. W. J., M. Dieckmann, C. Rhodes, and P. Berg. 1977. Labeling deoxyribonucleic acid to high specific activity in vitro by nick translation with DNA polymerase 1. J. Mol. Biol. 113:237-251.
40. Roizman, B., and W. Batterson. 1985. Herpesviruses and their replication, 497-526. In B. Fields (ed.), Virology. Raven Press, New York.
41. Sandri-Goldin, R. M., A. L. Goldin, L. E. Holland, J. C. Glorioso, and M. Levine. 1983. Expression of herpes simplex virus , and fy genes integrated in mammalian cells and their induction by an oxgene product. Mol. Cell. Biol. 3:2028-2044. 42. Silver, S., and B. Roizman. 1985.y2-Thymidine kinase chimeras
are identically transcribed but regulated as Y2 genes in herpes simplex virus genomes and as ,B genes in cell genomes. Mol. Cell. Biol. 5:518-528.
43. Spear, P. G. 1976. Membrane proteins specified by herpes simplex viruses.I. Identification of four glycoprotein precursors and their products in type 1-infected cells. J. Virol. 17:991-1008. 44. Spear, P., and B. Roizman. 1980. Herpes simplex virus, p. 615-647. In J. Tooze (ed.), DNA tumor viruses. Cold Spring Harbor Laboratory, ColdSpring Harbor, N.Y.
45. Sugawara, K., Z. Gilead, and M. Greene. 1977. Purification and molecular characterization of adenovirus type 2 DNA-binding protein. J. Virol. 21:338-346.
46. van derVliet,P.C., and J.S. Sussenbach. 1975. Anadenovirus type 5 gene function required for initiation of viral DNA replication. Virology 67:415-426.
47. Wagner, E. 1983. Transcription patterns in HSV infections, p. 239-270. In G. Klein (ed.), Advances in viral oncology, vol. 3. Raven Press, New York.
48. Watson, R. J., and J. B. Clements. 1980.Aherpes simplex virus type 1 function continuously required for early and latevirus RNA synthesis. Nature (London) 285:329-330.
49. Weller,S. K., K. J. Lee, D. J.Sabourin, and P. A. Schaffer. 1982. Genetic analysis of temperature-sensitive mutants which define the gene for the major herpes simplex virus type 1 DNA-binding protein. J. Virol.45:354-366.
50. Wolf, H., and B. Roizman. 1977. The regulation ofstructural polypeptide synthesis in herpes simplex virus type 1 and 2 infected cells, p. 327. In G. De The, W. Henle, and F. Rapp (ed.),Oncogenesis and herpesviruses III. International Agency forResearch on Cancer, Lyon.

![FIG. 4.centimeters)Panels:[3H]thymidine-labeled DNA replication in PAA-treated (+PAA) or untreated (-PAA) cells](https://thumb-us.123doks.com/thumbv2/123dok_us/1400973.93119/5.612.159.458.68.460/centimeters-panels-thymidine-labeled-replication-treated-untreated-cells.webp)


