0022-538X/90/020856-08$02.00/0
CopyrightC) 1990, AmericanSociety for Microbiology
Characterization
of the
Antigenic
Structure of
Herpes Simplex
Virus
Type
1
Glycoprotein C through DNA Sequence Analysis
of
Monoclonal
Antibody-Resistant
Mutants
CHIN-TUAN BROCADEWU,' MYRON
LEVINE,'
FREDHOMA,1t
STEVEN L. HIGHLANDER,2 ANDJOSEPH C. GLORIOSO2,3t*Departmentof Human
Genetics,'
UnitforLaboratoryAnimalMedicine,2 and Department of Microbiology and Immunology,3 The University of Michigan Medical School, Ann Arbor, Michigan 48109Received1August1989/Accepted 10 October1989
Earlier studiesofagroup of monoclonal antibody-resistant(mar) mutantsofherpes simplexvirustype 1 glycoprotein C (gC) operationally defined two distinct antigenic sites on this molecule, each consisting of numerousoverlappingepitopes.Inthisreport,wefurther defineepitopesofgC bysequenceanalysisof the mar mutant gC genes. In 18 mar mutants studied, the marphenotype was associated with a single nucleotide
substitution andasingle predicted aminoacidchange.The mutations were localized to tworegions within the
codingsequenceof the externaldomain ofgC and correlated with the twopreviouslydefined antigenicsites.
The predicted amino acid substitutions of site I mutants resided between residues Gln-307 and Pro-373,
whereas those of site II mutants occurred between amino acids Arg-129 and Glu-247. Of the 12 site II
mutations,9 inducedamino acid substitutionswithin anarginine-richsegment of 8 amino acidsextendingfrom residues143to151.Theclusteringofthemajority of substituted residuessuggeststhattheycontribute to the structureof theaffected sites. Moreover,the patternsof substitutionswhich affectedrecognitionbyantibodies
with similar epitope specificities provided evidence thatepitope structures arephysicallylinked and overlap
withinantigenicsites. Of thenine epitopes definedonthebasis ofmutations, threewerelocatedwithin siteI
andsix were locatedwithin siteIl. Substituted residuesaffectingthesite Iepitopesdidnotoverlapsubstituted
residues of siteII, supporting ourearlier conclusion that sites I and IIreside inspatially distinct antigenic domains. Acomputeranalysis ofthedistribution ofcharged residues and thepredicted secondarystructural
features ofwild-type gC revealed thatthe twoantigenic sites residewithin themosthydrophilic regionsof the molecule and that theantigenic residuesarelikelytobeorganizedas
0
sheetswhichloopoutfromthe surfaceofthemolecule.Together, these data and ourprevious studiessupporttheconclusion that themarmutations
identified bysequenceanalysis verylikely occurwithinor neartheepitopestructuresthemselves. Thus,two
highly antigenic regions of gC havenowbeenphysically and geneticallymappedtowell-defined domains of the proteinmolecule.
Thereare at least seven virally encoded envelope
glyco-proteins of herpes simplex virus (HSV) (1, 4, 25, 34)which
are alsopresentin the membranes of infectedcells.
Glyco-protein C (gC) isoneof themostimmunogenic glycoproteins andrepresents a majortarget antigen for antiviral immune
responses. gC elicits high-titer complement-dependent
neu-tralizing antibodies (5, 12, 31) and is a majortarget antigen
for cytotoxic T lymphocytes (6, 10, 24, 32). gC is also a
serotype-specific antigen, since the majority of antibodies
prepared against gC encoded by HSV type 1 (HSV-1 gC-1)
donotcross-reactwith gC encoded by HSV-2 (gC-2)(11). gC isnotessential for the production of active virusparticles in
cell cultures (35), althoughrecent evidence suggests that it may play a role in virus attachment (D. Wudunn and P.
Spear, personal communication). gCalso has the ability to
bind the C3b complement component (7), which can inter-fere with the alternate pathway for complement activation
(8) and thus inhibit both complement-mediated cytolysis of
HSV-infected cells and complement-dependent virus
neu-* Correspondingauthor.
tPresentaddress:
Upjohn
ResearchLaboratory,Kalamazoo,MI 49001.tPresent address: Department ofMicrobiology, Biochemistry, and Molecular Biology, University of Pittsburgh Medical School, 720Scaife Hall, Pittsburgh,PA 15261.
tralization (29, 30). These findings suggest that gC plays a role in viral pathogenesis in human hosts, although animal studies have notprovided consistent evidence fora direct role forgC in neuropathogenesis (23; 36).
Because of the important role of gC in type-specific antiviral immunity, studies to define its antigenic structure
by using genetic, immunologic, andbiochemical techniques were undertaken. Our approach was to generate a large panel of monoclonal antibodies which were used to selecta
library of neutralization escape mutants referred to as mono-clonalantibody-resistant,ormar,mutantsof gC (17). Marlin
etal.(28) described the neutralization resistancepatternsof 22 mar C mutants when tested against a panel of 30
gC-specific monoclonal antibodies. Nine gC epitopes were
op-erationally defined on the basis of the unique reactivity patterns observed. These epitopes were placed into two
groups, or antigenic sites, each composed of distinct but antigenically related epitopes. Site I consisted of three
epitopes, and site II contained six epitopes. The sites were
shown to betopographically distinct by the finding thatonly
antibodies which recognized the same antigenic site were
mutually competitive for antigen binding in antibody
com-petition assays. The physical locations of the two antigenic sites were approximated by analysis of the immunoprecipi-tation patternsof site I- and siteII-specific antibodies tested against a series of carboxy-terminal truncated gC
polypep-856
on November 10, 2019 by guest
http://jvi.asm.org/
L S~~~~~
(A,
p~~~~~~~~~~~
a.~~~~~~~~~~~~~~~~~~~~~~~~~~~~~c a Z
gC mRNA
ALG
Site
IIsite
I490
FH60
821
FH60-ESt65
FH60-St80 1407
1318_BW61 1739
BW75
821 1457 FH60-St7O
1494 2180
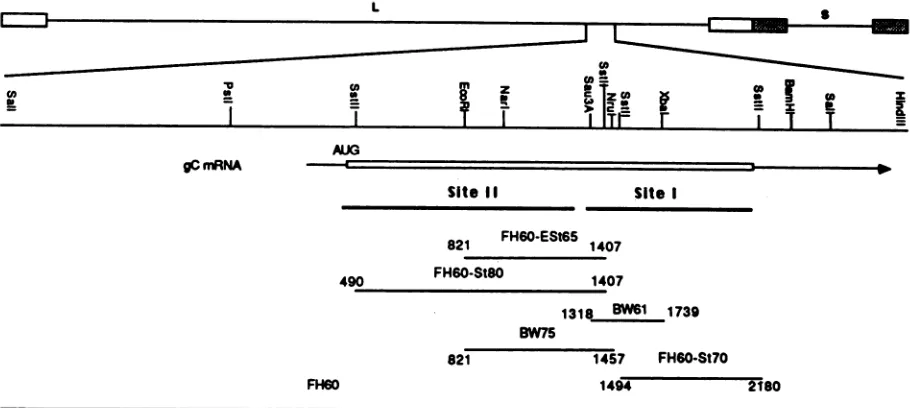
FIG. 1. HSV DNA fragments used for marker rescue of mar mutations. The HSV-1 genome is at the top. A SalI-HindIll fragment containing thecomplete gC gene is shown with restriction sites. Symbols: El,inverted repeats flanking the long unique element (L); 1 inverted repeats flanking the short unique element (S); -_, region coding for the gC message; riui, protein coding region with the approximate location of thetranslation start site; -, regions covered by the fragments used in the marker rescue experiments (the nucleotides encompassed by these fragments relative to the start of transcription areindicated); -, domains for antigenic sites I andII as predicted by Homa et al. (21).
tidessynthesized on infection with mutant viruses shown to
carrychain-terminating mutations. That study, in
combina-tion withbiochemical and genetic analyses of these
chain-terminating mutants, mapped site I between amino acid
residues298and 481 and siteIINterminal of residue 274 (16,
22, 28).
In this study, the mar C mutations of 18 mutants were localized within theirrespectivegC genes by marker rescue, and the appropriate regions weresequencedto identify the
specific base pair substitutions. By identifying the genetic
alterations which affected the antigenic activities of the
mutantgCmolecules, the structure and organization of the
gC epitopes of sites I and II were preciselymapped to two
well-separated regions within the external domain of gC
whicharepredicted to contain hydrophilic
p-turn
structures.These structures appeartorepresentthe mostimmunogenic
componentsof the gC molecule of HSV-1.
MATERIALS ANDMETHODS
Cells, viruses, and monoclonal antibodies. African green
monkeykidney (Vero) cellsgrowninEagle minimum
essen-tial medium supplemented with nonessential amino acids,
100 mgofstreptomycin per ml, 100 U ofpenicillin perml,
and5%fetal calfserum wereusedinallexperiments.HSV-1
KOS-321 andthemarCmutants wereisolated and
charac-terized as described previously (16, 21, 28). Virus stocks
were prepared by infection at a low multiplicity in Vero
cells. The monoclonal antibodies used in this
study
weredescribedpreviously byourlaboratory (16, 17, 28).The gC
gene nucleotide sequence was reported
previously
(9, 21).ThenumberingofnucleotidesinthegCgene
begins
with themessage capsite. Theinitiationcodon for translation of the
gCmessageisatnucleotide (nt)381. Aminoacid
numbering
starts with the first residue of the signal sequence in thenascent protein. The nomenclatures for
gC-specific
mono-clonal antibodies andmarCmutantswere thoseofHollandetal.
(17).
Marker rescue and DNA sequencing. Viral DNA was
prepared by the method ofGoldin et al. (13). Large-scale
preparation ofplasmid DNA was done by the method of
Birnboim and Doly (2) as modified by Maniatis et al. (26).
Minipreparations of plasmid DNA were isolated by the
boiling
methodofHolmes andQuigley(19), asdescribed byManiatis et al. (26). The calcium phosphate precipitation
protocol describedby Homaetal.(20) wasadaptedfrom the
method ofGraham and vander Eb (14)for use in
cotrans-fectionmarkerrescueexperiments. Linearized DNAofthe
rescue plasmids was individually cotransfected into Vero
cells with viral DNA extracted from the mar C mutants.
Progeny virusfromthecotransfectionswereplatedonVero
cells forplaque formationandscreened bythe
immunoper-oxidaseblack-plaqueassaydescribed by Hollandetal.(18).
Blackplaquescontainrecombinantvirus inwhichwild-type
antigenic activity has been restored, indicating
positive
rescue. The fragments of the wild-type gene used in the marker rescue experiments to localize the mar mutations
within thegCgenearedepictedin Fig. 1. Therecombinant
plasmids
pFH60-St8O,
pFH60-ESt65,pFH60-St7O,
andpFH60, whichcontaindifferent segments of thewild-type
gC
sequenceinsertedinpBR322,wereconstructedasdescribed byHollandetal. (16).pFH60-St80carriesanSstII
fragment
(nt 490to 1407) whichencompassesthe 5'
coding
sequenceofthegCgene.
pFH60-ESt65
carries an EcoRI-SstIIpiece
(nt 821to 1407) which spans the middle
region
ofthe gene,and
pFH60-St70
containsanSstIIfragment (nt
1494to2180)
at the 3' end of the gCcoding
sequence.pFH60
has a3.6-kilobase SalI fragmentinsert (map coordinates0.620 to
0.645) whichincludes the full
coding
sequenceof thegC
geneas well as some
5'-flanking
sequences. ThepBW
plasmids
were constructed by inserting
pieces
of thewild-type
gC
geneinto M13 vectors.
pBW61
hasaSau3A-XbaIfragment
(nt 1318to1739)in theAccI-XbaI sites of M13
mpl8,
whilepBW75 containsanEcoRI-NruI insert(nt821to
1457)
in theM13 mp8 vector.
pBW75
covers the middleregion
ofthe [image:2.612.80.535.75.279.2]L
... 8
on November 10, 2019 by guest
http://jvi.asm.org/
TABLE 1. Results of marker rescueexperiments as percentages of blackplaques'
Mutant % Black plaques from rescue fragments:
DNA
FH60-St8O
FH60-ESt65 BW75 BW61FH60-St7O
FH60 NoneSite II
C16.1 1.7 0 0
C9.6 2.0 0 0
C13.1 2.0 0 0
C10.3 3.5 0 0 0
C13.2 3.5 0 0
C7.1 2.0 0 0 1.2 0
C9.1 2.2 0 0 1.0 0
C16.2 1.4 0 0 0
C17.2 0.2 0 0
C17.3 4.0 0 0
C3.1 0.5 0 0 0
C1o.1 0.6 0 2.0 0
SiteI
C11.1 1.8 0 0
C14.1 0.01 0 2.8 0
C15.1 0.1 0 6.3 0
C2.1 0 0.5 0
C4.4 0 0 1.8 0
C15.4 0 0 0 0
a The recombination frequency is expressed as thepercentage of black recombinant plaques in thetotal number ofplaques. Ifnovalue isgiven, markerrescue wasnotattempted.
codingsequence andoverlaps withthepFH60-St80 fragment
on its 3' side. The pBW61 fragment bridges the regions
coveredby pFH60-St80 andpFH60-St7O.
Forsequencing, DNA wasextracted frommutant
virions,
and thegC coding sequencefrom eachmutant was purified
as a 3.6-kilobase SalI fragment (map coordinates 0.620 to
0.645), extractedby electroelution froman0.8%agarosegel,
and cloned intopBR322. Recombinant clonescontainingthe
gC sequences were cut further with different restriction
enzymes to yield fragments of appropriate sizes forDNA
sequencing. Fragments covering the whole gC protein
cod-ing regionweregel purified and subcloned intoM13 vectors.
Thesequencingwasperformedasdescribed by Sangeretal.
(33).Thenucleotide mix and primer kitwerepurchased from
Pharmacia, Inc. (Piscataway, N.J.).
[a-355]dATP
wasob-tained from Dupont, NEN Research Products (Boston,
Mass.).
RESULTS
Physical mapping of mar mutations. The physical location
ofeach marC mutationwasdeterminedby marker rescue of
the mutant phenotype with cloned wild-type sequences
spanning differentparts of the gC gene (Fig. 1). The
appear-ance ofblack plaques among the plated progeny indicated thatrecombination had occurred between the plasmid DNA
and the viral chromosome to rescue the mutation to the
wild-typesequence. The results of all marker rescue
exper-iments are shown in Table 1.
With theexceptionof mar C15.4, each mutant tested was rescuedby pFH60.This DNA fragment contained sequences
covering
theentire gC codingsequence. Physical mapping of the mar C15.4 mutation was not possible, because thereaction of the mutant gC with the C15 antibody was not
easily
distinguishable from that of wild-type gC in theblack-plaqueassay. For this mutant, it was assumed that the
alterationresided inaregion ofthe gC gene where mutations
affecting
similarepitopes
weremapped,
aprediction
con-firmed
by
sequencing (see below).
The mutations of all site IImutants, except the untested mar
C10.1,
were rescuedby
pFH60-St80.
ThepFH60-St80 fragment
coversthe 5'part
of thegC-1
coding
sequence between nt 490 and 1407 andaccordingly mapped
the mutations to thatregion.
ThemarC10.1
mutation, however,
wasrescuedby plasmid pBW75,
which
mapped
it within themiddleregion
of thegene. This mutation alsowasrescuedby pFH60
butnotpBW61,
which ruledouttheregion
from nt1318to1457 and narrowed thelocation ofthe mutation to between nt 821 and 1318.
At-temptswere made to rescuemany of the site II mutations,
including
thoseofmarC16.1, C16.2, C9.6, C13.1, C13.2,
andC17.3,
withpFH60-St7O.
Asexpected,
no blackplaques
wereobserved,
since thepFH60-St70 wild-type fragment
coversthe 3'part of the gene, which is distal to thepredicted
location of site IIinthe 5' end of the gene. In
addition,
themutationsof site II marmutants
C10.3, C7.1, C9.1,
C16.2,C17.2,
and C3.1 were notrescuedby pFH60-ESt65.
Since thepFH60-ESt65 fragment
wascontained in thepFH60-St8O
fragment,
the lack ofrescue withpFH60-ESt65 suggested
that thesemarmutationsliewithin the
region
fromnt490to821 andnotin the downstream
region
fromnt 821 to1407,
which is covered
by
both thepFH60-St80
and thepFH60-ESt65
fragments.
The mutations in mutants marC7.1 andmarC9.1were notrescuedwith
pBW61;
thiswasconsistentwith our
hypothesis.
The mutations in site I mutants mar
C11.1, C14.1,
andC15.1wererescued
by pBW75,
whichmapped
the mutationstothe
fragment
fromnt821to1457. Twoofthese mutationswere notrescued
by pBW61,
however,
making
itlikely
thatthey
were within the segment from nt 821 to 1318. Themutationin marC14.1 was rescued
by pBW75,
albeit atalow
frequency,
but notby pFH60-St70,
suggesting
that themutationwaswithinthe
region
fromnt821to1457,
which iscovered
by
thepBW75
fragment.
In contrast toother site Imutations,
the mar C2.1 mutation was localized to the segment inthe 3'endofthegene(nt
1318to1739) by
rescuewith
plasmid
pBW61.
The mutation was not rescuedby
pBW75, however,
whichsuggested
that themutationmight
liein the smallerregion,
fromnt1457to1739.ThemutationinmarC4.4wasrescued
only
withpFH60
andtherefore
wasnot
mapped
toa smallerregion
of thegC
gene. Since mar C4.4 has the samereactivity
patternagainst
gC-specific
monoclonalantibodiesas marC2.1
does,
this mutationwasthought
likely
to reside in the same area as the mar C2.1mutation.
Thelowrescue
frequency
of themarC14.1andmarC15.1mutations with
pBW75
may beattributedtothelocations ofthe mutations relative to the ends of the
fragment.
Asrevealed
by
thesequencing data,
the sites ofthemarC14.1and C15.1mutationsare
only
about 150ntfrom the 3' limitof the EcoRI-NruI
fragment,
which may result inareducedfrequency
of recombination.Supporting
thisargumentis thefinding
that both mutations were rescued withhigh
fre-quency
by
the 3.6-kilobase Sallfragment
inpFH60.Simi-larly,
the small size ofthe Sau3A-XbaI fragment used inmapping
themarC4.4mutationmayexplainrescuefailure.Of 18
mutations,
16 were rescuedby
afragment
corre-sponding
toasmallerregion
ofthegCgene. The mutationsin the mar mutant viruses selected
by
site II monoclonalantibodies were
generally
localized to the 5' part of theprotein coding region
of the gene, whilemutations in site I mar mutant viruses weremapped
to the 3'region.
The resultsofthephysical mapping
confirm ourprevious findings(28)
that the locations of theepitopes
definedby
site IIon November 10, 2019 by guest
http://jvi.asm.org/
EoRl Nal
2:
los
g
Sit.II
SmuSA NnX
I
I-Sit I C16.1
C13.1 C9.6
CIO.3
C13.2
C9.1
C7.1
C16.2
C17.2
C17.3
C3.1 c10.1 c1 1.1
C14.1
c15.1 C2.1 C4.4
CIS.4
765 C-_ T
6060-,W A
6060--lo A
614 -- A
614 a-*A
610 C- T
6109 C- T 619 C-* T
figC- T
632 0 * A 906 -o A
1527C- T 1527C-T
1120 -_ A
1 1300A-*0
1300 A-0o
1315
Iasa
0 * A1400 C-4 A 140 C- A
140 C- A
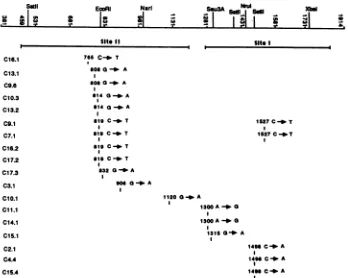
FIG. 2. Nucleotide changes in the mar C mutants. A partialrestrictionmapof the protein coding region of the gC gene and the numbering of the nucleotidesin the region areshown at the top. The thick solid lines below the map denote the domains for sites I andIIas predicted byHoma et al. (21). The position of each mutation, as well as the nucleotide change, is indicated.
monoclonalantibodies are toward the amino-terminal end of
the protein molecule, whereas those of site I epitopes are
nearerthe carboxy terminus of the molecule.
Nucleotide changes in mar mutants. The nucleotide
substi-tutions in marC mutant DNA were identified by sequence
analysis of the respective gC genes. The Sall fragment
containing the gC gene was purified from each mar mutant
and was subcloned into M13 for DNA sequencing. The
correspondingwild-type gC gene fragments were sequenced
in parallel to ensure the accuracy of sequence reading.
Selected regions outside those indicated by marker rescue
weresequenced to determine whether other mutations might
also bepresentinthe mar mutant genes. In the case of mar
C4.4and marC15.4, for which the physical mapping data
were inconclusive,the3' regionwas sequenced from nt 821
to2180, where the site I mutations are most likely toreside.
Nucleotide changes were found in all the mar mutants
(Fig. 2). The locations of the base changes confirmed the
predictions of the physical mapping. In mutants that had
been rescuedsuccessfully,thebasechangesweresituated in
theregions implicated bybothpositive andnegativerescue
results. Since all of these base changes were the only
mutations found within the region identified by marker
rescuefor each mutant, weconcluded that these mutations altered the antigenic properties of the gC mar mutants. In
twomutants,marC7.1 andC9.1,twomutationswerefound
(Fig. 2). The base change in both mutants at nt 1527 was
outside the region implicated by marker rescue data and
awayfrom thepositions of the restof the site II mutations.
Thisindicates that thechangeat nt1527wasunrelatedtothe
mar phenotypes of the two mutants. As
previously
indi-cated,markerrescuefailedtolocalize themarC4.4 andmar
C15.4 mutationsto asmaller
region
ofthegC
gene.Thebasechanges identified within the sequenced portion of the gC geneof thesemarmutants werewithin thepredicted domain
forantigenic site I. Althoughthesechanges are likely to be
the cause of the mar phenotypes of the two mutants, the
possibility that additional base changes elsewhere in the
geneexistandcontribute to the antigenic alterations cannot be ruled out until these mutant gC genes are completely
sequenced.
Thenucleotide changes of all site II mutants, except for
marC10.1,aresituated ina150-base-pairsegment(nt 765 to
906)toward the 5'end ofthegCopenreading frame. Nine of thesechanges cluster withina24-base-pair region. In
agree-mentwith thephysical mapping data, the marC10.1
muta-tion resides in the middle part of the gene, downstream from the othersiteII mutations.The siteI mutations are distrib-uted withina200-base-pairsegmenttoward the 3' end of the
coding sequence. Among the 18 base changes identified
which affectantigenicity,those ofmarC2.1,C4.4,and C15.4
aretransversions, whichchange pyrimidinestopurines.The
rest aretransitions. MutantsmarC9.6,C9.1, C10.3, C11.1, and C14.1 were selected from virus stocks that had been treated with5-bromodeoxyuridine, which
specifically
causestransition mutations(16).Our resultsareconsistent with the presence of this expected class of mutation and correlate
well with the presenceof transition mutations in sixgBmar mutants(15, 27).
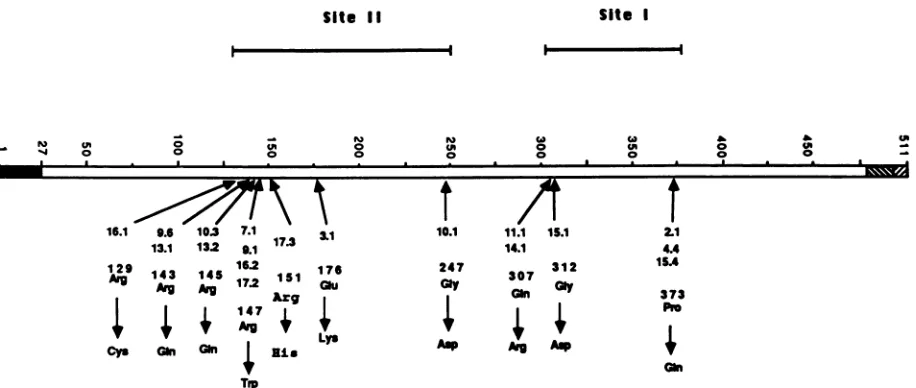
Amino acid substitutions. Theamino acid substitutions in the gC mutant molecules (Fig. 3) were predicted from the
indicated nucleotidechangesin themarC genes(Fig. 2).All the mutations induced amino acid substitutions. Of the
changes at the 10 mutated sites which caused antigenic
changes, six affected charged residues and nine amino acid
substitutions resulted in charge differences (either gain or Satil
C'
.!a 0 .
ON'.
aWI 7-1
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.137.485.69.347.2]Site I I Site I
I
-. 0 0 fU"o 0a (Ata 0a cii0 00 CR0
16.1 9.6 10.3 7.1
.I1
13.1 132 0.1 173
129 13 1516.2 176
Arg 143 145 17.2 151 Gi
X
~~~~~arg
LYCY GI Gin Ri
Tip
Io. 11.. Is.1 14.1
247 307 312
SP Gin Gt
IAm g Am
FIG. 3. Predicted amino acid substitutionsin themarCmutants.A schematicof thegCmolecule isshown. The numbers representthe
amino acid residuesin the sequence, with 1as the initiation methionine. Symbols: _, signalsequence of theprotein; =,extracellular
domain; ,,transmembrane region;0,intracellular domain. Shown above themaparethepredicted regionsfor sites I and IIasdetermined
by the datapresented in thisreport.Thepositions of the affected residues andtheidentities of the amino acid substitutions areindicated.
loss of charge). Charged residues are more likely to be surface exposedand arehence more accessibleto
antibod-ies. The large number of charged amino acids among the residuesidentifiedby thesequenceanalysis ofmarmutants is consistent with the role of these residues in theantigenic structureofgC. FiveArgresidueswerealtered in siteII,and
four were clustered within an 8-amino-acid segment. How these residues are involved in the antigenic activity of gC
andhowthesubstitutions affectthephysicalstructureof the
proteinarediscussed below. None of the amino acid substi-tutionsoccurred within thepredicted asparagine-linked gly-cosylation sites.
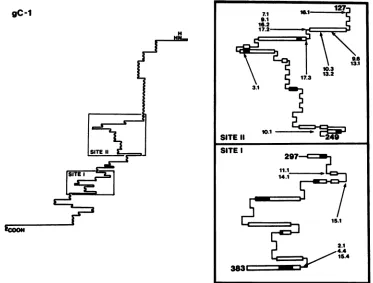
Computer analysesofpredicted structural features ofgC. Computer analyses were performedto assess theantigenic potential of the regions of gC identified in our studies. Candidate structures for potential antigenicity are n-turn structures flankedby P sheetsora-helicalregionswithhigh probabilities for flexibility and surface exposure (37, 38). Computer analysis ofthe primary sequenceofgC with the MSEQprogram (3) showed thatmost ofthe residues iden-tifiedasimportanttothe antigenicstructureresideator near oneof thetwo mosthydrophilic structuresin the molecule,
residues 120to 150 and300to 310. The amino acid
substi-tutions in the mar mutants often resulted in increases in hydrophobicity at the mutated positions (data notshown).
Thesecondarystructurepredictions for the gC moleculeand for sites I and II are shown in Fig. 4. There are several potential loop structures in both regions which feature a turnsflanked by a sheetsora-helicalstructures.Mostof the
substituted residues identified in the mar mutants are
situ-ated inthese potential loops. The localization of mutations within these loop structures supports the proposed role of sites I andIIasmajor antigenicdomains of gC.
DISCUSSION
Marker rescue and DNA sequencing data for the 18 mar
mutants presented in this report extend the analysis of the
antigenic structure of gC. Mutations in site II marmutants
wererescued withDNAfragmentsspanning the5'endof the
gCgene,physically mappingthemutationstothatregion of
the codingsequence. Similarly, mutations in site I mutants werephysically mappedtothe3' codingsequenceof thegC
gene. For the 16 mutations that were marker rescued to a
subfragmentof the gC codingsequence, asingle nucleotide substitutionwasdetected in theregioncorrespondingto the rescuing fragment(s).All siteII mutationswerelocated 5'to nt 1121, and all site I mutationswerelocated 3' to nt 1299. Allmutations werepredicted tocauseamino acid substitu-tions in the external domain of thegCmolecule. The amino acids affected by site I mutations spanned residues 307 to 373, while those affected by site II mutations spanned
residues129to247.Thus,themarkerrescueandsequencing
data confirm our earlier findings of two nonoverlapping antigenicdomainsongC (16, 21, 28)and further definetheir dimensions(Fig. 3 and4).
Fivemonoclonal antibodies wereusedtodefineantigenic
site I. Twoantibodies,C2 andC4, probablyrepresent sister
clones, since they were isolated in the same hybridoma
fusionexperiment, showed similarisoelectricfocusing
pro-files (17), and were indistinguishable by their reactivities withthe panel ofmarmutants(28).Themutations carried by
the mar C2.1 and mar C4.4 mutants selected by these
monoclonal antibodies both contained substitutions ofGln for Pro at residue 373. We conclude that monoclonal anti-bodies C2 and C4recognizethesamesite Iepitope, EpC2/4. The remaining site I antibodies were unique in their
isoelectric focusing patterns. Monoclonal antibodies Cll and C14 appear to recognize the same epitope, since mar C11.1 andmarC14.1 carriedidenticalArg-for-Gln
substitu-tions at residue 307 and exhibited identical patterns of
neutralization with thepanel of monoclonalantibodies (28).
The analysis of additional mar mutants selected by these
antibodies could provide evidence that these antibodies recognizedifferent butoverlapping epitopes.Forthis discus-sion, however,wewillconsiderthatantibodies Cll and C14 recognizea single epitope, EpC11/14.
Monoclonal antibody C15 interacts with an epitope, EpC15, defined by residues Gly-312 and Pro-373. The
sub-stitution inmarC15.1atresidue312 isclosetotheEpC11/14 at residue 307. In addition, the mar C15.4 alteration at
UN
21
4.4
ISA
373 Pr
Gin
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.90.548.71.265.2]gC-1
FIG. 4. Secondary structures of sites I andII in gC. A cartoon of the gC-1 molecule as drawn by the computer protein analysis program MSEQ (3) is shown. The boxed regions labeled SITE II (residues 127 to 249) and SITE I (residues 297 to 383) depict the predicted secondary structures of sitesII andI,respectively, and show the locations and identities of the mutations within each region. Symbols: _,ahelix;
=, p sheet;
-,
randomcoil;], 3 turn.position373 is identicalto the EpC2/4 alteration. However,
in immunoprecipitation studies with truncated gC
polypep-tides, monoclonal antibody C15, along with Cll and C14,
immunoprecipitated a protein which terminated at residue
359 but not one carrying frame shift-induced substitutions
carboxy terminaltoresidue297 (28). In contrast, antibodies
C2 and C4 failed to precipitate either polypeptide but did
precipitate another gCpolypeptide carrying a frame shift at
residue 480, a finding consistent with the location of a
substitution atresidue 373.This suggests thatEpC15 exists
entirely within the first 359 amino acids of gC and that
residue 373 is not required for C15 recognition of gC.
Therefore, it is likely that the residue change seen in mar
C15.4 results in the steric or electrochemical inhibition of
antibody bindingat amoreamino-terminallocation,
presum-ablyator nearresidue 312. We conclude that antigenic site
I ofgC consists of at least three epitopes, clustered in a
restrictedregion of thecarboxy-terminal half ofthe external
domain ofthemolecule.
EpC11/14
andEpC15
are likelytobeoverlapping, while
EpC2/C4
liesnearby. These antibodiesaremutually competitive forbindingto the intact gC
mole-cule(28),and the mar mutantreactivity
patterns
withtheseantibodies also indicate that the epitopesare
closely
related(28). Thus,itmightbespeculatedthat residues307,312,and
373 arebrought into close promixity by the
folding
ofgC,
although theimmunoprecipitation data described above
in-dicate that the stabilities ofEpC11/14 and EpC15 are not
strictly dependenton thisfolding. Nevertheless, alterations
in residue 373 can disrupt EpC15 and
EpC2/4
but notEpC11/14, suggestingthatthe integrity ofepitopes in siteI
relies on distinct butlocalizedstructureswithinthis segment of thegCmolecule.
Seven monoclonal antibodies were used to define the
epitopes ofantigenic site II. Exceptfor C9 and C17,which
had similarisolectric focusing patterns, the other
monoclo-nal antibodies had unique banding patterns and represent
distinct clonotypes(26). Thus far, all theepitopes in siteII
can be distinguished from one another by the residues
identified in the mar mutants; these residues are likely to
represent contact points for the monoclonal antibodies
which recognize overlapping site II epitopes. This
conclu-sion is consistent with the distinct but similar reactivity
patternsof the antibodies with site IImar mutants (28). For
instance, while EpC9 and EpC17 share Arg-147 (mar C9.1
and C17.2) as an antibody contact point, they are
distin-guishablebyasecondcomponent residue in eachepitopeas
derived from the sequence analysis of mar mutants C9.6
(Arg-143)andC17.3 (Arg-151).
A striking feature of site II is the clustering of altered
residueswithinasegment ofeightamino acids(residues143
to151). Fourof these aminoacids arecharged
arginines,
allof which were substituted in different mar mutations
af-fecting thissegment.Ofthe 12siteIImutations
sequenced
inthis study, 9 induce amino acid substitutions within this
arginine-rich segmentofthe gC molecule. Six of the seven
monoclonal antibodies recognize the arginines in this
8-amino-acid segment as components of their
epitopes,
sug-gestingthepresence ofmultiple,
overlapping
epitopes. Fourmonoclonalantibodies, C7, C9,C16,and
C17,
overlapped
intheir
recognition
ofArg-147.
Twoantibodies,
C9 andC13,
recognizedArg-143; anothertwo, C10and
C13,
overlapped
at Arg-145. Two of the
remaining
mar mutations affectedresidues outside of the 8-amino-acid segment, but these
changes were selected
by
antibodies which also selecton November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.125.498.74.357.2]changes
within thesegment.
EpC16
ischaracterizedby
marC16.1,
which is altered atArg-129,
andby
the marC16.2mutation at
Arg-147.
EpC1O
is characterizedby
the mar C10.1mutationatGly-247
andby
themarC10.3 mutation atArg-145.
BothEpC16
andEpC1O
appear to beconforma-tional
epitopes, composed
ofmoredistant amino acids.Thelast site II marmutant, mar
C3.1,
characterizesEpC3
withGlu-176 as a
binding
site. Additional mutants selectedby
monoclonal
antibody
C3 shouldbe studiedtofurther definethis
epitope.
It ispossible
thatanassociationof thisepitope
with the8-amino-acid
segment
wouldthenbedemonstrated. On the basis of thesefindings,
we conclude that the8-amino-acid
segment
is theantigenic
coreof site IIonthegC
molecule.
Computer analysis
of most mar C mutations failed toproduce
noticeable effectsonthepredicted
secondary
struc-tureof
gC.
Thiswasalsotrueformostmarmutations inthegB
glycoprotein
(S.
Highlander,
Ph.D.disseration,
Univer-sity
ofMichigan,
AnnArbor, 1988).
In these cases, it islikely
that contactpoints
forantigen-antibody
interactionshave been
disrupted
or madesterically
undesirable.How-ever, some of the mar mutations are
predicted
to inducestructuralalterationsin
gC.
Themostdramaticchange
wasaconsequenceofthe substitutionof residue
Arg-147
withTrp
within the 8-amino-acid core
antigenic
structure describedabove.
Secondary
structureanalysis
indicated thatthissub-stitutioncouldboth deletea aturnandaltera
loop
structureof
site II.
This structuralchange might
be the basis of theantigenic
alterationaffecting
EpC7,
-9,
-16,
and -17. Thesubstitution of
Arg-143 by
Gln in marC9.6and marC13.1mayextendan
existing
,Bsheetinapotential loop
structure.Outside thiscore
region,
thechange
ofGly-247
toAsp
inmarC10.1mayresult in the
disruption
ofanahelixwhich isalsopart
ofapotential
loop
structure.The role of localsecondary
structure in formation oftheseepitopes, particularly
in the8-amino-acid
segment,
willrequire
furtheranalysis
by
site-directed
mutagenesis.
ACKNOWLEDGMENTS
This workwas
supported
by
Public Health Service grantsA17900,
A18228,
RR00200, and GM34534 from the National Institutes of Health.Wethank
Judy
Worley
forhelp
inpreparing
themanuscript.LITERATURECITED
1.
Ackerman,
M., R.Longnecker,
B. Roizman, and L. Pereira. 1986.Identification, properties,
and gene location ofanovelglycoprotein specified by herpes simplex
virus 1. Virology150:207-220.
2.
Birnboim,
H.C.,andJ.Doly.
1979.Arapid
alkaline extractionprocedure
forscreening
recombinantplasmid
DNA. Nucleic Acids Res. 7:1513-1523.3.
Black,
S.D.,andJ.C.Glorioso. 1986.MSEQ:
a microcomputer-based approach to theanalysis, display,
and prediction ofprotein
structure.BioTechniques
4:448-460.4.
Buckmaster,
E. A., V.Gompels,
and A. C. Minson. 1984.Characterizationand
physical mapping
ofanHSV-1glycopro-tein of
approximately
115 x 103molecularweight. Virology139:408-413.
5. Eberle, R.,andS. W. Mou. 1983.Relativetitersofantibodiesto
individual
polypeptide antigens
ofherpessimplexvirustype 1in humansera. J.Infect. Dis. 148:436-444.6.
Eberle,
R.,R.G.Russell,andB. T. Rouse. 1981.Cell-mediatedimmunity
toherpes simplex
virus:recognitionoftype-specificandtype-common surface
antigens
bycytotoxicTcellpopula-tions. Infect. Immun. 34:795-803.
7.
Friedman,
H. M.,G. H. Cohen,R. J.Eisenberg, C. A.Seidel, andD.B.Cines. 1984.Glycoprotein
Cofherpes
simplexvirus1acts as a receptor for the C3b complement component of infected cells. Nature(London)309:633-635.
8. Fries, K. R.,H. M. Friedman, G. H. Cohen, R. J. Eisenberg, C. H. Hammer, and M. M. Frank. 1986. Glycoprotein C of herpes simplexvirustype 1 isaninhibitor of the complement cascade.J. Immunol. 137:1636-1641.
9. Frink,R.J.,R.Eisenberg,G.Cohen,and E. K.Wagner.1983. Detailedanalysisoftheportionof theherpessimplexvirustype 1 genomeencoding glycoproteinC. J. Virol.45:634-647. 10. Glorioso, J., U. Kees, G. Kumel, H. Kirchner, and P. H.
Krammer. 1985. Identificationofherpes simplex virustype 1 (HSV-1) glycoprotein gC as the immunodominant antigenfor HSV-1specificmemorycytotoxicTlymphocytes. J.Immunol. 135:575-582.
11. Glorioso,J.C.,andM. Levine. 1985. Monoclonal antibodies and herpes simplexvirus infections, p. 235-260. In S. Ferone and M. P. Dierich (ed.), Handbook on the use of monoclonal antibodies in biologyand medicine. NoyesPublications, Park Ridge,N.J.
12. Glorioso,J. C.,M.Levine, T. C.Holland,andM. S. Szczesiul. 1980. Mutant analysis of herpes simplex virus-induced cell surface antigens: resistance to complement-mediated immune cytolysis.J. Virol. 35:672-681.
13. Goldin, A. L., R. M. Sandri-Goldin, M. Levine, and J. C. Glorioso. 1981. Cloning of herpes simplex virus type 1
se-quencesrepresentingthewholegenome.J. Virol.38:50-58. 14. Graham,F.L.,andA. J.vander Eb. 1973. Anewtechniquefor
the assay ofinfectivityofhuman adenovirus5SDNA.Virology 52:456-467.
15. Highlander,S.L.,D.J.Dorney,P.J. Gage,T.C.Holland,W. Cai,S.Person,M.Levine, andJ. C. Glorioso. 1989. Identifica-tion of marmutations inherpes simplexvirustype1 glycopro-tein B which alter antigenic structure and function in virus penetration.J. Virol.63:730-738.
16. HoUand,T. C., F. L. Homa, S. D. Marlin, M. Levine,andJ. Glorioso. 1984. Herpes simplex virus type 1 glycoprotein C-negativemutantsexhibitmultiple phenotypes,including
secre-tion of truncatedglycoproteins. J.Virol. 52:566-574.
17. Holland,T.C.,S.D.Marlin,M.Levine,andJ.Glorioso. 1983. Antigenicvariants ofherpes simplexvirus selected with glyco-protein-specificmonoclonal antibodies. J. Virol. 45:672-682. 18. Holland, T. C., R. M. Sandri-Goldin, L. E. Holland, S. D.
Marlin,M.Levine,andJ. C.Glorioso. 1983.Physicalmapping of themutation in anantigenicvariant ofherpes simplexvirus type 1 by use of an immunoreactive plaque assay. J. Virol. 46:649-652.
19. Holmes,D.S.,andM.Quigley.1981. Arapid boiling method for the preparation of bacterial plasmids. Anal. Biochem. 114: 193-197.
20. Homa, F. L., T. M. Otal,J.C. Glorioso, and M. Levine. 1986. Transcriptional control signalsof aherpes simplexvirustype1 late (82) gene liewithin bases -34 to +124 relative to the 5' terminus of the mRNA. Mol. Cell. Biol.6:3652-3666. 21. Homa, F.L.,D.J. M.Purifoy, J. C.Glorioso,and M. Levine.
1986. Molecular basis of the glycoprotein C-negative pheno-types ofherpes simplex virus type 1 mutants selected witha
virus-neutralizing monoclonal antibody. J. Virol.58:281-289. 22. Kikuchi, G. E., J. E. Coligan, T. C. Holland, M. Levine, J. C.
Glorioso, and R. Nairn. 1984. Biochemicalcharacterization of peptides from herpes simplex virus glycoprotein gC: loss of CNBrfragmentsfrom the carboxy terminusof truncated,
se-cretedgC molecules. J. Virol. 52:806-815.
23. Kumel, G., H. C. Kaerner, M. Levine, C. H. Schroder, and J. C. Glorioso. 1985. Passive immuneprotection by herpes simplex virus-specificmonoclonalantibodiesand monoclonal antibody-resistant mutantsalteredinpathogenicity.J.Virol.56:930-937. 24. Lawman, M.J. P., R. J. Courtney, R. Eberle, P. A. Schaffer, M. K.O'Hara, and B. T. Rouse. 1980.Cell-mediatedimmunity toherpes simplex viurs: specificity of cytotoxic T cells.Infect. Immun.30:451-461.
25. Longnecker,R., S. Chatterjee, R. J. Whitley, and B. Roizman. 1987. Identification of a herpes-simplex virus 1 glycoprotein gene within a gene cluster dispensable for growthin cellculture.
on November 10, 2019 by guest
http://jvi.asm.org/
Proc. Natl. Acad. Sci. USA 84:4303-4307.
26. Maniatis, T.,E.F. Fritsch,and J.Sambrook. 1982.Molecular cloning: alaboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
27. Marlin, S. D., S.L. Highlander, T.C.Holland, M. Levine, and
J. C. Glorioso. 1986. Antigenic variation (mar mutations) in herpes simplex virus glycoprotein B caninduce
temperature-dependent alterations in gB processing and virus production. J. Virol. 59:142-153.
28. Marlin, S. D., T. C. Holland, M. Levine, and J. C. Glorioso. 1985. Epitopes of herpes simplex virustype1glycoprotein gC
areclustered intwodistinctantigenic sites. J. Virol. 53:128-136. 29. McNearney, T. A., C. Odeli, V. M. Holers, P. G. Spear, and J. P. Atkinson. 1987. Herpessimplexvirus glycoproteins of gCl and gC2 bind to the thirdcomponentof complement and provide protection against complementmediated neutralization of viral infectivity. J. Exp. Med. 166:1525-1535.
30. Muller-Eberhand, H. 1986. The membrane attack complex of complement. Annu. Rev. Immunol. 4:503-528.
31. Norrild, B. 1985. Humoral response to herpes simplex virus infections,p. 69-82. In B. Roizman (ed.), The herpesviruses,
vol. 4. Plenum Publishing Corp., New York.
32. Rosenthal, K. L., J. R. Smiley, S. South, and D. C. Johnson. 1987. Cells expressing herpes simplex virus glycoprotein gC but
not gB, gD, or gE are recognized by murine virus-specific cytotoxic T lymphocytes. J. Virol. 61:2438-2447.
33. Sanger, F., S. Nicklen, and A. R. Coulson. 1977. DNA
sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA74:5463-5467.
34. Spear, P. G. 1985. Antigenic structure ofherpes simplex
vi-ruses, p. 435-445. In M. H. V. Van Regenmortal and A. R. Neurath (ed.), Immunochemistry of viruses. Elsevier/North-HollandPublishingCo., Amsterdam.
35. Spear, P. G. 1985. Glycoproteins specified by herpes simplex viruses, p. 315-356. In B. Roizman (ed.), The herpesviruses,
vol. 3. Plenum PublishingCorp., New York.
36. Sunstrum, J.C., C. L. Chrisp, M. Levine, and J. C. Glorioso. 1988. Pathogenicity of glycoprotein C negative mutants of herpes simplex virus type 1 for the mouse central nervous
system.Virus Res. 11:17-32.
37. Tainer, J. A., E. D. Getzoff, H. Alexander, R. A.Houghten, A. J. Olsen, R. A. Lerner, and W. A. Hendrickson. 1984. The reac-tivity of anti-peptide antibodies is a function of the atomic mobility of sites inaprotein. Nature (London) 312:127-134. 38. Teichen, E., E. Maron, and R. Arnon.1973. The role ofspecific
amino acid residues in the antigenic reactivity of the loop peptide oflysozyme. Immunochemistry 10:265-271.