Copyright ©1970 American Society for Microbiology Printed in U.S.A.
Isolation and Characterization of Two
Basic Internal
Proteins
from the T-Even
Bacteriophages1
KENNETH R. STONE AND DONALD J. CUMMINGS
DepartmentofMicrobiology, University of Colorado Medical Center,Denver, Colorado80220
Receivedfor publication9 June 1970
Two species of basic internal proteinswerefound in osmotic shocksupernatant
solutions of bacteriophages T4B, T4D, T2H, T2L, and T6. The major species of protein isolated hada molecular weight of approximately 21,000 daltons, whereas
theminor proteinmolecular weightwas near9,500daltons. Thetwoprotein species exhibited unique isoelectric points and amino acid compositions. The
21,000-daltonprotein of T2L showed major electrophoretic and compositional differences from the other dalton proteins isolated. Similarities between the 21,000-daltonproteins and phage lysozymearediscussed.
Studies on the role ofdeoxyribonucleic acid (DNA) in the infective process of the T-even
bacteriophages led to the discovery of certain substances whichappeared to be associated with
the viral nucleic acid and which were detected only after rupture of the head membrane by osmotic shock (23, 24). These substances in-cluded: (i) anacid-soluble fractionwhichderived
mostof its carbon from arginine and yetyielded
no arginine upon acid hydrolysis; (ii) an
acid-soluble peptide which yielded predominantly
lysine, glutamicacid, andaspartic acid uponacid hydrolysis; and (iii) an acid-insoluble protein
fraction whichcouldbedistinguishedfromghosts
andwhole phages byimmunologicalmeans. The
first two substances have now beenwell charac-terized.Amesand co-workershave demonstrated the presence of the polyamines, putrescine and spermidine, in T4
phage
(3),
whereasChampe
et al. have found two acid-solublepeptides
in T2H, T4D, and T6phage (10, 18,41).
The acid-insolublefraction,however,
has notbeenaswell characterized. Levine and co-workers reportedthe presence of an internalantigenin T2 and T4
phage (31, 35) which they called the internal protein. Minagawa
(34) attempted
to correlate this internal antigen withHershey's
(23)
acid-insolublematerial.The internal
protein
has been estimated toaccount for 3 to 7% of thetotal
phage
protein
(23, 31, 34) andits function remains unknown.
Several laboratories have
attempted
isolations of theinternalproteins
from T-evenbacteriophages
(7,8, 11,29, 31,34;M.L.
Coval,
V.M0ller,
andIPresented bythesenior author inpartialfulfillment of the
requirements forthe Ph.D.degreetotheDepartmentof
Micro-biology,Universityof Colorado Medical Center.
H. Van Vunakis, Fed. Proc. 19:253, 1960).
Coval's group reported a partial amino acid
compositionin their abstract (M. L. Covalet al.,
Fed. Proc. 19:253, 1960 ). The protein from T2 washigh inlysine and histidine, andno cysteine was detected. Minagawa (34), however,
sug-gested the presence of more than one internal protein in T2. The recent reports of Bachrach et al. (8) and Kokurina and Tikhonenko (29) would tend to confirm thisfinding.
Theobject of the present study was to isolate and characterize the internal proteins Evidence
is presented which demonstrates the presence, in all of the T-evenphage examined,oftwo species
of basic internal proteins which can be distin-guished by a number of such parameters as
molecular weight, electrophoreticcharacteristics,
and amino acid composition.
MATERIALS AND METHODS
Bacteriophage growth and purification.
Bacterio-phages T4B, T4BO1, T4D, T2H, T2L, and T6 were prepared and purifiedunder standardconditions (1).
(All ofthesebacteriophageswith theexceptionof T6 have been used in our laboratoryfor several years;
T6 was obtained from M. Jesaitis.) The mu-tant phage AmN85 (G48) and AmH21 (G54)
were obtained from R. S. Edgar. Escherichia coli B was grown in 70-liter volumes of pH 7.3 Casamino
Acids-glycerolmedium similartothatof Kozloff and Lute(30), which contained per liter: 1.2gofNH4CI,
1.0 g ofNaCl, 0.5 g ofKCI, 2.4 g of tris(hydroxy-methyl)aminomethane, 0.1 g of gelatin, 24 ml of
glycerol, 1.0ml of 37% HCl, 1.0 g of MgSO4, and 23.5 mg of CaCl2. Growth of bacteriophage T4B required the addition of 0.2 g of L-tryptophan per liter for adsorption (4, 5). The medium was
supple-mented with30mgofthymineperliter 10min prior
tobacteriophage infection; in some cases, especially
445
on November 11, 2019 by guest
http://jvi.asm.org/
STONE AND CUMMINGS
with T6, better yields of bacteriophage were obtained in the presence of thymine. When the bacterial cell density was 2 X 108 to 3 X 108cells/ml,the bacteria were infected with phage, prepared 2 days prior touse,
at a multiplicity of three phage per bacterium. The phage growth was then allowed to proceed with aeration for another 5 hr.
At the endof thebacteriophage growth period, the phage were concentrated by the polyethylene glycol-dextran two-phase system (2) as modified by S. Ward (personal communication). Chloroform (1 ml/
liter) and a small quantity ofdeoxyribonuclease and
ribonuclease were added to thelysate. Thefollowing
materials were then added, inorder: 17 g of sodium chloride per liter, 2.3 g of sodium dextransulfate 500 (Pharmacia Fine Chemicals) per liter, and 71 g of polyethylene glycol 6000 (Carbowax, Union Carbide) per liter. Each compound was dissolved completely beforethenext wasadded. Thelysatewasthenplaced
at4Cfor 1 to2days toallowphaseseparation.The
bulk of the upper polyethylene glycol phase was removed bysuctionanddiscarded. The lowerdextran phase was pouredinto a4-liter beaker andallowedto separateagainovernightat4C. Theremaining poly-ethylene glycol wasremoved, and the dextran phase wasdiluted threefold with saline stock solution (0.15 MNaCl, 1 mM MgSO4, and 1 mMP04,pH7.5).The dextran wasthen precipitated by the addition of0.2 volumes of 3 M KCI withstirring.Thebacteriophage
were purified by two successive cycles of differential
centrifugation (2,000 X g for 10 min, followed by 15,000 X g for 1.5 hr) and finally resuspended in
minimal volumes ofsaline stock solution.
Bacterio-phageyields were in the order of 1015 to 2 X 1018 particlesper70liters.
Glycerol osmotic shock procedure. Osmotic shock
ofthe phage particles by using high salt concentra-tions (22)wasfoundtoliberatemoderateamountsof
phage structural proteins. Consequently, osmotic
shock was induced by rapid dilution of phage in
glycerol. Purified phage, resuspended from pelletsin
minimal volumesof saline stocksolutionat3 X 10's to5X 1013phage permlweremixed with 0.43volumes
ofglycerol (4 M final concentration). After 1 hr of
equilibration in the presence ofa small amount of
deoxyribonuclease, the phage were osmotically rupturedbyrapid dilutionwith 18volumes ofwater at25 Ccontainingthedeoxyribonuclease medium of Cummings (12). After 2 hr of deoxyribonuclease
digestion, the phagecapsids [or "ghosts" (22)] were removed by sedimentation at 40,000 X gfor 3 hr. The supernatant solution was again centrifuged at
40,000X gfor 15 hrto removeanyremainingphage substructures (14). This final supernatant solution,
which contained the internal proteins, was then
analyzed by chromatography on carboxymethyl cellulose (CM-cellulose).
Chromatography on CM-cellulose. CM-cellulose (CM23, Whatman) wasequilibratedwith ammonium acetate buffer (0.1 M acetate, pH 5.0) and dried by
suction. The supernatant solution containing the
internal proteins wasadjusted to pH 4.8 with acetic acid and mixed with sufficientCM-cellulose to make acolumn2.5 by10 cm (39). Themixture wasgently
stirred for 30 min and poured into the column.The columnwaswashed withstarting buffer (0.1 Macetate,
pH5.0)until alldigested DNA and unbound materials were eluted. All buffers were 0.1 M in acetate and
adjusted to the desired pH with concentrated am-monium hydroxide. Two linear gradients were then used in sequence to elute basic proteins (buffer at pH 5 tobufferatpH 10;followedbybufferatpH 10 to the same buffer containing 0.2 M Na2CO3). The flowrate was 2.5 ml/min, and 5 ml fractions were
collected. Protein peaks were monitored by fluore-scencewith an Aminco-Bowman Spectrophotofluor-ometer(excitation,280 nm;emission,340nm).
Frac-tionswerepooled in each peak andprecipitated with 5% trichloroacetic acid (final concentration). The pH 5 to 10gradient was very steep across the pH 6 to 8region since itbufferedverypoorlyinthis pH range. Chromatographyon6%agarose.Molecularweights
weredetermined fromthepartition coefficients ofthe
proteinson6%agarose(100to200mesh,controlno.
6470, Bio-Rad Laboratories) asdescribed by Davison
(16). Protein samples were dissolved in purified 6 M
guanidine (36) containing 3 mm Cleland's reagent
(dithiothreitol, A grade, Calbiochem). The column
(2 by 95 cm) was equilibrated with 5 M guaiiidine (Sigma Chemical Co.),0.05 MLiCl,0.01 M
ethylenedi-aminetetraacetic acid, and 3 mm Cleland's reagent. Thisguanidine solutionwaspreparedasfollows.The
ingredients,lessCleland's reagent, were mixed, heated to45to 50 Cfor solution, cooled overnight at4 C, filtered, and stored at room temperature. Cleland's reagent was added tothe guanidine solution, at the time it was used, for column elution. The flow rate was approximately 8 ml/hr, and 4-ml samples were
collected. Protein elution was monitored by fluor-escence in an Aminco-Bowman
Spectrophotofluor-ometer(excitation,280 nm;emission,340nm).Dextran
blue and dinitrophenyl-alanine were used as void
volume and internal volume markers, respectively.
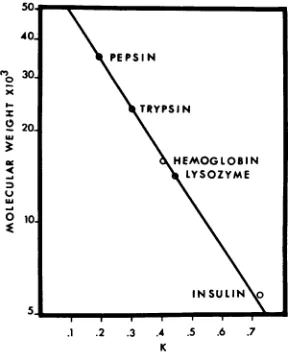
The following molecular weight (MW) standards
(40) were used tocalibrate the column: pepsin (2X crystallized; Sigma Chemical Co., MW = 35,500); trypsin (type III, Sigma Chemical Co., MW = 23,800); horse hemoglobin (Pentex, MW = 16,000); egg white lysozyme (isoelectric enzyme, Schwarz BioResearch Inc., MW = 14,400); insulin (Sigma
Chemical Co., MW = 5,773).Asshown inFig. 1,a
linearrelationshipexists between the partition coeffi-cient[K = (Ve-
Vo)/(Vi
- V.)whereVe = elution volume of theprotein sample, Vi = internalvolume,and V. = void volume] and the log MW.
Isoelectrofocusing in polyacrylamide gels.
Isoelec-trofocusing was performed in a disc electrophoresis unit (DE102, Hoefer Scientific Instruments, San
Francisco, Calif.) byamethodmodified from that of Dale and Latner (15). Gels (7 cm) were poured in glass tubes (0.4 by 12 cm). Thegel solution contained 8.0%acrylamide (w/v, Eastman OrganicChemicals),
0.2% N,N'-methylenebisacrylamide (w/v, Eastman
Organic Chemicals), 0.2%
N,N,N',N'-tetramethyl-enediamine (w/v, Eastman Organic Chemicals), 8 M urea, 0.01 MCleland's reagent, and 1.5%
ampho-line, pH ranges 3 to 10 (LKB Instruments). Acryla-mide was recrystallized from chloroform by the
446 J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
to3030 \
I- \TRYPSIN
20
HEMOGLOBIN
LYSOZYME
U \
10\
INSULIN
.1 .2 .3 .4 .5 .6 .7
K
FiG.1. Chromatography ofMW standardson a6% agarose column. Pepsin (35,500 daltons), trypsin (23,800 daltons), horse hemoglobin (16,000 daltons),
egg white lysozyme (14,400 daltons), and insulin (5,773 daltons) (40) were dissolved in 6M guanidine hydrochloride containing 3mM Cleland'sreagent and were chromatographedon a 6% agarose column.
procedure ofBishop et al. (9). No difference in the electrophoretic profile of the proteins was observed by using purified acrylamide. Protein samples dis-solved in not morethan 0.05 ml of 10M urea plus 0.1 MCleland's reagentwereplaced inthe bottomof gel tubes. Ammonium persultate (E-C Apparatus Corp.) wasaddedat 1.4mg/10mlofgelsolutionas
catalyst.Thegelswerequickly pouredwiththorough mixing of the protein sample throughout the gel. Usuallysix tubescould bepoured with 10 mlofgel
beforepolymerization began. Thegelswere given 15
minto setproperlyandwere placedinthedisc elec-trophoresis unitsothatthe 7-cmgelswerecompletely
immersed in the lower water-jacketed chamber. The bottom cathode solutionwas 1% ethylenediaminein water,and theupperanodesolutionwas1.4%
ortho-phosphoric acid. Electrophoresis was for 6 hr with
an initial currentof 5 mamp per tube.
Thegelswerestained withbromphenol blue bythe method of Awdeh(6).Minor bandswere moreevident if the gels were placed in distilled water for a few hours after destaining by the Awdeh method. Also, the bands were more stable when stored in water
than in thedestaining solution.
Numerous attempts were made to calibrate the
isoelectrofocusing gels for accurate determination of isoelectric points. This was not possible for two reasons. First, the ampholinecarrier ampholytes did
not give a perfectly linear gradient and there were
minor variations between different batches of ampho-line.Second,therewerenotenoughproteinsavailable with defined isoelectric points to span the gradient. At best, only an approximation could be made for isoelectricpHvaluesby usingthefollowing proteins as standards: lysozyme (pl - 10.5 to 11.0),
chymo-trypsinogen A (pI - 9.2), trypsin (pI - 10.0), bovine serum albumin (pl -. 5.1), and horse
hemo-globin (pI-6.9).ThepHgradientwascalibratedby
alinear plotofpH versustheratio ofthepositionto which a protein speciesmigratedfrom the anode end of the geloverthe total length of thegel.This ratio for agivenproteinwasreproducible, but the isoelec-tric pointsof the standard proteins were not defined under theconditions used here.Thus,the accuracyof the estimated pH gradient with these standards was probably no greater than i 0.5pH units.
Isoelectrofocusing in a sucrose gradient. The LKB
electrofocusing column (no. 8101) was used for measurement of the isoelectric points of the basic
internal proteins. Protein samples were dissolved in 10 M ureaandappliedin themiddle ofthe sucrose step
gradient ofthecolumn.Ampholine carrierampholytes
withapH range of 7to10 wereused. Theexperiments
wereperformedinthenormalmannerwith the excep-tion that 8 M urea was used in thesuspending medium.
Electrophoresis was for 24 hr at 400 v with the cathodeasthelower electrode. Atthecompletion of
the electrofocusing experiment, 2-ml fractions were
collected andanalyzed forpH andfor fluorescencein an Aminco-Bowman Spectrophotofluorometer (exci-tation,280 nm;emission, 340nm).
1251I
labeling of proteins.Proteins werelabeled with125I by the method ofMcConahey and Dixon (33). Amounts(mg) of theproteinsampleswere dilutedin 1 mlof0.05 Mphosphatebuffer (pH7.0) in a 10-ml beakerandstirredgentlyinanice bath; 0.05 mCi of
1251 in 0.05 M phosphate buffer was added.Labeling
wastheninitiated by theaddition of 50 ,ug of
chlor-amineTinphosphate buffer.Thereactionwasallowed to progressfor 10min and wasthenstopped by the addition of 50 ,ug ofsodium metabisulfite in
phos-phate buffer. Guanidine (1.0 g/ml) and a small
amountofCleland's reagentwereusedtodissolvethe
labeledproteins, which werethenfractionated bygel filtration on the 6% agarose column. The free 1251 was eluted with the internal volume (Vi) of the
column. Portions (1 ml) of eluted fractions were counted in an Automatic Well Gamma Counter
(Nuclear-Chicago Corp.).
Amino acidanalyses.Amino acidanalyseswere
per-formedinaBeckman-Spinco model 120Amino Acid Analyzer equipped with sensitive cuvettes and a
model CRS-110A Automatic Digital Integrator
(Infotronics Corp., Houston).
Quantities (mg) of the protein samples were hydrolyzed invacuo at110Cfor 24 hr in 2-mlvolumes
of three timesdistilled6NHCIcontaining
2-mercapto-ethanol and phenol (10mlof 6NHCI plus5 ,liters
eachof2-mercaptoethanolandliquified phenol)to
re-tarddestruction of tryptophan(J.M.Stewart, personal communication). By using egg-white lysozyme as a
standard, approximately 75 to 85% recovery of tryptophan was achieved by this method.
Half-cystine was determined as cysteic acid by the
per-formic oxidation method of Hirs (25). RESULTS
Isolation of the basic internalproteins. The
non-sedimentableproteins released fromthephage by
on November 11, 2019 by guest
http://jvi.asm.org/
[image:3.493.79.223.72.249.2]STONE AND CUMMINGS
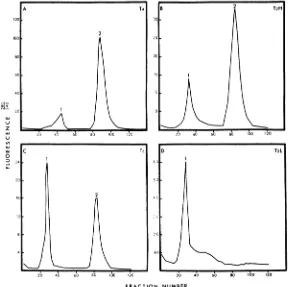
osmotic shock were fractionated on carboxy-methyl cellulose. Elution profiles for these pro-teins from phages T4, T2H, T6, and T2L are
showninFig. 2. Allphage supernatant solutions
examined, with the exception of T2L, resulted in
two peaks ofproteins which differed inpH and
ionic strength required for elution from this resin. Bacteriophage T2L contained both com-ponents found in the other viruses, but they eluted too closelytogether tobeseparatedby the method used here (Fig. 2D). The material from the T2L peak was further fractionated by gel
filtration on 6% agarose.
Fractions across peaks 1 and 2 wereseparately
pooled, and the proteins were precipitated with
5% trichloroacetic acid. The proteins in these peaks appeared to contain all of the internal proteins released by osmotic shock. No other
trichloroacetic acid-insoluble material was de-tected across theelution profileother than
occa-A
120
2
100
00
20
1.)
z
ui 20 4, 600 80 00o 20
n
O 24
sional trailing from one of these peaks. In all casesexamined, the materialfrom peaktrailswas
identical to that of the preceding peak. No further trichloroacetic acid-insoluble material
was released from the CM-cellulose upon the addition of0.5 N NaOH solution containing0.5 M NaCi. The void volume of the column
con-tained only minor amounts ofphage structural proteins and small amounts of peak 1 material. Thus, no acid-insoluble acidic internal proteins
werefound in any of thephageexamined. As will become evident in the electrophoretic studies, the CM-cellulosecolumn did notalways
fractionate the peaks cleanly. As a result, small amountsof the peak2 proteinscould be demon-strated inpeak 1 and very minor amountsof the peak 1 material were recovered from the void
volume. Somewhat better separations occurred
byusing step gradients, but splitting ofeach of
thetwo peakswas observedwith this method.
FRACTION NUMBER
FIG.2.Chromatography oftheinternalproteins on carboxymethyl cellulose. Supernatant solutions from osmotic shockofthebacteriophageswere equilibratedwithsufficientcarboxymethyl cellulose to pour a column 2.5 by 10 cm. Thecolumn waswashedwithstartingbuffer (0.1Macetate, pH 5.0)until all unbound materials and digested DNA
wereeluted. Separationwas thenachieved with two linear gradients. The first gradient(bufferat pHS to buffer
atpHJO) coveredapproximatelyfractions I to 60, followed by the second gradient (buffer at pH 10 to the same
buffercontaining0.2AfNa2CO3). Variationsoccurred in the gradient volumes, resulting in displacement of peaks
indifferentexperiments.
448 J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
[image:4.493.104.393.285.572.2]The mass ratio of the trichloroacetic
acid-insolubleproteins ofphagesT4B,T4D,and T2H
recovered by trichloroacetic acid-precipitation
from the CM-cellulose peaks was about 5:1
(peaks2/1). Phage T6,ontheotherhand,gave a ratio of 1:2 (peaks 2/1). The reason for this differencewas notclear,butitmay havereflected preferential losses of the peak 2 material during purification of the T6 proteins. Losses of this material were often observed during dialysis,
when performed; some loss is attributed to adherence to glassware. The best protein yields
were obtained when their concentrations were
kept high. The growth ofphage T6 was
repro-ducibly poor, and, consequently, less starting materialwasavailable. Sincethe samenumberof
pieces ofglasswarewas used in each experiment, itwould appear that the materialseen inpeak 2 of the CM-cellulose profile is that amount of
protein remaining after losses onthe glassware.
Further work withisotopically labeled proteinis necessary before thisdifficulty canbe resolved.
Molecular weight determinations.
Samples
of thetrichloroaceticacid-precipitated proteinsfrom peaks 1 and 2 ofCM-cellulose were washed in 0.05 M phosphate buffer (pH7.0)
and labeledwith
"2I.
Purifiedguanidine
(1.0 g/ml) and asmall amountof Cleland's reagentwereaddedto
dissolve the labeled
protein
suspension, and thiswas combined with unlabeled molecular weight standards (Fig. 1) also dissolved in6Mguanidine containing 3 mm Cleland's reagent. Molecular weights oftheinternal
proteins
werethendeter-minedbygel filtrationonthe 6% agarosecolumn
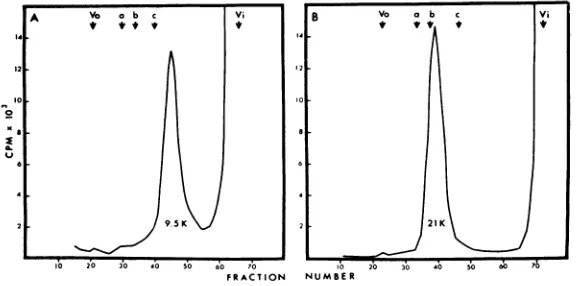
(Fig. 3).CM-cellulose peak1 material fromphage
T4 eluted from the agarose column as a
single
peak (Fig. 3A) with a molecular weight of
approximately9,500daltons. The peak 2 material
from phage T4 also showed a single peak (Fig.
3B) on the agarose column with an average molecular weight of about 21,000 daltons.
Both 21,000- and 9,500-dalton proteins were found in each of the T-even bacteriophages examined. The amount of these proteins recovered varied somewhat with the phage studied. High yieldsofT4B, T4D, and T2Hpermitted recovery of 1 to 2 mgof the 9,500-dalton protein and 5 to 15 mg of the 21,000-dalton protein per 2 x 1015 phage. Thetrichloroacetic acid-insoluble material from CM-cellulose peak 1 of T2L phage gave both a21,000- and a 9,500-dalton protein when further fractionated on a 6% agarose column. Detection of these proteins was by trichloroacetic acid precipitation of the fractions across the
elution profile since these proteins yielded only low levels of fluorescence (Fig. 2), especially in the presence of the high background of 5 M guanidine. Only minute amounts of the 9,500-dalton proteins were recovered from T2L phage
supernatant solutions. The isolation procedure for the proteins from this phage involved more steps, and the reduced recovery may have been a reflection of lossesincurred by the extra steps. T6 phage yielded only small quantities of the
21,000-dalton protein, as mentioned earlier,
which may have been the result of preferential
loss onglassware.
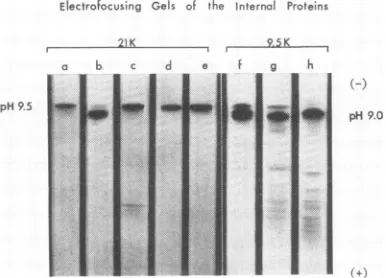
Electrophoretic studies. Electrofocusing ofthe
isolated proteinson polyacrylamide gels demon-strated the presence of two, or at most three, species of basic internal proteins (Fig. 4). The
21,000-dalton proteins from T4B (Fig. 4, d)
FRACTION NUMBER
FIG. 3. MWdererminationofthe internalproteinsofphageT4by
chromatography
on6% agarose.125I-labeledinternalproteinsdissolvedin6M guanidineand3mm Cleland's reagent werecombinedwith theunlabeled MW standards pepsin (a), trypsin (b), andlysozyme (c) andfractionatedotn 6% agarose. Thearrows designate the orderofelutionofthe threeMWstandardsalongwith thevoid volume(Vo)and internal volume (Vi) ofthe column. (A) ChromatographyofpeakIfromthecarboxymethylcellulosecolumn. (B) Chromatography ofpeak2fromthe
carboxymethylcellulose column.
449
on November 11, 2019 by guest
http://jvi.asm.org/
[image:5.493.113.401.445.588.2]STONE AND CUMMINGS
Electrofocusing Gels of the internal Proteins
21K r 9.5K
pH9.5
FIG. 4. Electrofocusing ofthe internal proteins on polyacrylamide gels. Electrophoresis of the protein samples mixed throughout the gelswasfor6hrat400 vwith pH3 to10rangeampholine. Cathode (-) was
at the top. Gels of the 21,000-dalton proteins are (a) T2H, (b) T2L, (c) T4BO0, (d) T4B,and(e)T4lysozyme.
Gels of the 9,500-daltons proteins read (f) T2H, (g) T6, and (h) T4D. Variations in gel lengths occurred resulting in displacement ofsome minor bands. The gels were aligned by isoelectric points of the major bands.
and T2H (Fig. 4, a) gave single bands by this method at approximately pH 9.5. The 21,000-dalton protein from T2L phage (Fig. 4, b), however, exhibited a minor band atthispH and
a major band at about pH 9.0. As will be dis-cussed later, major differencesin theamino acid
compositionwerealso noted between the
21,000-dalton protein of T2L and those of the other
phages. The 21,000-dalton protein of T6 phage was notexaminedongels since thesmall amount isolatedwas used for amino acidanalysis.
Included inFig.4 is agelof the21,000-dalton
protein from phage T4B01 (Fig. 4, c). This
isolate was prepared by direct trichloroacetic acidprecipitation ofthephagesupernatant
solu-tion after sedimentasolu-tion of the phage capsids. It was thus contaminated by smallamounts of the more acidic 18,000-dalton structural proteins of the phage head (21; G. Forrest and D. Cum-mings, unpublished data), illustrating the necessity for identification of other minorproteinsreleased by osmotic shock.
Finally, anelectrofocusing gelofT4lysozyme (lot 802025, Calbiochem) has been included (Fig. 4, e).Itis of interest that this protein witha molecular weight similar to the 21,000-dalton protein should also have the same isoelectric point. This similarity will be considered in
greater detail later.
Figure 4 also shows representative electro-focusing gels of the 9,500-dalton internal
pro-teins. It is clear that in many of the cases ex-amined, the 9,500-dalton proteins exhibited both a major and several minor bands. The major
band ofall ofthe9,500-dalton proteins (Fig. 4; f, g, h) was at approximately pH 9.0, whereas the minor bands of T2H and T6 wereintheregion of the21,000-dalton protein.By
"2I
labeling and gel filtrationon 6%agarose, these minor bands havebeen shown to betheresult of contamination of theCM-cellulose peak 1 with the 21,000-dalton materialfrom peak 2. The21,000-dalton protein ofT2L (Fig. 4,b), ontheotherhand,was at the same positionasthe 9,500-dalton proteins. This would be expected from its behavioronthe
CM-cellulose column.
The electrofocusing gel of the 9,500-dalton
protein from T6 phage (Fig. 4, g) illustrates again howminor contaminants can beidentified by this method. In thiscase,several ofthe struc-tural capsid proteins eluted inCM-cellulosepeak 1 (13, 14, 21; G. Forrest and D. Cummings, unpublished data). Only the head structural
11,000-dalton protein had an isoelectric point nearthe internal proteins (21; G. Forrest and D.
Cummings, unpublished data). It gave an
iso-electric point of approximately pH 7, near the
middle ofthe gel.
Structural proteins isolated fromthe heads of theT4ambermutants AmN85(G48) andAmH21
(G54) were also examined onisoelectrofocusing gels. Both ofthese mutantphages produced free headsandtailplateswhen grown under restrictive
conditions(R. S. Edgar, personal communication). Freeheads devoid of DNA were separated from
tail plates and isolated as described previously (13, 14, 21) and were examined without osmotic
shock treatment. Both the 9,500- and 21,000-dalton internal proteins were present in these
heads. It is of interest that these empty heads
contained the internal proteins and may indicate
either thatthepresenceofDNA is not necessary
for their occurrence or that the heads originally contained DNA which was digested by deoxy-ribonuclease treatment during purification. The major point to be made is that these internal
proteins could not have originated from tail substructures released during osmotic shock.
The internal proteins were also electrofocused in sucrose gradients by using the LKB
electro-focusing column to determine more precisely
their isolectric points. This instrument allowed directmeasurement ofthe pH across the
ampho-linegradient, whereas the pH gradient of thegels
wasestimated from positions to which proteins of known isoelectricpointsmigrated. The results
obtained for the 21,000- and 9,500-dalton pro-teinsofT4Bfrom the electrofocusingcolumnby
usingpH 7 to 10 rangeampholine arepresented
450 J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
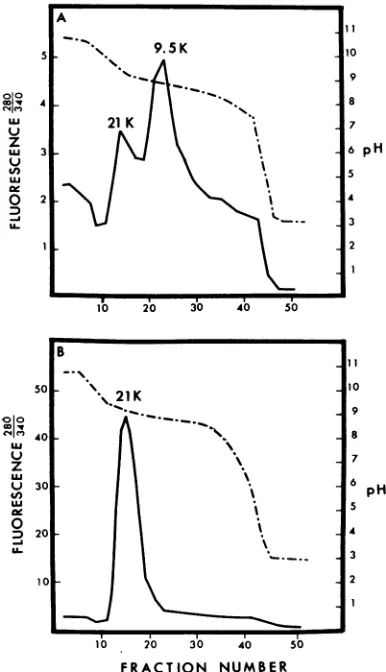
[image:6.493.43.236.71.210.2]VOL.6,1970T-EVENBACTERIOPHAGE PROTEINS45
in
Fig.
5.Figure
5Ademonstrates that the21,000--dalton
protein
gave asingle peak
atpH
9.2,
corresponding closely
with the isolectricpoint
ofpH
9.5 estimated for thisprotein
from theelec-trofocusing gels.
The isoelectric
point
for the9,500-dalton
protein, by
thismethod,
waspH
8.86 whichcor-responds closely
with thegel
results which gavepH
9.0. This isolate of the9,500-dalton protein
was contaminatedwith the heavier
21,000-dalton
protein, resulting
in the secondpeak.
Thisexperi-ment
clearly
demonstrated the distinctiveisoelec-A
9.5 K 10
9
4
~~~~~~~~~~~~~~8
ui ~~21K 7
U.' 3 6p
0 2 4
2
10 20 30 40 50
B
50 I10
\..21
Ku 7
LU
~~~~~~6p
U 30pH
4,,
ui
~~~~~~~~~~~~~~~5
0 4
D 20
U. 3
10 2
A02 30 4~0 50
[image:7.493.60.253.212.548.2]FRACTION NUMBER
FiG. 5. Electrofocusing ofthe internal proteins of phage T4 in sucrose gradients. Protein samples dis-solvedin 10 m urea were appliedat thecenter ofthe
sucrose step gradient. Ampholine (pH 7 to 10) was used;electrophoresiswasfor24 hrat400 v. Fractions
(2 ml) were collected and analyzed for pH (broken line) and fluorescence (solid line). (A) The
9,500-daltoninternalprotein,contamninatedbysmallamounts
ofthe 21,000-dalton protein. (B) The 21,000-daton
internalprotein.
tric characteristics of the two
proteins
and thereproducibility
of the method.Amino acid
analysis.
Amino acidanalyses
ofthe internal
proteins
verified the basic nature of theseproteins.
The basic amino acid content(lysine, arginine,
andhistidine)
of all of the21,000-dalton proteins
examined varied from18.2to 19.4%
(Table 1).
Thecompositions
ofthe9,500-dalton proteins
ofT4B3
andT4D3(Table
2)
yielded
17.5 and 17.1%,
respectively,
for thesameamino acids.
Although
the9,500-dalton
proteins
of
T2H, T2L,
and T6are notpresented here,
thepreliminary
amino acidcompositions
of theseproteins
indicated thatthey
also werecomposed
of
relatively high
amounts of the basic aminoacids.
The amino acid
compositions
of the 21,000-daltonproteins
ofT4B,
T41D, T2H4,
and T6werein close agreement. Allwere
high
in alanineandtryptophan,
but none had detectablehalf-cystine.
Theseproteins
also containedapproxi-mately
three times morephenylalanine
thantyrosine.
The21,000-dalton
protein
of T2Lphage,
ontheotherhand,
wasdifferent from theother
21,000-dalton
proteins
in a number ofam-inoacids. TheT2L,
protein
had neithertrypto-phan
norproline
andnearly
equal
amounts oftyrosine
andphenylalanine.
It washigh
inglutamic acid-glutamine
andlow inalanine.The
9,500-dalton proteins
of the T-evenbacte-riophages
have notbeenaswell characterized asthe
21,000-dalton
proteins. Only
those of T4B3and T4D3have as yet beenobtained free of
con-taminating
21,000-dalton proteins (Fig. 4).
The9,500-dalton
proteins
of thesephages,
however,
exhibited an amino acid
composition
distinct from that of the21,000-dalton proteins (Table 2).
T'he
9,500-dalton proteins
hadonly
about halfthe
aspartic acid-asparagine
content of the21,000-dalton
proteins.
Thetyrosine-phenyl-alanine ratio was near
unity
in the9,500-dalton
proteins,
whereas the21,000-dalton proteins
contained about three timesmore
phenylalanine
than
tyrosine.
The9,500-dalton
proteins
werehigh
in histidine and low in
tryptophan.
Amino acidanalyses
of the9,500-dalton
proteins
ofT2H4,
T2L,
andT6,
whichweresimilartothoseproteins
isolated from T413 andT41D,
have been omitted from Table 2 since thedegree
ofcontamination from the21,000-dalton proteins
has notyetbeen ascertained.DISCUSSION
Evidence has been
presented
which indicates that there are two basic internalproteins
in the T-evenbacteriophages.
Themajor
internal pro-teinsofT4B3, T4D3,
T2H,
andT2L,werefoundtohavea molecular
weight
of21,000
daltons.Phage
VOL.
6,
1970451
on November 11, 2019 by guest
http://jvi.asm.org/
STONE AND CUMMINGS
TABLE 1. Amino acid composition of the 21,000-dalton internal proteinsa
Amino acid T4B T4D T2H T2L T6
Aspartic acid +
as-paragine... 13.4 it0.3 13.5 4- 0.5 13.0 it0.5 12.9 it0.5 13.0 i0.5
Threonine... 3.2 4t0.1 3.0 1-- 0.1 3.5 ±- 0.3 6.6 it0.1 3.2 i0.1
Serine... 4.7 i0.3 4.8 -- 0.2 4.9 it0.3 6.6 -0.3 5.0 ± 0.9
Glutamic acid +
glutamine ... 6.3 i 0.2 6.5 -- 0.2 6.5 it 0.2 9.8 i 0.4 6.7 ±4 0.1
Proline... 2.0 - 0.5 2.1 -- 0.3 2.3 it0.4 0 2.1 it0.5
Glycine... 7.3 i0.2 7.4 ±i 0.3 7.3 it 0.3 8.2 i0.2 8.0 ±t0.3
Alanine... 14.6 i 0.3 14.5 ±t 0.5 14.0 ± 0.5 9.7 ±t 0.3 14.6 it 0.2 Valine... 5.5 -0.3 6.0 -- 0.5 6.0 i0.3 6.2 -- 0.2 5.1 -t0.7
Half-cystineb... 0 0 0 0 NDc
Methionine... 1.0 4t 0.1 1.2 -- 0.2 1.4 it 0.3 2.2 it0.2 1.3 4- 0.1
Isoleucine... 3.7 4- 0.1 3.9 4t 0.1 3.8 ±t0.1 5.8 it0.2 3.8 -- 0.1
Leucine... 6.1 it 0.2 6.3 -- 0.2 6.2 4t 0.2 4.2 -0.2 6.4 -0.1
Tyrosine... 2.0 4- 0.2 2.1 -- 0.2 2.3 it0.2 3.9 i0.1 2.0it 0.1
Phenylalanine... 7.0it 0.3 7.1 it0.2 6.7 ±t0.3 5.4 it0.3 6.9 -t 0.1
Lysine... 13.5 4- 0.5 13.7 ± 0.3 13.2 -- 0.5 13.0 ± 0.7 13.8 i- 0.3
Histidine... 1.0 it0.2 1.2 it0.4 1.2 -0.2 1.7 -0.1 1.1 -0.1
Tryptophan... 4.1 i 0.3 3.0 i 0.5 1.3 i 0.1 0 2.5 i 0.4
Arginine... 4.2 + 0.1 4.3 i 0.3 4.5 4 0.1 3.5 -h 0.2 4.5 ± 0.2 aValues given inmicromole per cent.
Determined ascysteicacid. cNot determined.
TABLE 2. Aminoacid composition of the9,500-dalton internal proteinsa
Amino acid T4B T4D
Aspartic acid +
as-paragine... 6.9 ± 0.4 7.0 4 0.2
Threonine... 6.3 i 0.2 5.9 A1 0.1
Serine... 6.1 ± 0.2 5.8i 0.3
Glutamic acid +
glutamine... 8.8i 0.3 8.7i4 0.3 Proline... 2.3 ± 0.3 2.7 ± 0.2 Glycine... 9.5 i 0.4 9.0 i 0.2 Alanine... 13.5 i1 0.5 13.0 i: 0.4
Valine... 4.6 i0.3 5.5i 0.4 Half-cystineb... 0 0
Methionine... 1.2 i 0.2 1.4 4 0.2
Isoleucine... 9.5 i 0.4 9.6 i 0.4 Leucine... 9.5 ±t0.2 9.5 ± 0.4 Tyrosine... 1.2 ±t0.2 1.4i 0.1
Phenylalanine... 1.1 i4 0.1 1.5 ± 0.3
Lysine... 13.6 i 0.4 12.9i 0.5 Histidine... 2.5 4± 0.3 2.5 i 0.1
Tryptophan... 1.0 ± 0.4 0.8 4 0.2
Arginine... 1.4 -t0.1 1.7 i 0
aValues given in micromole per cent.
bDeterminedas cysteic acid.
T6mayhavecontained the21,000-dalton protein
in major amounts also, but losses in handling perhaps reduced the final yield. The 21,000-dalton proteins of T4B, T4D, and T2H were shownto
haveanisoelectricpointof aboutpH 9.2,whereas
thatofT2Lwasslightly lower.Theminorinternal proteins ofT4B, T4D, T2H, T2L, and T6 phage
were found to have molecular weights in the range of9,500 daltons. The isoelectric points of the 9,500-dalton proteins of T4B, T4D, T2H, and T6werefoundtobenearpH8.86, as was the
21,000-dalton protein of T2L phage.
The procedures used in the present study to
isolate the internal proteins resulted in good recovery of the 21,000-dalton proteins in chro-matographically and electrophoretically pure
form. The 9,500-dalton proteins, however, were
usually
contaminatedbysmall amounts of otherphage proteins, including the 21,000-dalton internal proteins. The 9,500-dalton proteins,
therefore, will necessarily have to be further
fractionated by gel filtration before they can be characterized in more detail. Since undetermined losses of each of the proteins occurred during
isolation, noattempt has been made to calculate theamountof eachspecies ofinternalprotein per phageparticle.
Amino acid analyses of the two species of internal proteins supported the basic nature of theproteinsexhibitedbytheelectrofocusinggels. Theproteinswerecomposed of nearly 20% basic amino acids (lysine, arginine, and histidine).
Majordifferencesbetween the 21,000-and 9,500-dalton proteins were observed in several amino acids which added support to the premise that
thesetwo protein species wereunique.
452 J. VIROL.
on November 11, 2019 by guest
http://jvi.asm.org/
[image:8.493.46.240.345.586.2]One of the major problemsencountered in the present study has been identification of the
internalproteins. This is evident in Fig. 4, which
shows that the isolates ofthese proteins can be
contaminated by a variety of phage structural
proteins. Simultaneous investigations of other phage structures in this laboratory (13, 14, 21) have been invaluable in this respect. Thus,
con-tamination of theinternalproteinpreparations by 18,000- and 11,000-dalton head structural
pro-teinswereeasily monitoredontheelectrofocusing gels. Three procedures employed in the present
studyhavegreatlyreduced the level of
contamina-tion with other phage proteins. Osmotic shock
with glycerol, instead of high salt, greatly de-creased the amount ofhead structural proteins
released.Atthe sametime,osmotic shockinduced by the glycerol method resulted inionicconditions suitable for chromatography of the proteins on the carboxymethyl cellulose. Use of this resin
allowed recovery of the 21,000-dalton protein virtually free of contamination from the more
acidic 18,000-dalton head proteins. Finally, centrifugation of the osmotic shock supernatant solution for 15 hr after removal of the capsids resulted in quantitative removal of free tail
components and unsedimented capsids which would have accounted forasignificant proportion of theproteins isolated.
Additionalsupport forthe beliefthat the two
proteins isolated were indeed internal proteins of thephage headcamefrompreliminary studies
of
proteins
isolated from two amber mutants ofT4.Electrofocusing gels of proteins isolatedfrom
purified heads of AmN85 (G48) and AmH21
(G54) clearly
demonstrated the presence of thetwo internal proteins. It should also be pointed
outthatBlack
(L. Black,
personalcommunication)
found that there were two, and
possibly three,
internal proteins in T4D, having properties similar to those
properties
reported
here.Also,
Champe (S.
Champe,
personalcommunication)
has indicated that these internal
proteins
werephage specific and
accumuilated
duringthe time-course of infection.Correlation of theprevious
findings
with those ofotherlaboratories hasbeendifficult sinceit isnot known ifthe other groups
isolated
both oftheinternal proteins
together
oronly
one ofthe twoproteins.Levineetal. (31) reportedisolationofthe internal antigen in an
electrophoretically,
chromatographically, and
immunochemically
pure form. Covaletal.(Fed.
Proc.19:253,1960)
statedthat this proteinhad an aminoacidcom-position which was
high
inlysine
and histidine and contained nocysteine.
Both the9,500-dalton
protein isolated in the present
study
and the11,000-dalton
headstructuralprotein
(G.
Forrestand D. Cummings, unpublished data) would fit
this brief description. The composition of the
21,000-dalton internal protein, however, was
clearly not high in histidine.
Fourstudies reported thepossibleexistenceof
multiple internal proteins. Levine (32) reported that the 31S-labeled nonsedimentable proteins
released by osmotic shock were separated by
chromatography and yielded label in several fractions, only one of which possessed antigenic activity. Minagawa (34) also found that 20% of
the 35S-labeled nonsedimentable proteins of T2 were not antigenically active against antibody
produced against ruptured phage. Recently
Bachrach et al. (8) found one major and two minorproteins could be isolated with phage DNA
released by Sarkosyl rupture of the phage head
membrane. Kokurina and Tikhonenko (29) fractionated ruptured phage onSephadex G-200
and found four fractions which reacted to anti-body prepared against ruptured phage. Onlytwo of these four fractions lacked cross-reactivity to
capsid proteins.
Earlier in this report, the close similarities of
the 21,000-dalton internal protein and T4
lyso-zyme werenoted.Thetwoproteinspossesssimilar molecular weights andidenticalisolectric points. The aminoacid compositionsofthe twoproteins
are quite different, however (27, 42). Several earlier reports suggested the presence of a lyso-zyme-like activity in phage lysates and internal protein preparations (26, 32, 34, 37, 38). Panijel (26,37,38) founda "prolysine" activity, capable oflysingacetonepowders ofbacteria, whichwas
released from purified phage by osmotic shock. Levine (32), however, reported that the lytic activity ofPanijel and theinternal antigen were
separable by chromatography. Minagawa (34) found that lysozyme activity was present in
internalproteinpreparations prepared
by
osmotic shock with high salt, but were absent when glycerol was substituted. In the latter case, the lysozyme activity was found associated with the capsids. Emrich and Streisinger (20) recently found lowlevels ofa newlytic
activity
inpurified
phagecarrying deletions of theegene. The
phage
used were es mutants[s
=spackle;
thespackle
mutation allowedegene mutants to
lyse, releasing
free phage (19)]. The low level of lytic activityobserved was not blocked by
antibody
to the egene lysozyme, suggesting the presence of a
second species of
lysozyme
inpurified
phage.
Severalother
possible
functions fortheinternal proteinshavebeenproposed;
theseincludearoleinorganizationof thephageDNA
during
matura-tion (28), possible involvement in
regulation
of transcription ofthephageDNA(11),
andrestora-tion of themembrane functionafter
phage
infec-453
on November 11, 2019 by guest
http://jvi.asm.org/
STONE AND CUMMINGS
tion (17). Several of these theories, and other
possibilities,willbe thesubjectof futurestudyby
using thetwoproteinsisolated here.
ACKNOWLEDGMENTS
WeareindebtedtoSelina Janion for translationof the Koku-rinaandTikhonenkopaper.
This investigation was supported by Public Health Service
grants Al-06472 and AI-08265 from the National Institute of Allergy and Infectious Diseases and GM-01379 from the Na-tionalInstituteofGeneralMedicalSciences.
LITERATURE CITED
1. Adams, M. H. 1959.Bacteriophages. Interscience Publishers Inc., New York.
2. Albertsson,P.-A. 1960.Partitionof cellparticles and macro-molecules. JohnWileyand Sons, NewYork.
3. Ames, B. N., D.T.Dubin, andS. M.Rosenthal. 1958.
Pres-ence of polyamines in certain bacterial viruses. Science (Washington) 127:814-816.
4. Anderson, T. F. 1945.The role oftryptophaneinthe adsorp-tionoftwobacterial virusesontheirhost,E. coli.J.Cell.
Comp. Physiol. 25:17-26.
5. Anderson, T. F. 1948.Theactivationofthebacterialvirus T4by L-tryptophan. J. Bacteriol. 55:637-649.
6. Awdeh, Z. L. 1969. Stainingmethod forproteinsafter iso-electricfocusinginpolyacrylamide gel. Sci. Tools 16:42-43. 7.Bachrach, U., and A. Friedmann. 1967. Purification andsome possible functionsofinternalproteinsfromcoliphage T2. Biochem. Biophys. Res. Commun. 26:596-601.
8.Bachrach, U.,R.Levin,and A. Friedmann. 1970.Studieson phage internal proteins: isolation ofprotein-DNA
com-plexes fromT2phagesandfromphage-infectedbacteria. Virology 40:882-892.
9. Bishop, D. H. L., J. R. Claybrook, and S. Spiegelman. 1967. Electrophoreticseparation of viral nucleic acidson poly-acrylamide gels. J.Mol.Biol. 26:373-387.
10. Champe, S. P., and H.L.Eddleman.1967.Polypeptides asso-ciated with morphogenic defects in bacteriophage T4, p. 55-70. In J. S. Colterand W.Paranchych (ed.), The molecular biology of viruses. Academic Press Inc., New York.
11. Chaproniere-Rickenberg, D. M., H. R. Mahler, and D. Fraser. 1964. The interaction of DNA and internal protein from coliphage T2. Virology 23:96-102.
12. Cummings, D. J. 1963.Subunit basis of headconfigurational changes in T2 bacteriophage. Biochim. Biophys. Acta 68:472-480.
13. Cummings,D.J., A. R. Kusy, V.A.Chapman,S. S.DeLong, and K. R. Stone. 1970. CharacterizationofT-even bac-teriophage substructures. I. Tailfibers and tailtubes. J.
Virol. 6:534-544.
14. Cummings, D. J., V. A. Chapman, S. S. DeLong, A. R. Kusy, andK.R. Stone. 1970. Characterization ofT-even bacteriophagesubstructures.II.Tailplates.J. Virol.
6:545-554.
15. Dale, G., and A. L. Latner. 1968. Isoelectric focusing in polyacrylamide gels. Lancet 1:847-848.
16. Davison,P. F. 1968.Proteinsindenaturingsolvents:gel
ex-clusion studies. Science 161:906-907.
17. Duckworth, D. H. 1970. The metabolism of T4 phage ghost-infected cells. I. Macromolecular synthesis and transport ofnucleic acid andproteinprecursors.Virology40:673-684. 18. Eddleman, H. L., and S. P. Champe. 1966. Components in
T4-infected cells associated with phageassembly.Virology 30:471-481.
19. Emrich, J. 1968. Lysis of T4-infected bacteria inthe absence
oflysozyme. Virology 35:158-165.
20. Emrich,J., and G. Streisinger. 1968. The role of phage
lyso-zymein the life cycle of phage T4. Virology 36:387-391. 21. Forrest, G. L., and D. J. Cummings. 1970. Head proteins
from T-evenbacteriophage. I. Molecular weight charac-terization. J. Virol. 5:398-405.
22.Herriott, R. M., and J. L. Barlow. 1957. The proteincoats
or'ghosts' of coliphage T2. J. Gen. Physiol. 40:809-825. 23. Hershey, A. D.1955. Anupperlimittothe proteincontentof
the germinal substance of bacteriophage T2. Virology 1:108-127.
24. Hershey, A. D. 1957. Some minorcomponents of bacterio-phage T2 particles. Virology 4:237-264.
25.Hirs, C. H. W. 1956. The oxidation of ribonuclease with performic acid. J. Biol. Chem. 219:611-621.
26.Huppert, J., and J. Panijel. 1956. Recherchessurles proly-sines.II.Lecasgen6ral dela synthese des prolysines. Ann. Inst.Pasteur 90:711-727.
27. Inouye,M.,and A.Tsugita. 1968.Amino acidsequenceof T2phagelysozyme.J. Mol. Biol.37:213-223.
28. Kellenberger, E. 1961. Vegetative bacteriophage and the
maturationof thevirusparticles. Advan.Virus Res. 8:1-61. 29. Kokurina,N.K., and T. 1. Tikhonenko. 1969.Isolationand fractionation of inner proteins of T2 andDDVI bacterio-phages. Vop.Virusol. 2:224-228.
30.Kozloff, L. M., and M. Lute. 1960. Calciumcontentof bac-teriophageT2.Biochim.Biophys. Acta 37:420-424. 31. Levine,L., J. L. Barlow, and H. Van Vunakis. 1958. An
inter-nal protein in T2 and T4bacteriophages. Virology 6:702-717.
32. Levine, L. 1961. Immunochemical nature of the internal material in the T-evencoliphages,p.171-182. In M. Heidel-berger and0.J.Plescia(ed.), Immunochemical approaches
to problems in microbiology. The Rutgers University Press, New Brunswick.
33.McConahey,P.J.,and F. J. Dixon.1966. Amethodof trace
iodinationofproteinsforimmunologic studies.Int.Arch. Allergy 29:185-189.
34.Minagawa, T. 1961.Somecharacteristicsoftheinternal pro-teinphageT2.Virology13:515-527.
35.Murakami, W. T., H. VanVunakis, and L. Levine. 1959. SynthesisofT2internalprotein in infectedEscherichia coli, strainB.Virology9:624-635.
36.Nozaki, Y., andC. Tanford. 1967. Acid-basetitrations in
concentrated guanidine hydrochloride. Dissociation
con-stantsof theguanidiniumion and ofsomeaminoacids. J. Amer. Chem. Soc. 89:736-742.
37.Panijel,J., and J. Huppert. 1956.Recherchessurles
proly-sines. 1. La prolysinedu phageFcz. Ann. Inst. Pasteur
90:619-636.
38. Panijel, J. 1959. Lesactivites enzymatiques lieesauxphages
etalasynthese phagioue.Ann.Inst. Pasteur 97:198-217.
39.Rhodes, M. B.,P. R.Azari,and R. E.Feeney. 1958.Analysis, fractionation, andpurificationofeggwhiteproteins with
cellulose cationexchanger,J.Biol. Chem.230:399-408.
40. Sober, H.A. 1968.Handbook ofbiochemistry. The Chemical RubberCo., Cleveland.
41. Stemnberg,N., and S. P.Champe. 1969.Genetic determinant ofaninternal peptideofbacteriophageT4.J. Mol. Biol. 46:377-392.
42. Tsugita,A., and M. Inouye. 1968. Complete primary
struc-tureofphagelysozymefromEscherichiacoli T4. J. Mol. Biol. 37:201-212.
454 J.VIROL.