JOURNAL OFVIROLOGY, Jan. 2011, p. 189–199 Vol. 85, No. 1 0022-538X/11/$12.00 doi:10.1128/JVI.01838-10
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Persistent Friend Virus Replication and Disease in
Apobec3
-Deficient
Mice Expressing Functional B-Cell-Activating Factor Receptor
䌤
Mario L. Santiago,
1,2,3* Diana S. Smith,
1Bradley S. Barrett,
1Mauricio Montano,
4Robert L. Benitez,
4Roberta Pelanda,
3,6Kim J. Hasenkrug,
7and Warner C. Greene
4,5Departments of Medicine,1Microbiology,2and Immunology,3University of Colorado Denver, Aurora, Colorado 80045; Gladstone Institute of Virology and Immunology4and Departments of Medicine, Microbiology and Immunology,5
University of California San Francisco, San Francisco, California 94158; National Jewish Health, Denver, Colorado 802066; and Rocky Mountain Laboratories, National Institute for Allergy and
Infectious Diseases, NIH, Hamilton, Montana 598407
Received 30 August 2010/Accepted 14 October 2010
Rfv3is an autosomal dominant gene that influences the recovery of resistant mice from Friend retrovirus (FV) infection by limiting viremia and promoting a more potent neutralizing antibody response. We previously reported thatRfv3is encoded byApobec3, an innate retrovirus restriction factor. However, it was recently suggested that the Rfv3 susceptible phenotype of high viremia at 28 days postinfection (dpi) was more dominantly controlled by the B-cell-activating factor receptor (BAFF-R), a gene that is linked to but located outside the genetically mapped region containingRfv3. Although one prototypicalRfv3susceptible mouse strain, A/WySn, indeed contains a dysfunctional BAFF-R, two otherRfv3susceptible strains, BALB/c and A.BY, express functional BAFF-R genes, determined on the basis of genotyping and B-cell immunophenotyping. Furthermore, transcomplementation studies in (C57BL/6 [B6]ⴛBALB/c)F1and (B6ⴛA.BY)F1mice revealed that the B6Apobec3gene significantly influences recovery from
FV viremia, cellular infection, and disease at 28 dpi. Finally, theRfv3phenotypes of prototypic B6, A.BY, A/WySn, and BALB/c mouse strains correlate with reportedApobec3mRNA expression levels. Overall, these findings argue against the generality ofBAFF-Rpolymorphisms as a dominant mechanism to explain theRfv3recovery phenotype and further strengthen the evidence thatApobec3encodesRfv3.
Following the discovery of Friend virus (FV) in 1957 (15), studies of different mouse strains infected with FV provided numerous insights into host genetic control of retroviral infec-tions (9, 18, 32). FV infection of susceptible mice, such as BALB and A strains, results in the rapid proliferation of eryth-roblast precursors, leading to erythroleukemia and death. On the other hand, two related mouse strains, C57BL/6 (B6) and C57BL/10 (B10) (3), contain both immunological and nonim-munological resistance genes and do not develop leukemia. One resistance gene isRecovery fromFriendVirus gene3, or
Rfv3. Rfv3 was discovered by Chesebro and Wehrly while studying the phenotypes of crosses and backcrosses between a resistant strain (B10) and susceptible strains (A.BY, A/WySn, and BALB.B) (10) (Table 1). Rfv3 was determined to be a single autosomal gene that acted in a dominant manner to promote the recovery of mice from splenomegaly and leuke-mia by stimulating the development of neutralizing antibody (NAb) responses and inhibiting viremia (10, 12, 14).
Three mapping studies have been performed to determine the molecular identity ofRfv3(19, 22, 45) (Table 1). Plasma viremia at 1 month postinfection was used to determine the
Rfv3 phenotypes of intercrosses and backcrosses between B10.A (Rfv3r/r) and A/WySn (Rfv3s/s) mice, and microsatellite markers were used to map associated chromosomal regions. The first two studies mappedRfv3to a 0.83-centimorgan
re-gion on chromosome 15 that contained approximately 130 genes (19, 45). A third study by the Miyazawa group (22) further narrowed this region to a 61-gene segment and con-firmed an association betweenRfv3 action and the develop-ment of NAbs.
The sequencing of the mouse genome (49) facilitated direct analysis of the gene composition of the Rfv3 chromosomal region. Comparison of theRfv3regions of B6 (Rfv3r/r) and A (Rfv3s/s) mice revealed the enrichment of polymorphisms and signals of positive selection in theApobec3gene in contrast to the 60 other genes (42). Because Apobec3 exhibits broad an-tiretroviral activity (as reviewed in reference 41), B6Apobec3 -deficient mice were developed to investigate whetherRfv3was encoded byApobec3. TheApobec3-deficient mice displayed an
Rfv3susceptible phenotype, but it was theoretically possible thatApobec3deficiency might simply mimic theRfv3 suscep-tible phenotype. To exclude this possibility, transcomplemen-tation experiments mice were performed in F1 mice. Both
Apobec3and Rfv3act in a dominant manner to confer resis-tance. If they were distinct genes, then F1offspring from B6
Apobec3⫺/⫺⫻ A.BY (Rfv3s/s) crosses would have an Rfv3r/s/
Apobec3⫺/⫹genotype and a resistant phenotype. In fact, these
mice had anRfv3susceptible phenotype (42). The failure of B6
Apobec3⫺/⫺to transcomplement the susceptibleRfv3allele of
A.BY provided strong evidence of identity betweenApobec3
andRfv3.Subsequent to this report, Takeda et al. correlated
Apobec3 polymorphisms with Rfv3 genotypes and suggested thatRfv3was encoded byApobec3(46).
Recently, the Miyazawa group called into question whether theRfv3phenotype was due toApobec3(48). They confirmed
* Corresponding author. Mailing address: Division of Infectious Diseases, University of Colorado Denver, Mail Stop B-168, 12700 E. 19th Avenue, Aurora, CO 80045. Phone: (303) 724-4946. Fax: (303) 724-4926. E-mail: [email protected].
䌤Published ahead of print on 27 October 2010.
189
on November 7, 2019 by guest
http://jvi.asm.org/
the ability of the wild-type B6Apobec3gene to promote FV-specific NAb responses in the (B6⫻A/WySn)F1background. However, they suggested the involvement of a linked gene, B-cell-activating factor receptor (BAFF-R). BothBAFF-Rand
Apobec3were located within theRfv3region in the first two mapping studies (19, 45). However, the latestRfv3 mapping study excludedBAFF-R, since it is located⬃300,000 bp outside
D15Mit118, the proposed telomeric boundary of theRfv3 re-gion (22). The recent contention that BAFF-RencodesRfv3
was prompted by the finding that A/WySn mice displayed 8.4-fold higher plasma viremia at 28 days postinfection (dpi) than A/J mice (48). A/J and A/WySn mice have a common origin but exhibit critical differences in B-cell phenotypes: A/WySn mice contain lower B-cell numbers, a decreased percentage of B cells in the spleen, and defects in B-cell maturation in the bone marrow (31). A/J and A/WySn intercrosses mapped this phenotype to the codominant Bcmd locus (21), which was subsequently namedBAFF-R. The defective B-cell phenotype in A/WySn mice was later attributed to the replacement of 8 amino acids in the C terminus of BAFF-R with 21 amino acids encoded by an intracisternal A-particle (IAP) element (2, 47, 50). Current evidence suggests that this modification results in aberrant BAFF-R signaling, leading to decreased B-cell mat-uration and survival (28).
The Miyazawa group reported that B6 BAFF-R⫺/⫺ mice
exhibited ⬎18.5-fold higher viremia at 28 dpi than B6
Apobec3⫺/⫺mice (48). On the basis of these data, the authors
concluded that the BAFF-R defect better accounted for the
Rfv3 recovery phenotype than Apobec3. However, the Rfv3
recovery phenotype was not originally defined in highly resis-tant pure B6 or B10 strains (27) but was defined in F1strains that are susceptible to splenomegaly (Table 1) (9, 13). In fact, the authors demonstrated that (B6Apobec3⫺/⫺⫻A/WySn)F
1 mice have higher viremia at 28 dpi and weaker NAb responses than (B6Apobec3⫹/⫹⫻A/WySn)F
1mice (48), a result that is completely consistent with our prior findings in (B6⫻BALB/ c)F1and (B6⫻A.BY)F1backgrounds (42). Surprisingly, the authors still concluded that BAFF-R accounts for the Rfv3
recovery phenotype, despite the fact that the key comparison with (B6BAFFR⫺/⫺⫻ A/WySn)F
1 mice was not performed. Thus, the role ofBAFF-Rin exacerbating theRfv3 recovery phenotype in (B6⫻A/WySn)F1mice remains unclear.
Aside from A/WySn mice, A.BY and BALB strains were also utilized extensively for definingRfv3(10). Thus, ifBAFF-R
polymorphisms primarily account for theRfv3recovery phe-notype, these strains should also have the same defect ob-served in A/WySn mice. The Jackson Laboratory describes the A.BY strain as having been backcrossed once to the A/WySn line during its derivation (http://jaxmice.jax.org/strain/000140 .html). Thus, the A.BY and A/WySn strains could have similar B-cell phenotypes due to sharedBAFF-Rpolymorphisms.
We therefore evaluated the A.BY and BALB/c strains and show that both these strains express a functional BAFF-R. Thus, while BAFF-R deficiency may mimic theRfv3 suscepti-ble phenotype of high viremia because of defective B-cell re-sponses, the BAFF-R defect does not fit the strain distribution ofRfv3. As further evidence thatRfv3is encoded byApobec3, we now show that, in addition to influencing neutralizing an-tibody responses (42), the B6Apobec3gene also significantly influences recovery from viremia and disease at 1 month postinfection in F1crosses with both BALB/c and A.BY mice. Finally, in contrast to BAFF-R functional differences,Apobec3
mRNA expression levels fit the strain distribution of Rfv3.
Thus, the preponderance of evidence strongly indicates that
Rfv3is encoded byApobec3, and while BAFF-R defects may exacerbate viremia in A/WySn mice, it is notRfv3.
MATERIALS AND METHODS
Mice.B6, BALB/c, A.BY, A/WySn, A/J, and B10.A mice were obtained from the Jackson Laboratory (Bar Harbor, ME). Strain 129/Ola was obtained from
Harlan (Indianapolis, IN). B6Apobec3-deficient mice were derived from the
XN450 cell line (44) and backcrossed for 9 generations. All studies were per-formed in accordance with institutional policies for animal care and use at the University of Colorado Denver and the University of California San Francisco.
BAFF-RandApobec3genotyping.To determineBAFF-Rgenotypes, we
em-ployed a PCR scheme based on a forward primer anchored to exon 3 ofBAFF-R
[image:2.585.42.542.80.237.2](primer 1, 5⬘-CCTCCTCAGAAACCCCTCAT) and a reverse primer anchored
TABLE 1. FV infection phenotypes and genotypes of mouse strains relevant toRfv3
Type Strain Fv2
genotypea Splenomegaly
Rfv3
genotype Viremia NAb
e H-2
genotypea
Cellular
immunity Recovery Reference(s)
Inbred B6b r/r Low r/r Low High b/b High High 27, 40, 42, 48
B10b,c r/r Low r/r Low High b/b High High 9, 13
B10.Ac r/r Low r/r Low High a/a Low High 9, 13
A/WySn s/s Intermediate s/s High Low a/a Low Low 10, 48
A.BY s/s Intermediate s/s High Low b/b High Low 10, 42d
BALB.Bc s/s Severe s/s High Low b/b High Low 10, 13
BALB/cc s/s Very severe s/s High Low d/d Low Low 13, 27, 42
F1hybrids B10.A⫻A/WySn r/s Severe r/s Acutef High a/a Low Low 10, 19, 22, 45
B6⫻A/WySn r/s Severe r/s Acute High a/b Medium Medium 48
B10⫻A.BY r/s Severe r/s Acute High b/b High High 10, 13
B6⫻A.BY r/s Severe r/s Acute High b/b High High 40, 42d
B10⫻BALB.B r/s Severe r/s Acute High b/b High High 10, 13
B6⫻BALB/c r/s Severe r/s Acute High b/d Medium Medium 42d
aFv2facilitates splenomegaly induction, whileH-2is the major histocompatibility complex and controls cellular immunity (9, 18, 32).
bB6 and B10 strains were separated from a nearly inbred C57BL line in 1921 (3).
cB10 and B10.A and BALB.B and BALB/c are congenic strains, respectively, that differ only in theH-2locus.
dData to support these designations are also presented in this study.
eNAb response.
fThe level of viremia is high during acute infection but resolves.
on November 7, 2019 by guest
http://jvi.asm.org/
either to wild-typeBAFF-R(primer 2, 5⬘-CGGCTAGAACAGCACACAAA) or
the IAP insertion characterized in A/WySn mice (primer 3, 5⬘-ACGTTCACGG
GAAAAACAGA) (2, 28). To facilitate multiplexing, the primers were designed to amplify different sizes depending on the genotype (Fig. 1A). To detect the
presence of a 531-bp retroviral insertion in the B6Apobec3exon 2 splice donor
site (43), we utilized a forward primer in exon 2 (5⬘-CCTTCACCATGGGGTC
TTTA) and a reverse primer in exon 3 (5⬘-GATGTCCAGGCTCAGGTTGT) of
Apobec3. Tail-clip DNA samples from different mouse strains were extracted,
and 50 to 100 ng was added into a 40-l PCR mixture containing 1⫻Sweet PCR
mix (SA Biosciences, Frederick, MD) and 20 pmol of each of the three BAFF-R
primers described above. ForBAFF-Rgenotyping, the samples were subjected to
a 15-min hot start at 95°C, followed by 40 cycles at 94°C for 30 s, 58°C for 30 s,
and 72°C for 2 min. The extension time was increased to 4 min forApobec3
genotyping. PCR products were visualized in a 1.5% agarose gel.
Apobec3mRNA quantification.TotalApobec3mRNA levels were quantified as described previously (40). Briefly, total RNA was extracted from normal B6, A.BY, and BALB/c mice using an RNEasy kit (Qiagen, Valencia, CA), and 10 ng
was used for cDNA synthesis using random hexamers in an RT2
reverse tran-scription (RT) kit (SA Biosciences). Real-time PCR was performed using
dif-ferent primer sets (see Fig. 4). TotalApobec3copy numbers were quantified
using primers mA3.F (5⬘-CTGCCATGGACCTATACGAA) and mA3.R (5⬘-T
CCTGAAGCTTAGAATCCTGGT) and a TaqMan probe, mA3.P (5⬘-FAM-C
CAAGGCCTGAATCGCCTGC-TAMRA-3⬘, where FAM is
[image:3.585.44.542.69.484.2]6-carboxyfluores-cein). Exon 2-positive (Exon2⫹) transcripts utilized primers mA3x2.F (5⬘-CGG
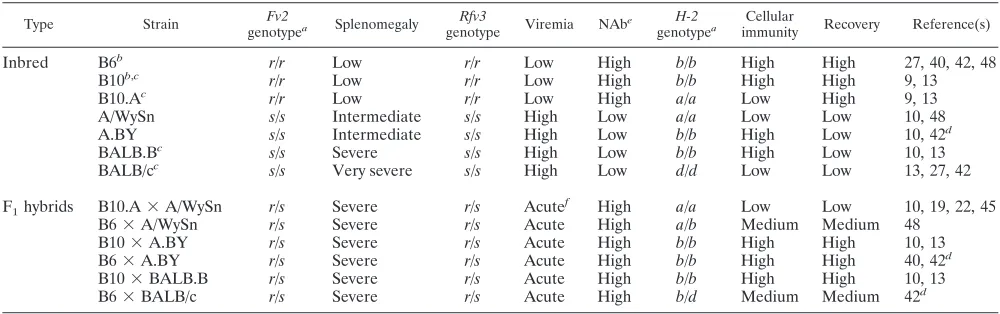
FIG. 1. Functional BAFF-R in A.BY and BALB/c mice. (A) BAFF-R genotyping. Primers anchored to BAFF-R exon 3 that are identical (primer 1) and distinct (primers 2 and 3) between A/J and A/WySn mice were designed for multiplex PCR. Primer 3, in particular, is specific for the IAP insertion that functionally modified A/WySn BAFF-R. Tail-clip DNA samples from several mouse strains were used for genotyping. The amplification of a 511-bp band but not a 260-bp band in A.BY and BALB/c mice indicates that these strains lack the IAP modification and encode a functional BAFF-R. (B) B-cell proportions in the spleen. (Upper panel) Representative flow cytometry plots, gated from the live spleen population, show reduced B-cell percentages in A/WySn mice; (lower panel) A.BY and BALB/c strains have normal B-cell percentages, consistent with a functional BAFF-R. (C) Bone marrow B-cell immunophenotyping. (Lower left panel) B-cell percentages do not vary between the mouse strains; (upper panels) representative flow cytometry plots showing the gating strategy for quantifying mature, IgMlowIgDhighB cells in the bone
marrow; (lower right panel) in contrast to A/WySn mice, A.BY and BALB/c mice maintained normal levels of mature B cells, indicative of BAFF-R function. Data sets were analyzed by Student’sttest, with exactPvalues being noted. Due to their common origin, the differences between A/WySn and A.BY strains were highlighted.
VOL. 85, 2011 Apobec3 AND FRIEND RETROVIRUS RESISTANCE 191
on November 7, 2019 by guest
http://jvi.asm.org/
AAAGATACCTTCTTGTGC) and mA3x2.R (5⬘-CGTGGATGTTGTCCTTG
TTC) and a TaqMan probe, mA3x2.P (5⬘FAM-TGCGATTCACCCGTCTCCC
TT-TAMRA-3⬘, where TAMRA is 6-carboxytetramethylrhodamine). Total and
Exon2⫹Apobec3quantifications were performed in a 22-l reaction mixture
containing 2l cDNA with 20 nM primers and probe in 1⫻TaqMan universal
master mix (Applied Biosystems, Foster City, CA), using a 95°C 15-min hot start, followed by 40 cycles of 94°C for 15 s and 60°C for 60 s. Beta-actin levels were quantified using mouse beta-actin primers (catalogue no. PPM02945A; SA Bio-sciences). Beta-actin levels were estimated using SYBR green (Applied
Biosys-tems). RelativeApobec3levels were interpolated from in-plate standards of
known copy number, using a power equation (r2⬎0.98). The ratio ofApobec3
total or Exon2⫹levels and beta-actin levels were normalized against the lowest
value.
Detection ofApobec3⌬2 transcripts.Spleen cDNA samples from B6, BALB/c, A.BY, and A/WySn mice were subjected to an end-point PCR with primers
positioned in exon 1 (5⬘-AGCCATCGCAAATGCTATTC) and exon 4 (5⬘-TC
CTGAAGCTTAGAATCCTGGT) using the conditions outlined above in the “BAFF-R and Apobec3 genotyping” section. Primers biased for detecting
Apobec3transcripts lacking exon 2 (⌬2) transcripts included mA3⌬2.F (5⬘-ATG CTATTCACCGATCAGGACA), which is based on the exon 1/exon 3 junction,
and mA3⌬2.R (5⬘-TGAAGATGTCCAGGCTCAGGT).Apobec3⌬2 levels were
estimated using SYBR green (Applied Biosystems), as outlined above.
FV infection.Virus stocks were prepared and titers were determined as
de-scribed previously (42). To ensure relevance for the historicalRfv3phenotype
(10), we utilized the original B-tropic FV stock that contained Friend murine leukemia helper virus, spleen focus-forming virus, and lactate dehydrogenase-elevating virus (38). This virus complex was used in the original studies defining
Rfv3(10) and in all three mapping studies ofRfv3(19, 22, 45). In addition, all
mouse strains infected in these studies areFv1b/band are therefore susceptible to
B-tropic FV infection. (B6⫻BALB/c)F1mice were infected intravenously with
140 spleen focus-forming units (SFFUs) of FV stock, while (B6⫻A.BY)F1mice
were infected with 1,400 SFFUs. Plasma, spleen, and bone marrow were har-vested at the indicated time points.
Viremia and spleen infectious titers.Levels of viremia were quantified by
serially diluting plasma and incubating withMus dunnicells pretreated with 4
g/ml Polybrene, as described previously (42). Spleen infectious centers were
determined by cocultivation of serially diluted splenocytes withMus dunnicells.
After 2 to 3 days, theMus dunnicells were fixed with 100% ethanol, washed 3
times with TNE buffer (10 mM Tris, pH 7.4, 200 mM NaCl, 1 mM EDTA), and then incubated with monoclonal antibody (MAb) 720 supernatant for 3 to 4 h at room temperature. MAb 720 binds to the Friend virus gp70 protein even after cell fixation (37). Following two washes with TNE buffer, 1:500 goat anti-mouse IgG horseradish peroxidase conjugate (GE Healthcare, Piscataway, NJ) was incubated for 1 h. After two washes with TNE buffer, MAb 720-positive cells were developed with aminoethylcarbazole (Sigma, St. Louis, MO), and the num-ber of foci was counted. Plasma viremia was expressed as the numnum-ber of focus-forming units (FFUs) per ml of plasma, while spleen infectious centers were expressed as the number of FFUs per spleen.
Plasma viral load.FV RNA copy numbers were measured using a slight modification of a recently published quantitative real-time PCR protocol (20).
Viral RNA from plasma samples (10l) was extracted using an RNEasy kit and
eluted into 100l of buffer (Qiagen, Valencia, CA). Ten microliters of this
extract was combined with 15l of a master mixture containing 1⫻TaqMan
one-step RT-PCR mix (Applied Biosystems, Carlsbad, CA) and 10 pmol of
FV-specific primers (FLVsense, 5⬘-GGACAGAAACTACCGCCCTG;
FLVan-tisense, 5⬘-ACAACCTCAGACAACGAAGTAAGA) and probe (FLVprobe, 5⬘
-FAM-TCGCCACCCAGCAGTTTCAGCAGC-TAMRA). T7-transcribed RNA derived from the FV molecular clone pLRB302 (34) was quantified (Nanodrop Products, Wilmington, DE), serially diluted 10-fold, and included as a standard for each plate. The thermal cycling conditions in an ABI TaqMan 7300 machine included a reverse transcription step at 48°C for 15 min, a hot start for 10 min at 95°C, and 40 cycles of denaturation (95°C for 15 s) and annealing/elongation (60°C for 1 min). RNA copy numbers were computed from the in-plate standard curves using ABI 7300 software. Using this procedure, we routinely achieve
linearr2values of⬎0.98 within a 7-log range, down to 10 copies per reaction
mixture. With an equivalent of 1l input plasma per reaction mixture, the
current assay has a limit of detection of 10,000 copies/ml of plasma.
Flow cytometry.Quantification of FV-positive (FV⫹) cells was performed as described previously (40). Briefly, cells were stained with MAb 34 (8) hybridoma supernatant for 1 h at 4°C, followed by a rat anti-IgG2b antibody conjugated to allophycocyanin (Columbia Biosciences, Columbia, MD) to detect Glyco-Gag-positive cells. For B-cell immunophenotyping, splenocytes and bone marrow cells were stained with IgM-fluorescein isothiocyanate (clone II/41) (Biolegend, San
Diego, CA), IgD-phycoerythrin (11-26c.2a), and CD19-peridinin chlorophyll protein Cy5.5 (6D5) (eBioscience, San Diego, CA) and the corresponding iso-type controls. Cells were phenoiso-typed in a FACSCalibur II machine (BD Bio-sciences, San Jose, CA), collecting 80,000 to 120,000 events per sample, and data were analyzed using Flowjo software (Treestar, Ashland, OR).
Statistical analyses.Differences between means were compared using a
two-tailed Student’sttest. Survival was evaluated using the log-rank (Mantel-Cox)
test using Prism (version 5.0) software (GraphPad Software, San Diego, CA).
Differences were considered statistically significant when thePvalue was⬍0.05.
RESULTS
Prototypic Rfv3 susceptible strains BALB/c and A.BY ex-press functional BAFF-R.In order to determine theBAFF-R
status of A.BY and BALB/c mice, we generated genotyping primers spanning the known IAP insertion in exon 3 of the
BAFF-Rgene in A/WySn mice (2) (Fig. 1A). Our results con-firmed that B6 and A/J mice do not contain the IAP insertion found in theBAFF-Rgene of A/WySn mice (28, 47, 50). Con-sistent with the findings of prior studies suggesting normal BAFF-R function in the B cells of BALB/c mice (17, 39), this strain similarly did not contain the IAP insertion in the
BAFF-Rgene. Likewise, the IAP insertion was not detected in A.BY mice. Thus, there was no genetic evidence forBAFF-R
defects in eitherRfv3s/sBALB/c or A.BY mice.
The mouse strains were next tested for BAFF-R function. TheBAFF-R mutation in the A/WySn line reduces the per-centage of B cells in the spleens of adult mice to 15%. In contrast, the percentage of B cells in the spleens of A/J mice is ⬃50%. (A/J ⫻A/WySn)F1 offspring exhibit an intermediate phenotype, with the mice having 30% splenic B cells, consis-tent with the codominance of the BAFF-R/Bcmd phenotype (31). We therefore evaluated the B-cell percentages in the spleens of five mouse strains. Consistent with prior data (31), splenic B-cell percentages were significantly higher in A/J mice than in A/WySn mice (Fig. 1B, upper panel). BALB/c and A.BY mice also exhibited splenic B-cell percentages signifi-cantly higher than those in A/WySn mice (Fig. 1B, lower panel).
Low B-cell percentages in the spleens of A/WySn mice have been linked to decreased survival of mature B cells (31). Ma-ture B cells are characterized by cell surface expression of low IgM and high IgD (6, 17) (Fig. 1C, upper panels). No signifi-cant difference in the total B-cell percentages in bone marrow was observed between A/WySn and A.BY mice (Fig. 1C, lower left panel). In contrast, the mean percentage of mature IgMlowIgDhighB cells in A/WySn mice was significantly re-duced relative to the mean percentages for the A/J, B6, A.BY, and BALB/c strains (Fig. 1C, lower right panel). Altogether, these genetic and biological data provide strong evidence that
Rfv3s/s BALB/c and A.BY mice express functional BAFF-R. Thus, while defective BAFF-R may accentuate theRfv3 sus-ceptible phenotype of A/WySn mice, these findings are incon-sistent with the notion that theBAFF-Rlocus encodesRfv3.
B6 Apobec3 is critical for recovery from FV viremia and
disease in (B6ⴛ BALB/c)F1 mice. In their study suggesting
that Rfv3was encoded by the BAFF-R locus, the Miyazawa group stated that B6Apobec3deficiency did not fully recapit-ulate theRfv3susceptible phenotype of high viremia at 28 dpi (48). We previously reported a 14.3-fold increase in plasma viremia at 28 dpi in surviving (B6Apobec3⫺/⫺⫻BALB/c)F
1
on November 7, 2019 by guest
http://jvi.asm.org/
mice compared to that in (B6Apobec3⫹/⫹⫻BALB/c)F 1mice. However, because of the increased mortality of these (B6
Apobec3⫺/⫺⫻BALB/c)F
1mice, statistical significance was not achieved (42). We therefore performed additional FV infec-tions in a larger number of mice and analyzed the combined data set. As shown in Fig. 2A, B6Apobec3deficiency resulted in significantly increased mortality during the study period, with (B6Apobec3⫺/⫺⫻BALB/c)F
1mice exhibiting a median survival time of only 15 days. At 28 dpi, the infectious plasma viremia titers in the surviving (B6Apobec3⫺/⫺⫻BALB/c)F
1 mice were at least 8.8-fold higher than the average titers found in (B6 Apobec3⫹/⫹ ⫻ BALB/c)F
1 mice. This difference was highly significant (P⬍0.0001) (Fig. 2B).
It has also been shown thatRfv3controls virus production by leukemic cells late in FV infection by antibody-mediated down-modulation of cell surface envelope glycoprotein (4, 12). Since the ability of infected cells to produce infectious centers in the focal immunofluorescence assay is dependent on high virus production (11),Rfv3s/smice exhibit higher levels of in-fectious centers thanRfv3r/smice even if they contain similar numbers of infected cells. Such an effect is exactly what was
observed in the surviving (B6Apobec3⫺/⫺⫻BALB/c)F 1mice compared to (B6Apobec3⫹/⫹⫻BALB/c)F
1mice at 28 dpi. As expected for these F1 mice, the incidence of leukemia, as measured by gross splenomegaly at 28 dpi, was very high (Fig. 2C). Of note, the numbers of infectious centers were signifi-cantly higher in the (B6Apobec3⫺/⫺⫻BALB/c)F
1mice than (B6Apobec3⫹/⫹⫻BALB/c)F
1mice (Fig. 2D). These findings were again consistent with identity betweenRfv3andApobec3.
Persistent viremia, splenomegaly, and cellular FV infection in (B6ⴛA.BY)F1mice lacking the B6Apobec3allele.Plasma
viremia from more resistant (B6⫻A.BY)F1mice at 28 dpi was measured using the standard focal infectivity assay in Mus dunnicells (37). However, the levels in most of the samples were at or below the limit of detection (600 FFU/ml; data not shown), so a more sensitive quantitative RT-PCR (qRT-PCR) assay was employed (20). Viral RNA quantification of an FV stock with known titer in theMus dunniassay revealed that 1 focus-forming unit corresponded to approximately 16,000 viral RNA copies (or roughly 8,000 viral particles). Viral RNA was extracted from plasma samples from (B6 Apobec3⫹/⫹ ⫻
A.BY)F1and (B6Apobec3⫺/⫺⫻A.BY)F
[image:5.585.113.474.72.361.2]1mice at 28 dpi and
FIG. 2.Apobec3influences the recovery of BAFF-R-sufficient (B6⫻BALB/c)F1mice from FV. (A) B6Apobec3promotes survival from
FV disease. FV-infected mice (140 SFFUs) from three independent cohorts (wild-type F1mice,n⫽24;Apobec3knockout F1mice,n⫽23)
were monitored daily following infection. Kaplan-Meier survival curves were constructed and analyzed using the log-rank (Mantel-Cox) test. (B) Impaired viremia recovery in B6Apobec3-deficient mice. Plasma samples were obtained from four independent cohorts of surviving mice at 28 dpi, and the titer for FV viremia was determined using theMus dunnifocal infectivity assay. The dashed line corresponds to the assay’s 600-FFU/ml cutoff. (C) Persistent splenomegaly in (B6⫻BALB/c)F1mice regardless ofApobec3status. Spleens from (B6Apobec3⫹/⫹⫻
BALB/c)F1and (B6Apobec3⫺
/⫺⫻BALB/c)F
1mice were harvested at 28 dpi and weighed. A normal mouse spleen would weigh⬍150 mg.
(D) B6Apobec3deficiency is associated with higher spleen infectious centers at 28 dpi. Spleens from both cohorts were disaggregated at 28 dpi, and splenocytes were subjected to theMus dunniassay. The number of focus-forming units or infectious centers was normalized for the whole spleen. In panels B to D, the mean values are highlighted with solid lines. The values were compared using a two-tailed Student’st
test, with exactPvalues being noted.
VOL. 85, 2011 Apobec3 AND FRIEND RETROVIRUS RESISTANCE 193
on November 7, 2019 by guest
http://jvi.asm.org/
subjected to the qRT-PCR assay. Surprisingly, both cohorts exhibited substantial plasma viremia (Fig. 3A). However, the increased sensitivity of this assay allowed us to demonstrate that B6Apobec3deficiency impaired recovery from viremia in (B6 Apobec3⫺/⫺ ⫻ A.BY)F
1 mice, where a 121-fold higher plasma viral load was observed at 28 dpi compared to that in wild-type F1animals (Fig. 3A).
In contrast to (B6 Apobec3⫹/⫹ ⫻ A.BY)F
1 mice, (B6 Apobec3⫺/⫺⫻A.BY)F
1mice also did not recover from spleno-megaly at 28 dpi. The mean spleen weights in these animals were 9.0-fold higher (P⫽ 0.0082) (Fig. 3B) and the spleens were grossly enlarged in comparison to the findings for (B6
Apobec3⫹/⫹ ⫻ A.BY)F
1 mice (Fig. 3C). As expected, flow
cytometric studies revealed that the percentage of FV-positive splenocytes from (B6Apobec3⫺/⫺⫻A.BY)F
1mice at 28 dpi was 5.9-fold higher than that from (B6Apobec3⫹/⫹⫻A.BY)F
1 mice (Fig. 3D), translating to about a 53-fold higher level of cellular FV infection when splenic size was accounted for. Subpopulation analysis revealed that these changes were ac-companied by significantly increased infection of splenic eryth-roblasts (data not shown) and B cells at 28 dpi (Fig. 3E). Thus,
Apobec3was critical for recovery from both splenomegaly and cellular FV infection in BAFF-R-sufficient (B6 ⫻ A.BY)F1 mice.
Decreased totalApobec3mRNA levels and aberrant mRNA splicing correlate withRfv3susceptibility. We previously
re-FIG. 3.Apobec3and recovery of (B6⫻A.BY)F1mice from viremia, splenomegaly, and cellular FV infection. (A) Impaired plasma viremia
recovery in (B6Apobec3⫺/⫺⫻A.BY)F
1mice. The numbers of viral RNA copies in plasma samples obtained at 28 dpi were determined by TaqMan
real-time PCR and expressed as log10values. The values from two independent cohorts were combined. The gray dotted line corresponds to the
assay limit of detection (104copies/ml). (B) Spleen weights at 28 dpi. (B6 Apobec3⫹/⫹ ⫻A.BY)F
1mice exhibited higher spleen weights.
(C) Splenomegaly in (B6Apobec3⫺/⫺⫻A.BY)F
1mice at 28 dpi. Spleens from FV-infected (140 SFFUs) (B6Apobec3⫺
/⫺⫻A.BY)F
1mice are
larger than those from (B6Apobec3⫹/⫹⫻A.BY)F
1mice. As a reference, two spleens obtained from A.BY mice at 28 dpi are shown. A normal,
uninfected (B6⫻A.BY)F1mouse spleen would roughly correspond to the smallest spleen in this panel. (D) Cellular FV infection levels in (B6⫻
A.BY)F1spleens at 28 dpi. FV⫹splenocytes were detected using MAb 34, a monoclonal antibody against a cell surface FV antigen, Glyco-Gag.
B6Apobec3deficiency resulted in significantly higher FV infection of total splenocytes. (E) Splenic B cells were significantly more infected in the absence of B6Apobec3. In the relevant graphs, means were highlighted with a solid line. Values were compared using a two-tailed Student’sttest, with exactPvalues being noted.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.82.503.69.459.2]ported that Rfv3 susceptibility correlated with aberrant
Apobec3mRNA splicing in splenocytes (42). In addition to full-lengthApobec3 transcripts, we cloned aberrantly spliced
Apobec3mRNA transcripts lacking exon 2 from BALB/c and A.BY mice that resulted in a frameshift mutation. On the other hand, no⌬2 transcripts were found in B6 mice, which predominantly expressedApobec3mRNA transcripts lacking exon 5. In contrast to⌬2 transcripts, the⌬5 transcripts encode a fully functional protein (1, 5, 33, 42, 46). We did not detect any significant difference in total mRNA levels between B6, A.BY, and BALB/c strains (42). However, shortly after publi-cation of our report, the Neuberger, Ross, and Miyazawa groups reported that B6 mice expressed significantly higher totalApobec3mRNA levels in the spleen than BALB/c (23, 33,
46) and A/WySn (46) strains. Reinspection of the original data set (42) revealed inaccurate interpolation of Apobec3 levels that were multiplied with incorrectly computed beta-actin nor-malization values. When the same data set was reanalyzed by interpolating from an in-plate best-fit standard curve (r2 ⬎ 0.98), mRNA levels were higher in B6 mice than in A.BY and BALB/c mice (data not shown).
To further confirm this reanalysis, we extracted samples from two new donors from each strain and utilized newly designedApobec3primer sets and TaqMan probes. Consistent with subsequent observations and contrary to our initial result,
B6 mice expressed up to 38-fold more Apobec3 mRNA in
[image:7.585.134.448.67.379.2]spleen than BALB/c or A.BY mice, using primers designed in exons 3 and 4 (Fig. 4A). Using exon 2-specific primers and
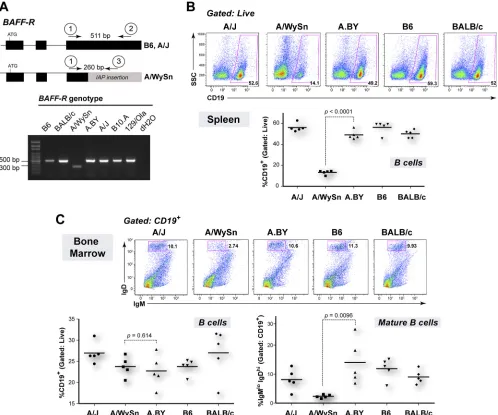
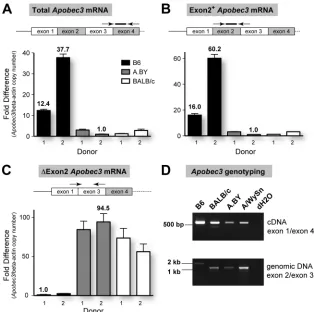
FIG. 4.Apobec3mRNA levels and aberrant splicing correlates with theRfv3phenotype. Total RNA was extracted from the spleens ofRfv3
resistant B6 andRfv3susceptible A.BY and BALB/c mice (two donors each) and reverse transcribed using random hexamers. cDNAs were subjected to multiple real-time PCR analyses, and beta-actin levels were determined for normalization of input cDNA. Serial 10-fold dilutions of plasmid standards with known copy numbers were included in each reaction plate to compute quantities. (A)Rfv3susceptibility correlates with decreasedApobec3mRNA levels. The ratios ofApobec3/beta-actin copy numbers were normalized against the lowest value (A.BY donor 2). B6 mice show higher totalApobec3mRNA expression than A.BY and BALB/c mice. (B) Enrichment of Exon2⫹Apobec3transcripts in B6 mice. Compared to the data in panel A, the fold difference between the B6 and theRfv3susceptible strains using this primer set was higher. This would suggest that the total mRNA pool inRfv3susceptible strains contains transcripts lacking exon 2. (C) Detection ofApobec3⌬2 transcripts inRfv3
susceptible strains. Using a forward primer in the exon 1/exon 3 junction,Apobec3⌬2 transcripts were amplified up to 95-fold more efficiently in A.BY and BALB/c mice than in B6 mice, despite having lower totalApobec3levels. In all panels, arrows and lines correspond to primers and probes, with their relative locations being highlighted. Note that for panel A, the forward primer crosses the exon 3/exon 4 junction, while in panel B, the reverse primer crosses the exon 2/exon 3 junction. Error bars correspond to standard deviations in triplicate determinations. (D) Detection of transcripts with an exon 2 deletion correlate with a corresponding intronic deletion inApobec3. (Upper panel) Amplification of cDNA using primers onApobec3exons 1 and 4 reveals a smaller product consistent with exon 2 deletion (148 bp) in BALB/c, A.BY, and A/WySn mice. While
Apobec3⌬2 transcripts have been previously cloned from A.BY mouse cDNA (42), the A.BY⌬2 product is faint due to lower overall amplification signal. (Lower panel) The genomic region spanning exons 2 and 3 was smaller for BALB/c, A.BY, and A/WySn mice, consistent with the lack of a 531-bp X-MLV LTR insertion in these strains.
VOL. 85, 2011 Apobec3 AND FRIEND RETROVIRUS RESISTANCE 195
on November 7, 2019 by guest
http://jvi.asm.org/
probe, we observed even higher fold differences, with B6 mice expressing up to 60-fold moreApobec3mRNA than BALB/c and A.BY mice (Fig. 4B). Of note,Apobec3mRNA levels in the two donors tested for each strain vary by 2.2- to 3.7-fold using either primer pair, but this intrastrain variation is still substantially lower than the mean 19.4-fold difference between B6 mice and BALB/c or A.BY mice.
The higher fold difference in Apobec3levels using primers located in exon 2 could be explained by the existence of
Apobec3transcripts that lack exon 2, which were readily cloned from BALB/c and A.BY strains in our previous study (42). In fact, normalizing for beta-actin levels, primers specific for
Apobec3⌬2 transcripts resulted in significant amplification sig-nals in A.BY and BALB/c strains in contrast to the sigsig-nals in B6 mice (Fig. 4C), despite the fact that the A.BY and BALB/c strains had significantly lower totalApobec3mRNA expression (Fig. 4A). The existence ofApobec3⌬2 transcripts was con-firmed by amplifying cDNA samples with primers spanning exon 2 (Fig. 4D, upper panel). In contrast to Exon2⫹ tran-scripts,Apobec3⌬2 transcripts represent only a small subset of the mRNA pool inRfv3susceptible mice and could be missed if the overall amplification signal is suboptimal (Fig. 4D, upper panel, A.BY sample). Thus, in the context of reduced total
Apobec3mRNA expression, the relative levels ofApobec3⌬2 transcripts detected inRfv3 susceptible mice are unlikely to significantly influence theRfv3phenotype. In fact,Apobec3⌬2 transcripts may be rapidly degraded (see Discussion).
Recently, a comprehensive study by Kozak and colleagues reported that B6 and other mouse strains encode a 531-bp genomic xenotropic murine leukemia virus (X-MLV) insertion at the exon 2 splice donor site ofApobec3that is associated with increasedApobec3mRNA levels (43). We confirmed this insertion in B6 mice but not BALB/c mice by sizing amplicons following PCR amplification with exon 2/exon 3 primers (Fig. 4D, lower panel) and DNA sequencing (data not shown). Im-portantly, A.BY and A/WySn mice also did not harbor this retroviral insertion (Fig. 4D, lower panel). Thus, in contrast to
BAFF-R polymorphisms, decreased Apobec3 mRNA expres-sion, aberrant splicing ofApobec3exon 2, and the lack of an X-MLV insertion in theApobec3in the exon 2 splice donor site correlated perfectly withRfv3susceptibility (Table 2).
DISCUSSION
The molecular identity of the Rfv3 gene, which controls viremia, virus-neutralizing antibody responses, and recovery from Friend retrovirus infection (10, 14), is of great interest to
anyone trying to understand immune responses to retroviral infections. The recent report that Rfv3 was encoded by
Apobec3(42), a deoxycytidine deaminase that can restrict a broad range of retroviruses, including HIV-1, generated sub-stantial interest and highlighted the general relevance of FV studies to retroviral immunology. However, the role of
Apobec3inRfv3-mediated recovery from FV viremia and dis-ease was recently challenged following the realization that one of the critical strains used in defining and mapping Rfv3, A/WySn, displayed a genetic defect inBAFF-R, a gene linked toRfv3(48).
TheBAFF-R mutation caused by an IAP retroelement in-sertion results in severe B-cell maturation and survival defects in A/WySn mice (2, 28). There is no argument that this muta-tion can affect FV-induced NAb development and recovery from viremia and disease in this strain. However, the strain distribution for Rfv3 susceptibility includes the A.BY and BALB strains (10), both of which express functional BAFF-R, as demonstrated here by genetic and B-cell immunophenotyp-ing methods. Thus, by analyzimmunophenotyp-ing key strains used to originally defineRfv3, we show that theBAFF-Rpolymorphism and its consequent impact on B-cell homeostasis are not well corre-lated with theRfv3phenotype (Table 2).
The study by Tsuji-Kawahara et al. (48) also questioned whether B6Apobec3deficiency results in increased viremia at 28 dpi, as previously reported forRfv3susceptible mice. Anal-ysis of Apobec3 deficiency in both the (B6 Apobec3⫺/⫺ ⫻
BALB/c)F1 and (B6 Apobec3⫺/⫺ ⫻ A.BY)F1 genetic back-grounds demonstrated significantly increased viremia at day 28 in the absence of the B6Apobec3gene. Although B6Apobec3
deficiency resulted in increased mortality in (B6Apobec3⫺/⫺⫻
BALB/c)F1mice, the surviving mice displayed significantly in-creased levels of viremia at 28 dpi. Inin-creased viremia was also detected in the more resistant (B6Apobec3⫺/⫺⫻ A.BY)F
1 mice using a qRT-PCR assay. In addition, B6 Apobec3 defi-ciency in (B6⫻A.BY)F1mice also significantly impaired re-covery from cellular FV infection and splenomegaly, making these normally resistant F1 mice more susceptible to FV-in-duced leukemia. The failure of the B6 Apobec3⫺/⫺ mice to
provide a transcomplementing Rfv3 resistance gene in F1 crosses to either A.BY or BALB/c provides strong evidence thatApobec3encodesRfv3, as previously concluded (42, 46).
[image:8.585.42.543.81.147.2]Further evidence thatApobec3encodesRfv3stems from the correlation betweenApobec3mRNA levels and theRfv3strain distribution. Previous reports from the Neuberger, Ross, and Miyazawa groups show thatRfv3susceptible strains BALB (23,
TABLE 2. BAFFRversusApobec3polymorphisms in relation toRfv3
Strain Rfv3
genotype
% spleen B cells
BAFF-RIAP
insertiona BAFF-R
Apobec3
mRNA levelsb Apobec3splicingc
Apobec3X-MLV
insertiond
B6 r/r Normal Absent Functional High ⌬5 Present
A/WySn s/s Reduced Present Defective Low FL,⌬2e Absente
A.BY s/s Normal Absent Functional Low FL,⌬2 Absente
BALB/c s/s Normal Absent Functional Low FL,⌬2 Absent
aThe IAP insertion results in replacement of the C-terminal 8 residues of BAFF-R with 21 novel amino acids (2, 47, 50).
bOn the basis of quantitative real-time PCR estimations on spleen RNA (23, 33, 46; this study).
c⌬5 is the dominant form in B6 (1, 5, 23, 33, 42); full-length (FL) transcripts are more abundant in the other strains (7, 33; this study).
dCorresponds to a 531-bp retroviral insertion between exons 2 and 3 of theApobec3genomic region (43).
eDetermined by sizing amplicons following PCR of genomic DNA or cDNA with exon-specific primers (this study).
on November 7, 2019 by guest
http://jvi.asm.org/
33, 46) and/or A/WySn (46) exhibitApobec3mRNA levels that are significantly lower than those in Rfv3 resistant C57BL strains (Table 2). Revisiting these experiments, we now concur with these observations. To extend these findings, we also attempted to generate a polyclonal antibody against mouse Apobec3. However, three attempts in rabbits and chickens (Pocono Rabbit Farm, Canadensis, PA; Aves Labs, Tigard, OR; SDIX, Newark, DE) have failed to generate a suitable antiserum. A commercially available anti-mouse Apobec3 an-tibody (catalogue no. 07-0723; Millipore, Billerica, MA) also did not recognize overexpressed Apobec3. The impact of in-creased mRNA levels on endogenous Apobec3 protein levels therefore remains to be determined. However, at least for the human homologue Apobec3G, there is good concordance be-tween mRNA and protein levels in primary cells (35).
The molecular basis for the higher expression of Apobec3
mRNA in B6 mice than in BALB/c, A.BY, and A/WySn mice remains unclear, but a recent study linked this phenomenon to an X-MLV insertion in theApobec3exon 2 splice donor site (43) (Fig. 4D). It was suggested that the long terminal repeat (LTR) region of this insertion may increase transcription levels through an enhancer effect and by affecting splicing (43). No-tably, the absence of this retroviral insertion correlated with the detection of Apobec3 transcripts lacking exon 2 in Rfv3
susceptible mice by our group (42) as well as others (7, 33, 43) (Table 2).Apobec3⌬exon 2 transcripts result in a frameshift mutation, leading to a premature stop at the 36th codon (42). Premature termination codons due to aberrant splicing of cel-lular mRNAs are powerful signals for nonsense-mediated mRNA decay (16, 24, 29), suggesting thatApobec3⌬2 tran-scripts may be rapidly degraded inRfv3susceptible mice. In other words, the X-MLV insertion at theApobec3exon 2 splice site in B6 mice may reduce or prevent aberrant mRNA splicing of exon 2, resulting in higherApobec3mRNA expression. If proven, this would indicate that retroviral insertions in this localized region in chromosome 15 were responsible for both positive (Apobec3) and negative (BAFF-R) effects on retroviral resistance. Of interest, mRNA transcripts from which exon 2 is deleted have also been detected in the human homologues,
Apobec3GandApobec3F(25). The potential role of alternative splicing in regulating endogenous levels of these biologically important restriction factors is currently being explored.
The B6 Apobec3 gene promotes the development of the FV-specific antibody and/or NAb response in inbred B6 mice and in outcrosses, including (B6 ⫻ BALB/c)F1, (B6 ⫻ A.BY)F1, and (B6 ⫻ A/WySn)F1 mice (42, 48). The results that we present in this report substantiate the notion that
Apobec3-dependent enhancement of virus-specific antibody re-sponses is critical for recovery from FV infection and disease. Thus, FV infection of mice with or without the B6Apobec3
gene may provide important clues on the nature of a protective humoral immune response against a pathogenic retrovirus in-fection. However, not all genetic backgrounds may be useful for probing this question. Pure B6 mice do not develop spleno-megaly due to anFv2r/rgenotype (Table 1), while the majority of (B6 Apobec3⫺/⫺⫻ BALB/c)F
1 mice do not survive to 28 dpi. These genetic backgrounds may therefore be more useful for exploring events surrounding acute FV infection. On the other hand, due to the codominant BAFF-R defect in A/WySn mice (31), the (B6⫻A/WySn)F1strain may be more relevant
for retrovirus pathogenesis studies in B-cell-compromised hosts. (B6 ⫻A.BY)F1 mice express functional BAFF-R, ex-hibit normal B-cell function, display susceptibility to FV infec-tion, develop splenomegaly, and survive to 28 dpi, despite the absence of the B6Apobec3gene. We have therefore adopted the (B6⫻A.BY)F1strain to probe how the B6Apobec3gene influences FV-specific humoral immunity.
To date, our studies in the (B6 ⫻ A.BY)F1 background suggest that two events likely synergize to weaken NAb re-sponses in (B6Apobec3⫺/⫺⫻A.BY)F
1mice. These include (i) delayed induction of germinal center B cells and plasmablasts and increased hypergammaglobulinemia during acute infec-tion (40) and (ii) progressive B-cell infecinfec-tion in the context of splenomegaly (Fig. 3). The Miyazawa group also reported in-creased B-cell activation and delayed B-cell maturation in (B6
Apobec3⫺/⫺⫻A/WySn)F
1mice during acute infection (48). In both studies (40, 48), the B6Apobec3gene protected B cells from acute FV infection. Thus, FV infection could directly alter B-cell function, leading to weaker NAb responses. The consequences of FV infection on antigen-specific B-cell devel-opment and function are currently being investigated.
Since FV infects B cells and HIV-1 does not, the relevance of the Apobec3/Rfv3phenotype toward improving HIV-1-spe-cific humoral immunity may be difficult to reconcile. However, it should be noted that CD4⫹T cells, the primary targets of HIV-1 infection, are critical for antigen-specific B-cell devel-opment (30). In germinal centers, which are the primary sites for affinity maturation, antigen-specific B cells form direct con-jugates with CD4⫹T cells (36). Acute HIV-1 infection is as-sociated with germinal center destruction, primarily in mucosal compartments (26). Thus, the relevance of the Apobec3/Rfv3
phenotype for HIV-1 infection has more to do with reducing similar virus-induced pathological events that precede weaker NAb development than with protecting identical cell types from infection. Documenting the sequence of events that result in poor NAb responses in (B6Apobec3⫺/⫺⫻A.BY)F
1mice and testing therapeutic interventions to halt or reverse this pathological course by inducing the expression of the A.BY
Apobec3gene may provide critical insights on modulating hu-man Apobec3G/Apobec3F expression levels in CD4⫹T cells and improving humoral immunity against HIV-1.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants R56 AI-0841230 and R01 AI090795 (to M.L.S.) and R01 A065329 (to W.C.G.).
We thank Brent Palmer and Michelle Dsouza (University of Colo-rado Denver) and Jason Neidleman (GIVI) for flow cytometry assis-tance and Jeffrey Wilusz (Colorado State University) for discussions of alternative splicing and mRNA decay.
REFERENCES
1.Abudu, A., A. Takaori-Kondo, T. Izumi, K. Shirakawa, M. Kobayashi, A. Sasada, K. Fukunaga, and T. Uchiyama.2006. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr. Biol.
16:1565–1570.
2.Amanna, I. J., J. P. Dingwall, and C. E. Hayes.2003. Enforced bcl-xL gene expression restored splenic B lymphocyte development in BAFF-R mutant
mice. J. Immunol.170:4593–4600.
3.Beck, J. A., S. Lloyd, M. Hafezparast, M. Lennon-Pierce, J. T. Eppig, M. F. Festing, and E. M. Fisher.2000. Genealogies of mouse inbred strains. Nat.
Genet.24:23–25.
4.Britt, W. J., and B. Chesebro.1983. Use of monoclonal anti-gp70 antibodies to mimic the effects of the Rfv-3 gene in mice with Friend virus-induced
leukemia. J. Immunol.130:2363–2367.
VOL. 85, 2011 Apobec3 AND FRIEND RETROVIRUS RESISTANCE 197
on November 7, 2019 by guest
http://jvi.asm.org/
5.Browne, E. P., and D. R. Littman. 2008. Species-specific restriction of
apobec3-mediated hypermutation. J. Virol.82:1305–1313.
6.Cariappa, A., M. Tang, C. Parng, E. Nebelitskiy, M. Carroll, K. Georgopou-los, and S. Pillai.2001. The follicular versus marginal zone B lymphocyte cell
fate decision is regulated by Aiolos, Btk, and CD21. Immunity14:603–615.
7.Casey, R. E.2006. Mouse strain-specific splicing of Apobec3. M.Sc. thesis. Worcester Polytechnic Institute, Worcester, MA. http://www.wpi.edu/Pubs /ETD/Available/etd-082206-113216/unrestricted/Casey.pdf.
8.Chesebro, B., W. Britt, L. Evans, K. Wehrly, J. Nishio, and M. Cloyd.1983. Characterization of monoclonal antibodies reactive with murine leukemia viruses: use in analysis of strains of Friend MCF and Friend ecotropic murine
leukemia virus. Virology127:134–148.
9.Chesebro, B., M. Miyazawa, and W. J. Britt.1990. Host genetic control of spontaneous and induced immunity to Friend murine retrovirus infection.
Annu. Rev. Immunol.8:477–499.
10.Chesebro, B., and K. Wehrly.1979. Identification of a non-H-2 gene (Rfv-3) influencing recovery from viremia and leukemia induced by Friend virus
complex. Proc. Natl. Acad. Sci. U. S. A.76:425–429.
11.Chesebro, B., and K. Wehrly.1978. Rfv-1 and Rfv-2, two H-2-associated genes that influence recovery from Friend leukemia virus-induced
spleno-megaly. J. Immunol.120:1081–1085.
12.Chesebro, B., K. Wehrly, D. Doig, and J. Nishio.1979. Antibody-induced modulation of Friend virus cell surface antigens decreases virus production by persistent erythroleukemia cells: influence of the Rfv-3 gene. Proc. Natl.
Acad. Sci. U. S. A.76:5784–5788.
13.Chesebro, B., K. Wehrly, and J. Stimpfling.1974. Host genetic control of recovery from Friend leukemia virus-induced splenomegaly: mapping of a
gene within the major histocompatibility complex. J. Exp. Med.140:1457–
1467.
14.Doig, D., and B. Chesebro.1979. Anti-Friend virus antibody is associated with recovery from viremia and loss of viral leukemia cell-surface antigens in leukemic mice. Identification of Rfv-3 as a gene locus influencing antibody
production. J. Exp. Med.150:10–19.
15.Friend, C.1957. Cell-free transmission in adult Swiss mice of a disease
having the character of a leukemia. J. Exp. Med.105:307–318.
16.Garneau, N. L., J. Wilusz, and C. J. Wilusz.2007. The highways and byways
of mRNA decay. Nat. Rev. Mol. Cell Biol.8:113–126.
17.Gorelik, L., A. H. Cutler, G. Thill, S. D. Miklasz, D. E. Shea, C. Ambrose, S. A. Bixler, L. Su, M. L. Scott, and S. L. Kalled.2004. Cutting edge: BAFF regulates CD21/35 and CD23 expression independent of its B cell survival
function. J. Immunol.172:762–766.
18.Hasenkrug, K. J., and U. Dittmer.2007. Immune control and prevention of
chronic Friend retrovirus infection. Front. Biosci.12:1544–1551.
19.Hasenkrug, K. J., A. Valenzuela, V. A. Letts, J. Nishio, B. Chesebro, and W. N. Frankel.1995. Chromosome mapping of Rfv3, a host resistance gene
to Friend murine retrovirus. J. Virol.69:2617–2620.
20.He, J. Y., H. J. Cheng, Y. F. Wang, Y. T. Zhu, and G. Q. Li.2008. Devel-opment of a real-time quantitative reverse transcriptase PCR assay for de-tection of the Friend leukemia virus load in murine plasma. J. Virol.
Meth-ods147:345–350.
21.Hoag, K. A., K. Clise-Dwyer, Y. H. Lim, F. E. Nashold, J. Gestwicki, M. P. Cancro, and C. E. Hayes.2000. A quantitative-trait locus controlling
periph-eral B-cell deficiency maps to mouse chromosome 15. Immunogenetics51:
924–929.
22.Kanari, Y., M. Clerici, H. Abe, H. Kawabata, D. Trabattoni, S. L. Caputo, F. Mazzotta, H. Fujisawa, A. Niwa, C. Ishihara, Y. A. Takei, and M. Miyazawa.
2005. Genotypes at chromosome 22q12-13 are associated with
HIV-1-ex-posed but uninfected status in Italians. AIDS19:1015–1024.
23.Langlois, M. A., K. Kemmerich, C. Rada, and M. S. Neuberger.2009. The AKV murine leukemia virus is restricted and hypermutated by mouse
APOBEC3. J. Virol.83:11550–11559.
24.Lareau, L. F., A. N. Brooks, D. A. Soergel, Q. Meng, and S. E. Brenner.2007. The coupling of alternative splicing and nonsense-mediated mRNA decay.
Adv. Exp. Med. Biol.623:190–211.
25.Lassen, K. G., S. Wissing, M. A. Lobritz, M. Santiago, and W. C. Greene.
2010. Identification of two APOBEC3F splice variants displaying HIV-1
antiviral activity and contrasting sensitivity to Vif. J. Biol. Chem.285:29326–
29335.
26.Levesque, M. C., M. A. Moody, K. K. Hwang, D. J. Marshall, J. F. White-sides, J. D. Amos, T. C. Gurley, S. Allgood, B. B. Haynes, N. A. Vandergrift, S. Plonk, D. C. Parker, M. S. Cohen, G. D. Tomaras, P. A. Goepfert, G. M. Shaw, J. E. Schmitz, J. J. Eron, N. J. Shaheen, C. B. Hicks, H. X. Liao, M. Markowitz, G. Kelsoe, D. M. Margolis, and B. F. Haynes.2009. Polyclonal B cell differentiation and loss of gastrointestinal tract germinal centers in the
earliest stages of HIV-1 infection. PLoS Med.6:e1000107.
27.Lilly, F.1970. Fv-2: identification and location of a second gene governing the spleen focus response to Friend leukemia virus in mice. J. Natl. Cancer
Inst.45:163–169.
28.Mayne, C. G., I. J. Amanna, and C. E. Hayes.2009. Murine BAFF-receptor residues 168–175 are essential for optimal CD21/35 expression but
dispens-able for B cell survival. Mol. Immunol.47:590–599.
29.McGlincy, N. J., and C. W. Smith.2008. Alternative splicing resulting in
nonsense-mediated mRNA decay: what is the meaning of nonsense? Trends
Biochem. Sci.33:385–393.
30.McHeyzer-Williams, L. J., N. Pelletier, L. Mark, N. Fazilleau, and M. G. McHeyzer-Williams.2009. Follicular helper T cells as cognate regulators of
B cell immunity. Curr. Opin. Immunol.21:266–273.
31.Miller, D. J., and C. E. Hayes.1991. Phenotypic and genetic characterization of a unique B lymphocyte deficiency in strain A/WySnJ mice. Eur. J.
Immu-nol.21:1123–1130.
32.Miyazawa, M., S. Tsuji-Kawahara, and Y. Kanari.2008. Host genetic factors
that control immune responses to retrovirus infections. Vaccine26:2981–
2996.
33.Okeoma, C. M., J. Petersen, and S. R. Ross.2009. Expression of murine APOBEC3 alleles in different mouse strains and their effect on mouse
mammary tumor virus infection. J. Virol.83:3029–3038.
34.Portis, J. L., F. J. McAtee, and S. C. Kayman.1992. Infectivity of retroviral
DNA in vivo. J. Acquir. Immune Defic. Syndr.5:1272–1273.
35.Refsland, E. W., M. D. Stenglein, K. Shindo, J. S. Albin, W. L. Brown, and R. S. Harris.2010. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction.
Nucleic Acids Res.38:4274–4284.
36.Reinhardt, R. L., H. E. Liang, and R. M. Locksley.2009. Cytokine-secreting
follicular T cells shape the antibody repertoire. Nat. Immunol.10:385–393.
37.Robertson, M. N., M. Miyazawa, S. Mori, B. Caughey, L. H. Evans, S. F. Hayes, and B. Chesebro.1991. Production of monoclonal antibodies reactive with a denatured form of the Friend murine leukemia virus gp70 envelope protein: use in a focal infectivity assay, immunohistochemical studies,
elec-tron microscopy and Western blotting. J. Virol. Methods34:255–271.
38.Robertson, S. J., C. G. Ammann, R. J. Messer, A. B. Carmody, L. Myers, U. Dittmer, S. Nair, N. Gerlach, L. H. Evans, W. A. Cafruny, and K. J. Hasen-krug.2008. Suppression of acute anti-Friend virus CD8⫹T-cell responses by
coinfection with lactate dehydrogenase-elevating virus. J. Virol.82:408–418.
39.Rowland, S. L., K. F. Leahy, R. Halverson, R. M. Torres, and R. Pelanda.
2010. BAFF receptor signaling aids the differentiation of immature B cells
into transitional B cells following tonic BCR signaling. J. Immunol.185:
4570–4581.
40.Santiago, M. L., R. L. Benitez, M. Montano, K. J. Hasenkrug, and W. C. Greene.2010. Innate retroviral restriction by Apobec3 promotes antibody
affinity maturation in vivo. J. Immunol.185:1114–1123.
41.Santiago, M. L., and W. C. Greene.2008. The role of the Apobec3 family of cytidine deaminases in innate immunity, G-to-A hypermutation and
evolu-tion of retroviruses, p. 183–206.InE. Domingo, C. R. Parrish, and J. J.
Holland (ed.), Origin and evolution of viruses. Academic Press, London, United Kingdom.
42.Santiago, M. L., M. Montano, R. Benitez, R. J. Messer, W. Yonemoto, B. Chesebro, K. J. Hasenkrug, and W. C. Greene.2008. Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection.
Sci-ence321:1343–1346.
43.Sanville, B., M. A. Dolan, K. Wollenberg, Y. Yan, C. Martin, M. L. Yeung, K. Strebel, A. Buckler-White, and C. A. Kozak.2010. Adaptive evolution of Mus Apobec3 includes retroviral insertion and positive selection at two clusters of
residues flanking the substrate groove. PLoS Pathog.6:e1000974.
44.Stryke, D., M. Kawamoto, C. C. Huang, S. J. Johns, L. A. King, C. A. Harper, E. C. Meng, R. E. Lee, A. Yee, L. L’Italien, P. T. Chuang, S. G. Young, W. C. Skarnes, P. C. Babbitt, and T. E. Ferrin.2003. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res.
31:278–281.
45.Super, H. J., K. J. Hasenkrug, S. Simmons, D. M. Brooks, R. Konzek, K. D. Sarge, R. I. Morimoto, N. A. Jenkins, D. J. Gilbert, N. G. Copeland, W. Frankel, and B. Chesebro. 1999. Fine mapping of the Friend retrovirus
resistance gene, Rfv3, on mouse chromosome 15. J. Virol.73:7848–7852.
46.Takeda, E., S. Tsuji-Kawahara, M. Sakamoto, M. A. Langlois, M. S. Neu-berger, C. Rada, and M. Miyazawa.2008. Mouse APOBEC3 restricts Friend
leukemia virus infection and pathogenesis in vivo. J. Virol.82:10998–11008.
47.Thompson, J. S., S. A. Bixler, F. Qian, K. Vora, M. L. Scott, T. G. Cachero, C. Hession, P. Schneider, I. D. Sizing, C. Mullen, K. Strauch, M. Zafari, C. D. Benjamin, J. Tschopp, J. L. Browning, and C. Ambrose. 2001. BAFF-R, a newly identified TNF receptor that specifically interacts with
BAFF. Science293:2108–2111.
48.Tsuji-Kawahara, S., T. Chikaishi, E. Takeda, M. Kato, S. Kinoshita, E. Kajiwara, S. Takamura, and M. Miyazawa.2010. Persistence of viremia and production of neutralizing antibodies differentially regulated by polymorphic
APOBEC3 and BAFF-R loci in Friend virus-infected mice. J. Virol.84:
6082–6095.
49.Waterston, R. H., K. Lindblad-Toh, E. Birney, J. Rogers, J. F. Abril, P. Agarwal, R. Agarwala, R. Ainscough, M. Alexandersson, P. An, S. E. Antonarakis, J. Attwood, R. Baertsch, J. Bailey, K. Barlow, S. Beck, E. Berry, B. Birren, T. Bloom, P. Bork, M. Botcherby, N. Bray, M. R. Brent, D. G. Brown, S. D. Brown, C. Bult, J. Burton, J. Butler, R. D. Campbell, P. Carninci, S. Cawley, F. Chiaromonte, A. T. Chinwalla, D. M. Church, M. Clamp, C. Clee, F. S. Collins, L. L. Cook, R. R. Copley, A. Coulson, O. Couronne, J. Cuff, V. Curwen, T. Cutts, M. Daly, R. David, J. Davies, K. D. Delehaunty, J. Deri, E. T. Dermitzakis, C. Dewey, N. J. Dickens, M.
on November 7, 2019 by guest
http://jvi.asm.org/
Diekhans, S. Dodge, I. Dubchak, D. M. Dunn, S. R. Eddy, L. Elnitski, R. D. Emes, P. Eswara, E. Eyras, A. Felsenfeld, G. A. Fewell, P. Flicek, K. Foley, W. N. Frankel, L. A. Fulton, R. S. Fulton, T. S. Furey, D. Gage, R. A. Gibbs, G. Glusman, S. Gnerre, N. Goldman, L. Goodstadt, D. Grafham, T. A. Graves, E. D. Green, S. Gregory, R. Guigo, M. Guyer, R. C. Hardison, D. Haussler, Y. Hayashizaki, L. W. Hillier, A. Hinrichs, W. Hlavina, T. Holzer, F. Hsu, A. Hua, T. Hubbard, A. Hunt, I. Jackson, D. B. Jaffe, L. S. Johnson,
M. Jones, T. A. Jones, A. Joy, M. Kamal, E. K. Karlsson, et al.2002. Initial
sequencing and comparative analysis of the mouse genome. Nature420:520–
562.
50.Yan, M., J. R. Brady, B. Chan, W. P. Lee, B. Hsu, S. Harless, M. Cancro, I. S. Grewal, and V. M. Dixit.2001. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell
deficiency. Curr. Biol.11:1547–1552.
VOL. 85, 2011 Apobec3 AND FRIEND RETROVIRUS RESISTANCE 199