Hepatitis B Virus P Protein Reveals Multiple Functions in Replication
and a Common Structure with the Primer Grip in HIV-1 Reverse
Transcriptase
Yong-Xiang Wang,a,bCheng Luo,cDan Zhao,cJürgen Beck,aand Michael Nassala
University Hospital Freiburg, Internal Medicine II/Molecular Biology, Freiburg, Germanya; Key Laboratory of Medical Molecular Virology, Institute of Medical Microbiology, Shanghai Medical College, Fudan University, Shanghai, Chinab; and State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, Chinac
Hepadnaviruses, including the pathogenic hepatitis B virus (HBV), replicate their small DNA genomes through protein-primed reverse transcription, mediated by the terminal protein (TP) domain in their P proteins and an RNA stem-loop,⑀, on the pre-genomic RNA (pgRNA). No direct structural data are available for P proteins, but their reverse transcriptase (RT) domains con-tain motifs that are conserved in all RTs (box A to box G), implying a similar architecture; however, experimental support for this notion is limited. Exploiting assays available for duck HBV (DHBV) but not the HBV P protein, we assessed the functional consequences of numerous mutations in box E, which forms the DNA primer grip in human immunodeficiency virus type 1 (HIV-1) RT. This substructure coordinates primer 3=-end positioning and RT subdomain movements during the polymerization cycle and is a prime target for nonnucleosidic RT inhibitors (NNRTIs) of HIV-1 RT. Box E was indeed critical for DHBV replica-tion, with the mutations affecting the folding,⑀RNA interactions, and polymerase activity of the P protein in a position- and amino acid side chain-dependent fashion similar to that of HIV-1 RT. Structural similarity to HIV-1 RT was underlined by mo-lecular modeling and was confirmed by the replication activity of chimeric P proteins carrying box E, or even box C to box E, from HIV-1 RT. Hence, box E in the DHBV P protein and likely the HBV P protein forms a primer grip-like structure that may provide a new target for anti-HBV NNRTIs.
H
epadnaviruses are small hepatotropic DNA viruses that infecthumans and select mammals and birds. Hepatitis B virus
(HBV), one of the most relevant viral pathogens of humans (20), is
their prototypic member. All hepadnaviruses replicate their⬃3.0-kb
genomes by chaperone-assisted protein-primed reverse transcription
(10), executed by their P proteins. These are unusual reverse
trans-criptases (RTs), which, beyond the common RNA-dependent and DNA-dependent DNA polymerase and RNase H (RH) domains, contain a unique terminal protein (TP) domain at their N termini (Fig. 1A). To initiate reverse transcription, the phenolic OH group of a specific Tyr residue in TP fills the role that conventionally is taken by
the 3=-hydroxyl end of a nucleic acid primer (30).
P proteins are translated from a greater-than-genome-length transcript, the pregenomic RNA (pgRNA), which also acts as mRNA for the viral core protein. The interaction of the P protein
with an RNA stem-loop,⑀, on the pgRNA is crucial for viral
rep-lication; it triggers the coencapsidation of pgRNA and the P pro-tein into newly forming nucleocapsids and the synthesis of a short
DNA oligonucleotide, which is templated by the bulge in⑀and, via
its 5=-terminal nucleotide, becomes covalently attached to the Tyr
residue in TP (“protein priming”). Upon transfer to a 3=-proximal
acceptor site on pgRNA, the oligonucleotide is extended into full-length minus-strand DNA, and the pgRNA template is concur-rently degraded by the P protein’s RH activity. Some 15 to 18
residues from the RNA 5=end are spared and, upon another
tem-plate switch, serve as primers for plus-strand DNA. The final product is a capsid-borne relaxed circular DNA (RC-DNA),
which still carries P protein covalently bound to the 5=end of the
minus strand (reviewed in reference10).
Duck HBV (DHBV) has as yet provided the deepest insights into the mechanism of hepadnaviral replication. Beyond
provid-ing a feasiblein vivoinfection system (41,42), DHBV is the only
hepadnavirus for which replication initiation has successfully
been reconstitutedin vitro. The DHBV P proteinin vitro
trans-lated in rabbit reticulocyte lysate (RRL) (51) or expressed as a
fusion protein inEscherichia coliand supplemented with RRL or
purified chaperones (Hsc70 and Hsp40, with further stimulation
by Hsp90 and Hop [46]) displays authentic protein-priming
ac-tivity when supplied with the cognate DHBV⑀ (D⑀) RNA and
deoxynucleoside triphosphates (dNTPs), as manifested by the
co-valent labeling of the protein if␣-32P-labeled dNTPs are used (8,
25,46). A simpler system exploits severely truncated P proteins
(miniPs) lacking part of TP, the spacer, the RH domain, and the C-terminal part of the RT domain, which exert
chaperone-inde-pendent priming activity (7,8,12,52). In any suchin vitroassays,
the HBV P protein shows, at most, specific binding to its cognate
⑀RNA (19,24) but no enzymatic activity.
The structural correlates to the diverse activities of hepadnavi-ral P proteins are not well defined, because it has been impossible
Received4 January 2012 Accepted4 April 2012
Published ahead of print18 April 2012
Address correspondence to Michael Nassal, [email protected].
Supplemental material for this article may be found athttp://jvi.asm.org/. Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.00011-12
on November 7, 2019 by guest
http://jvi.asm.org/
to generate sufficient amounts of homogeneous P protein for di-rect analyses. Due to its importance as a drug target (all five cur-rently approved chemotherapeutics for hepatitis B virus are
nucleos[t]idic RT inhibitors [NRTIs] [31]), several models for the
RT domain of the HBV P protein have been calculated by using
HIV-1 RT as the template (5, 16,17, 50). The rationale is the
presence in the RT domains of all P proteins of short motifs (boxes
A to E plus box F [or box II] and box G [or box I] [Fig. 1A]) that
are universally conserved in RTs; for an easier comparison of dif-ferent HBV isolates, a unified numbering system accounts
exclu-sively for the RT/DNA polymerase domain (47,61); individual
residues are identified as rtXn, where X denotes the specific amino acid and n its position number. In retroviral RTs, the conserved boxes constitute distinct structure elements that together form the catalytic core. Experimental support for the modeled HBV P pro-tein structures comes mainly from the similar locations of
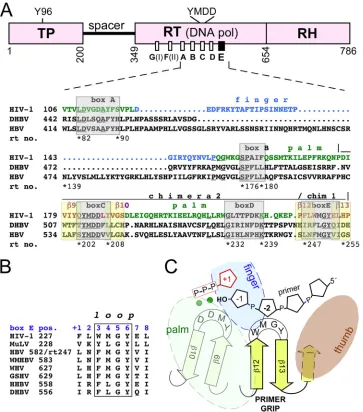
muta-FIG 1Relationship of hepadnaviral P proteins to other reverse transcriptases. (A) Domain structure, relative positions, and primary sequence of the conserved motifs box A to box E in P proteins versus HIV-1 RT. Beyond the RT (DNA polymerase) and RNase H (RH) domains found in all RTs, P proteins contain an extra terminal protein (TP) domain, in which a specific Tyr residue (Y96 for DHBV and Y63 for HBV) acts as an acceptor for the first nucleotide of minus-strand DNA. Numbers are amino acid positions for the DHBV P protein. Of the conserved boxes, box E constitutes the primer grip in HIV-1 RT. The alignment (determined by ClustalW) comprises the sequences in HIV-1 RT and the DHBV and HBV P proteins from boxes A to E; the position of the first amino acid is given in each line; the fourth line (rt no.) refers to HBV P positions in the unified RT numbering system (47). For HIV-1 RT, residues contributing the palm and finger subdomains are shown in green and blue lettering, respectively; the-strands9/10 (box C with the YMDD motif) and12/13 (box E with the primer grip) are highlighted by yellow boxes. HIV-1 RT sequences transplanted into the DHBV P protein are indicated as chimera 2 and chimera 1. For an extended version of the alignment, see Fig. S1 in the supplemental material. (B) Conservation of box E. Shown is an alignment of box E from HIV-1 RT and Moloney murine leukemia virus (MuLV) RT with the corresponding regions of the P proteins of various hepadnaviruses (WMHBV, woolly monkey HBV; WHV, woodchuck hepatitis virus; GSHV, ground squirrel hepatitis virus; HHBV, heron hepatitis B virus). Residues forming the loop between12 and13 in the HIV-1 RT primer grip are boxed. For comparison, box E positions have been numbered from⫹1 to⫹8. (C) The primer grip as a central hub in HIV-1 RT. The primer grip coordinates the relative positions of the palm, finger, and thumb subdomains; its loop interacts with the active-site YMDD motif and with the second-last residue of the primer, thereby properly positioning the primer 3=end toward the incoming dNTP (red pentangle). Green spheres, bivalent metal ions.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.112.475.66.478.2]tions conferring resistance to nucleoside analogs in and around
the conserved boxes as those in HIV-1 RT (61) and from cell
culture studies in which mutations, e.g., in the YMDD motif (36)
of the catalytic center (in box C) and in the proposed helix clamp motif (see Fig. S1 in the supplemental material) downstream of
box E (53–55,60), reduced or abolished viral replication.
How-ever, the limited experimental repertoire applicable to the HBV P protein’s function often does not allow one to distinguish whether such inhibitory mutations act specifically or simply induce global P protein misfolding.
The DHBV P protein shares about 30% sequence identity as well as susceptibility to several NRTIs with its HBV counterpart but offers broader experimental options. We therefore previously began a more systematic analysis of the functional consequences of mutations in the conserved boxes. For instance, the replace-ment of F451 (in box A, corresponding to rtF88 in HBV and ho-mologous to Y115 in HIV-1 RT) rendered the DHBV P protein capable of using ribonucleotide triphosphates as substrates, dem-onstrating the involvement of box A in dNTP versus nucleoside
triphosphate (NTP) discrimination, as in HIV-1 RT (11). Here we
focus on box E, the most C-proximal motif. Based on various crystal structures of HIV-1 RT-nucleic acid complexes, it has been
termed the “DNA primer grip” (48). The sequence forms a hairpin
(12-13) that acts as a connecting hub for all other subdomains
(fingers, palm, and thumb); its loop also contacts the second-last
residue from the primer 3=end (mostly via M230 and G231) and
thus helps in positioning the 3=-terminal OH group close to the
catalytic site for a nucleophilic attack on the incoming dNTP (Fig.
1C). Moreover, the primer grip forms part of a binding pocket for
nonnucleosidic HIV-1 RT inhibitors (NNRTIs), which cause a misalignment of important components at the polymerase active site and/or prevent the dynamic intersubdomain movements
re-quired during polymerization (reviewed in references38and43).
Proper geometry can also be disturbed by mutations in the primer grip, explaining the multiple phenotypes reported for such mu-tants, including impacts on polymerase activity, primer/template
utilization (21,29), dNTP binding (57), the fidelity of DNA
syn-thesis (14,22,56), and (via the thumb domain that connects to the
RH domain) also RNase H activity (21,34).
Characteristic residues for box E in retroviral RTs and in P
pro-teins from diverse hosts (Fig. 1B) are an aromatic residue at position
⫹3, a hydrophobic residue at position⫹4, and a GY dipeptide at
positions⫹5 and⫹6, as exemplified by DHBV P protein residues 556
to 563. Moreover, the box E motif (rt247 to rt254 in the unified HBV
RT numbering system) is almost invariably conserved in the⬎3,000
sequences in the HBV database (http://hivdb.stanford.edu/HBV
/DB/cgi-bin/MutPrevByGenotypeRxHBV.cgi). More directly, box E residue Y561 in the DHBV P protein must be close to the active site, because this residue can replace Y96, although less
efficiently and only in truncated DHBV miniP, in thein vitro
protein-priming reaction (7).
Therefore, we used the DHBV P protein as a model to reveal the potential presence in hepadnaviral P proteins of an actual primer grip element that is functionally and structurally related to that in HIV-1 RT. To this end, we extensively mutagenized the box E motif and subjected the variant P proteins to numerous
func-tional assays, from replication in transfected cells toin vitroD⑀
RNA binding. Together with HIV-1 RT homology-based model-ing and the functionality of chimeric P proteins carrymodel-ing box E, or even the sequence encompassing the catalytic YMDD motif in box
C to box E, from HIV-1 RT, these data demonstrate a common catalytic core architecture of the RT domains of hepadnaviral P proteins and retroviral RTs, in particular the formation of a primer grip-like structure by the box E residues in P proteins. Because the primer grip in HIV-1 RT is the target for multiple
approved and experimental NNRTIs (13,18), the presence of a
primer grip equivalent in P proteins suggests that related com-pounds might be developed into a new class of HBV inhibitors.
MATERIALS AND METHODS
Cell culture and transfection.LMH cells were cultured in Iscove’s mod-ified Dulbecco’s medium (IMDM; Invitrogen) supplemented with 10% fetal bovine serum, 100 U/ml penicillin G, 100g/ml streptomycin, and 0.2 mML-glutamine at 37°C in a humidified atmosphere of 5% CO2. Cells
were transfected by using TransIT-LT1 reagent (Mirus), as recommended by the manufacturer.
Compounds.Lamivudine (LAM) and phosphonoformic acid (PFA) were purchased from Sigma-Aldrich, and MG132 was purchased from Axxora Platform Biochemicals.
Plasmid constructs.All variant DHBV expression vectors were based on plasmid pCD16, which harbors a 1.1-fold DHBV16 genome (GenBank accession no.K01834) under the control of the cytomegalovirus immedi-ate-early (CMV-IE) promoter (33), followed by a T7 promoter in the reverse orientation. Mutations were introduced by standard mutagenic PCR. In the priming-defective variant pCD16-Y96D, the codon for Y96 was changed from TAT to GAT; in the replication-defective variant pCD16-YMHA, the codons for the two essential Asp residues in the YMDD motif were changed to codons for His and Ala (15). In pCD16-P-null, the P open reading frame (ORF) was prematurely terminated by the replacement of the codon for Y96 with TGA. Box E mutations (amino acids [aa] 556 to 563 in the P ORF) introduced into pCD16 or pCD16-Y96D included I556A (ATA⬎GCT), R557A (AGA⬎GCT), F558A (TTC⬎GCT), F558L (TTC⬎CTC), F558W (TTC⬎TGG), L559A (CTC⬎GCT), L559M (CTC⬎ATG), L559I (CTC⬎ATC), G560A (GGT⬎GCT), Y561A (TAC⬎GCT), Q562A (CAG⬎GCT), and I563A (ATT⬎GCT). Expression vectors for the wild-type (wt) and mutant DHBV P proteins with an N-terminal 3⫻FLAG tag were based on plasmid pcDNA3.1/ Hygro(⫺) (Invitrogen). For the chimeric P proteins (chimeras 1 and 2), the DHBV sequences encoding P protein aa 556 to 563 and 505 to 563, respec-tively, were replaced by fragments encoding aa 227 to 234 and 177 to 234 from HIV-1 RT (GenBank accession no.AAK08484.2), respectively. Vectors for thein vitrotranscription and translation of the DHBV P protein were based on plasmid pT7AMVpol16H6 (6), which encodes a His6tag between P
pro-tein aa 2 and 3. pAAV-Dcore contains the DHBV core ORF between the ClaI and XhoI sites in pAAV-MCS (Stratagene). All HBV vectors were based on plasmid pCH-9/3091, encoding a genotype D, subtype ayw, wt HBV genome (32). All plasmid constructs were confirmed by DNA sequencing.
Extraction of capsid-associated viral DNA from transfected cells and Southern blotting.Cells were lysed with NP-40 lysis buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 50 mM NaCl, and 0.5% Nonidet P-40) and incubated with 10 mM Mg acetate, 100 units/ml DNase I, and 100 g/ml RNase A to remove free nucleic acids. Extraction and detection by Southern blotting of intracellular capsid-associated viral DNA were con-ducted as described previously (54). Densitometry was performed by us-ing MultiGauge V2.2 software (Fujifilm).
RNA encapsidation assay.RNA encapsidation efficiency was deter-mined as reported previously (54). In brief, transfected cells were lysed with NP-40 lysis buffer. Lysates were centrifuged at 12,000⫻gfor 2 min, and 40-l supernatants were electrophoresed on a native agarose gel and transferred onto a nitrocellulose membrane by using TNE buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, and 50 mM NaCl). Capsids were de-tected by using anti-DHBV core monoclonal antibody (MAb) 2B9-4F8 (49), followed by peroxidase-conjugated anti-mouse secondary antibody (Jackson ImmunoResearch Laboratories) and ECL reagent (GE Health-care). For the detection of encapsidated viral RNA, the same membrane
on November 7, 2019 by guest
http://jvi.asm.org/
was subsequently treated with 0.2 M NaOH–1.5 M NaCl for 30 s, followed by neutralization with 0.2 M Tris-HCl (pH 7.4)–1.5 M NaCl for 5 min. RNAs were then detected by using a32P-labeled antisense riboprobe.
Western blotting.SDS-PAGE and Western blotting were conducted according to standard procedures. Primary antibodies used were anti-FLAG MAb M2 or MAbs against tubulin, actin, or lamin (all Sigma); a MAb against green fluorescent protein (GFP) (Roche); and mAb9 against the DHBV P protein (58). For detection, the blots were incubated with peroxidase-conjugated secondary antibodies, followed by ECL⫹reagent (GE Healthcare). Chemiluminescent signals were recorded on X-ray film or by using a Fuji LAS 3000 instrument.
Endogenous polymerase assay.For the enrichment of cytoplasmic nucleocapsids, NP-40 lysates were treated with 10 mM Mg acetate, 100 units/ml DNase I, and 100g/ml RNase A and then loaded onto a cushion solution (NP-40 lysis buffer plus 20% sucrose). After centrifugation with a TLS55 rotor at 55,000 rpm for 90 min at 20°C, the pellets were suspended in capsid buffer (20 mM Tris-HCl [pH 7.5], 50 mM NaCl, 1 mM EDTA, 0.01% Triton X-100, 0.1% NP-40, and 0.05%-mercaptoethanol) and then incubated overnight at 4°C. The suspension was briefly sonicated and centrifuged at 15,000⫻gfor 5 min to remove insoluble material. Cleared supernatants were treated with 15 U micrococcal nuclease and 5 mM CaCl2and then incubated with reaction buffer containing 50 mM
Tris-HCl (pH 7.5), 75 mM NH4Cl, 1 mM EDTA, 10 mM EGTA, 20 mM
MgCl2, 0.1% (vol/vol)-mercaptoethanol, 0.5% (vol/vol) Nonidet P-40,
0.4 mM dATP, 0.4 mM dCTP, 0.4 mM dGTP, and 10Ci [␣-32P]dTTP (3,000 Ci/mmol) at 37°C for various times. Labeled viral DNA was re-leased by the addition of 20 mM EDTA, 0.5% SDS, and 0.5g/l protei-nase K and incubation at 50°C for 2 h. After extraction by phenol-chloro-form and precipitation by ethanol, labeled DNAs were separated on 1% agarose gels in 0.5⫻Tris-borate-EDTA (TBE) buffer. Radioactive signals on the dried gels were recorded by using a Typhoon imager and quanti-tated by using ImageQuant software (both GE Healthcare).
DHBV P protein trans-complementation. One microgram of pCD16-P-null plasmid DNA was mixed with either 1 g pCDNA-3⫻FLAG-Dpol or its derivative plasmids and then transfected into LMH cells. Four days later, capsid-associated viral DNA was extracted and de-termined by Southern blotting.
In vitrotranscription.D⑀RNA as the template forin vitropriming was obtained from ClaI-linearized pD⑀1 (6), by using the Ampliscribe T7 high-yield transcription kit (Epicentre Biotechnologies).32P-labeled D⑀
RNA and antisense riboprobes were generated by using the Riboprobe T7 system (Promega) from ClaI-linearized pD⑀1 and NheI-linearized pCD16 plasmids, respectively.
D⑀RNA binding assay.A D⑀RNA binding assay was performed as previously described (6). In brief, using plasmid pT7AMVpol16H6 as the template, His6-tagged P proteins werein vitrotranslated in the presence of
[35S]Met by using the TNT T7 quick coupled transcription/translation
system (Promega). Three microliters of32P-labeled D⑀RNA
(approxi-mately 8 ⫻105 cpm/l) was added to the translated products. The
mixture was incubated at 30°C for 30 min to allow the formation of P protein-D⑀RNA complexes. Complexes were immobilized on Ni-nitrilo-triacetic acid (NTA) agarose beads (Qiagen) in 300l binding buffer (0.1 M sodium phosphate [pH 7.5], 150 mM NaCl, 0.1% NP-40, 20 mM im-idazole, 100g/ml yeast total RNA). After shaking for 2 h at 4°C, the beads were washed twice individually with 1 ml binding buffer and 1 ml TMK buffer (50 mM Tris-HCl [pH 7.5], 40 mM KCl, 10 mM MgCl2, 100
g/ml yeast total RNA). Bound complexes were released by the addition of 2⫻SDS loading buffer (100 mM Tris-HCl [pH 6.8], 4% [wt/vol] SDS, 20% [vol/vol] glycerin, 2% [wt/vol]-mercaptoethanol, and 0.2% [wt/ vol] bromophenol blue) containing 100 mM EDTA and subsequently denatured at 100°C for 5 min.35S-labeled P protein and32P-labeled RNA
were analyzed by SDS-PAGE, followed by phosphorimaging.
In vitroprotein-priming assay.In vitroprotein priming was per-formed as described previously (11). In brief, wt and mutant P proteins werein vitrotranslated in the presence of [35S]Met. Five microliters of the
translation mixture products was analyzed by SDS-PAGE to control translation efficiency. For priming, 10l of translated products was mixed with 0.6M D⑀RNA and 10Ci [␣-32P]dGTP (3,000 Ci/mmol;
Perkin-Elmer) in a 15-l volume containing 10 mM Tris-HCl (pH 7.5), 2 mM MnCl2, and 6 mM MgCl2. The mixture was incubated at 30°C for 1 h and analyzed by SDS-PAGE. After drying, the gels were covered with or with-out 3 layers of Inkjet films and exposed to a phosphorimage plate to separately monitor the32P and35S signals.
Molecular modeling.Molecular models of the RT domains of the wt DHBV P protein and chimeras 1 and 2 were constructed similarly to methods reported previously for the HBV P protein (17,50). BLASTP (1) was used to search for homologs; multiple-sequence alignments were per-formed by using ClustalW from EBI Tools (http://www.ebi.ac.uk/). Based on the results, chain A from the crystal structure (Protein Data Bank [PDB] accession number 1RTD) of the HIV-1 RT and DNA complex (28) was used as the template to model the three-dimensional (3D) structures of RT domains by using Insight II software (Accelrys, Inc., San Diego, CA). Energy minimizations were performed by using molecular mechan-ics in the SYBYL software package (version 6.8; Tripos Associates, St. Louis, MO), with the Kollman all-force field. All protein atoms were as-signed Kollman all-atom charges, and the energy convergence gradient value of the simulation systems was set to 0.005 kcal · mol⫺1· Å⫺1. The geometrical reasonability of the modeled structures was tested by a Pro-check analysis (http://nihserver.mbi.ucla.edu/SAVS/). DNA and dTTP were superimposed onto the RT structures in Pymol (http://www.pymol .sourceforge.net/), followed by energy minimization to remove unreason-able contacts. Graphic representations were generated by using DS View-erPro 5.0 (Accelrys). Distances between atoms were calculated by using 3D-Mol Viewer (a module of Vector NTI 8.0; InforMax, Inc.).
Drug sensitivity assays. LMH cells were transfected with equal amounts of plasmid pCD16, pCD16-F558A, or pCD16-L559A. Five hours later, different concentrations of LAM or PFA were added to the media. Four days later, cells were lysed, and intracellular capsid-associated viral DNA was determined by Southern blotting. All experiments were per-formed five times. The 50% inhibitory concentrations (IC50s) for LAM
and PFA were calculated by sigmoidal curve fitting, as implemented in Origin 8.0 software (OriginLab, Northampton, MA). Statistical signifi-cance was evaluated by a one-way analysis of variance (ANOVA) using Origin 8.0.
RESULTS
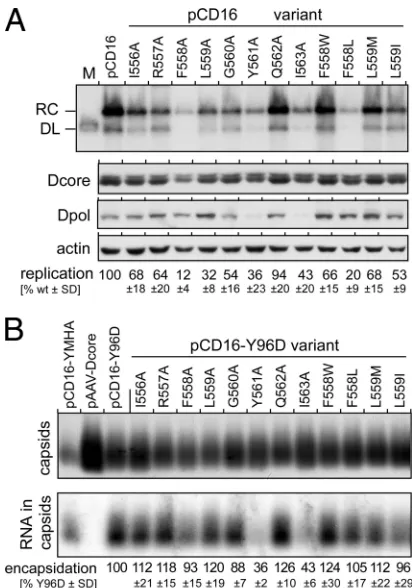
The box E motif in the DHBV P protein is crucial for viral rep-lication.Box E in the DHBV P protein comprises residues I556 to
I563 (Fig. 1B). To address a potential role in viral replication, all 8
residues were individually changed to alanines. All mutations were transferred into the DHBV expression vector pCD16, which harbors a 1.1-fold wt DHBV16 genome under the control of the CMV-IE enhancer/promoter and produces intact virions upon
transfection into the chicken hepatoma cell line LMH (33). Viral
DNAs from cytoplasmic nucleocapsids of cells transfected with the mutant genomes and the wt DHBV16 vector as a reference
were analyzed by Southern blotting (Fig. 2A). The amounts of
viral core protein and P protein, and of cellular-actin, in the
respective cytoplasmic lysates were assessed by Western blotting (Fig. 2A). For quantitation, all signals were first normalized to
equal loading by using the-actin signals. The signals for viral
DNA (RC-DNA plus some double-stranded linear DNA [DL-DNA], which is regularly formed as a by-product of reverse tran-scription) were correlated with the amounts of core protein in the same sample. Mean replication levels and standard deviations (from 5 to 7 experiments for the Ala-scanning mutants and 3 experiments for the other variants) relative to those of wt DHBV,
set at 100%, are indicated at the bottom ofFig. 2A; a graphical
on November 7, 2019 by guest
http://jvi.asm.org/
representation of interexperiment variation is shown in Fig. S6 in the supplemental material. Accordingly, the Q562A mutant rep-licated with wt-like efficiency, and the I556A, R557A, and G560A mutants displayed less than 50% reductions; the lowest replica-tion levels were seen for the F558A, L559A, Y561A, and I563A
variants. Hence, at the latter four positions (positions⫹3,⫹4,
⫹6, and⫹8 in box E), the replacement of the bulky hydrophobic
side chains by the small methyl groups of Ala had the strongest negative impact on P protein functionality. This position-specific
pattern of relative functional importance in box E strongly resem-bles that reported previously for Ala-scanning mutations in the
HIV-1 RT primer grip (21), with mutations W229A, M230A, and
Y232A but not the flanking residues producing the strongest im-pairments in DNA polymerase activity.
We also analyzed four mutants with more conservative ex-changes, namely, the F558W and F558L mutants and the L559M and L559I mutants. In all RTs, the position analogous to DHBV P
protein F558 (residue⫹3 in box E) harbors an aromatic residue
(F, Y, or W) but invariably F in hepadnaviral P proteins. Residue
⫹4 (L559 in DHBV) is L in all avian hepadnaviruses and M in the
mammalian hepadnaviruses and HIV-1 RT; an I residue here is uncommon but may be selected in HBV (rtM250I) in response to
entecavir treatment (50).
The F558W exchange had little impact (Fig. 2A), whereas the
F558L exchange reduced replication strongly, similarly to the
F588A mutation. Hence, an aromatic residue at the⫹3 position of
box E in the P protein appears to be as important as it is in the HIV-1 primer grip. Both the L559M and L559I mutations were
much better tolerated than the L559A exchange. Hence, the⫹4
position of box E exerts a strong preference for a large hydropho-bic side chain, again in line with the preference for L and M at this
position in the primer grip of retroviral RTs (Fig. 1B).
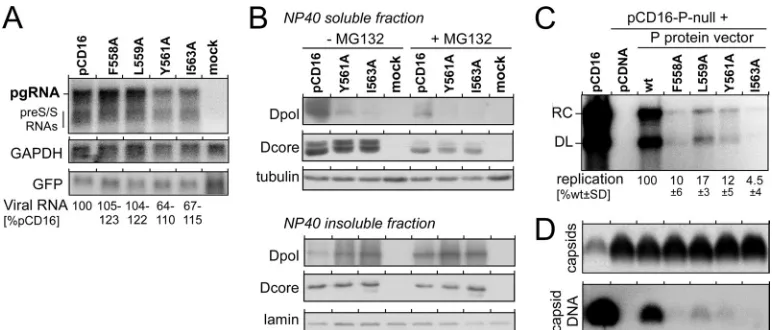
Differential effects of box E mutations on pgRNA packaging.
Because the formation of capsid-borne RC-DNA is a multistep process, multiple reasons for the pronounced negative impact on viral replication of some of the box E mutations could be envis-aged. Reverse transcription is preceded by the coencapsidation of
P protein and pgRNA, which depends on P protein binding to⑀
and the availability of core protein. We did not see major
differ-ences in the levels of core protein (Fig. 2A); potential effects on
transcript levels (Fig. 3A) or pgRNA functionality were excluded
in subsequent experiments by use of atrans-complementation
format (see below). Hence, either the amounts or the properties of the mutant P proteins were responsible for poor replication.
To address pgRNA encapsidation competence, we used native agarose gel electrophoresis (NAGE) of intact capsids to compare their relative pgRNA contents; in this assay, capsids can be de-tected by immunoblotting, and encapsidated viral RNA can be
detected by molecular hybridization with a32P-labeled probe. To
prevent a loss of pgRNA due to conversion into DNA, we analyzed the box E mutants in pCD16 vectors encoding a P protein lacking the protein-priming Tyr residue in TP (Y96D), which prevents reverse transcription but not pgRNA encapsidation. Another packaging-proficient but catalytically inactivated variant with an altered YMDD motif (YMHA) and a vector encoding DHBV core protein (pAAV-Dcore) as the sole viral component served as ad-ditional controls.
As shown inFig. 2B, cytoplasmic lysates of cells transfected
with all viral vectors produced similar amounts of capsids (the low capsid levels from the YMHA variant were specific for this partic-ular sample); enhanced capsid production from plasmid pAAV-Dcore appears to be an intrinsic feature of this vector (M. Nassal,
unpublished data). A visual inspection of the RNA blot (Fig. 2B)
already indicated clear reductions in encapsidated pgRNA levels for the Y561A and I563A variants. As expected, no signal was produced by the pAAV-Dcore vector. For semiquantitation, all RNA signals (determined by phosphorimaging) were normalized to the capsid signals from the same sample (by densitometry of the immunoblot signals), which confirmed about equal
encapsida-FIG 2Distinct impact of box E mutations on overall replication and pgRNA encapsidation of DHBV. (A) Relative replication levels. LMH cells were trans-fected with wt DHBV expression vector pCD16 and its indicated derivatives, and viral DNAs from cytoplasmic nucleocapsids were then analyzed by South-ern blotting. RC, relaxed circular DNA; DL, double-stranded linear DNA; M, marker for DL-DNA. Aliquots from the same cell lysates were analyzed by immunoblotting for-actin and the DHBV core protein (Dcore) to normalize for loading and transfection efficiency. The presence of soluble DHBV P pro-tein was monitored by using an anti-DHBV P propro-tein antibody (Dpol). DNA signal intensities were determined by phosphorimaging and, after normaliza-tion, were used to calculate replication efficiencies relative to that of pCD16, which was set at 100%. Mean values and standard deviations (SD) were ob-tained from 5 to 7 experiments (Ala-scanning mutants) and 3 experiments (non-Ala mutants). A graphical representation of interexperiment variation is shown in Fig. S6 in the supplemental material. (B) Relative pgRNA encapsi-dation efficiencies. LMH cells were transfected with the protein-priming-de-ficient but pgRNA encapsidation-proprotein-priming-de-ficient DHBV vector pCD16-Y96D or its indicated box E derivatives. Vectors for a polymerase-defective DHBV genome (pCD16-YMHA) and for DHBV core protein only (pAAV-Dcore) served as controls. Aliquots from cytoplasmic lysates were subjected to native agarose gel electrophoresis; capsids were detected by immunoblotting, and capsid-borne viral RNA was detected by molecular hybridization using a32P-labeled
DHBV-specific riboprobe. RNA signals were quantified by phosphorimaging and normalized to the capsid signals from the same sample to derive relative encapsidation efficiencies. Mean values⫾SD are based on data from four independent experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.59.265.64.358.2]tion efficiencies for all other variants; the slightly enhanced pgRNA levels per capsid for some variants were not significant. Hence, none of the mutations, save the mutations Y561A and
I563A, affected the mutant P proteins’ ability to interact with⑀
and core protein. Their reduced replication activities (Fig. 2A)
therefore likely resulted from impaired DNA polymerase activity. Notably, low-level pgRNA encapsidation by the Y561A and I563A variants correlated with reduced amounts of detectable P
protein (Fig. 2A), suggesting that these mutations affected P
pro-tein solubility and/or stability. The immunoblot samples had been obtained from the soluble fraction of cell lysates produced by use of NP-40 detergent, which selectively solubilizes the plasma mem-brane. To distinguish between reduced production, increased proteolytic turnover, or insolubility, we directly compared the P protein contents in the soluble and the insoluble fractions (con-taining nuclei and insoluble aggregates) of the NP-40 lysates; in addition, the cells were treated, or not, with the proteasome
inhib-itor MG132 to reduce potential proteasomal degradation (Fig.
3B). DHBV core protein and tubulin or lamin B (for the soluble
and insoluble NP-40 fractions, respectively) were assessed as a
reference. Consistent with the data shown inFig. 2A, much less of
the mutant P proteins than of the wt protein was detectable in the soluble NP-40 fraction, whereas the reverse was true for the insol-uble fraction. Proteasome inhibition for 24 h, starting on day 3 posttransfection, caused no major change in this distribution, ex-cept that the overall core protein levels appeared reduced com-pared to those of the nontreated cells; similar results were
ob-tained after 7 h of MG132 treatment (not shown). We have not investigated whether this reflects a specific effect on the core pro-tein or is due to the known cytotoxic effects of prolonged MG132 treatment. Clearly, however, the P protein Y561A and I563A vari-ants displayed a reduced solubility regardless of the presence or absence of MG132, in line with a negative impact of these muta-tions on protein folding.
Provision of P proteins intransexcludes that altered proper-ties of box E sequence-modified pgRNAs reduce replication.
Al-though not significantly altering pgRNA steady-state levels (Fig.
3A), the nucleotide exchanges required for the box E mutations
could still have affected the quality of the modified pgRNAs as encapsidation and replication templates, thereby contributing to the reduced replication of the F558A, L559A, Y561A, and I563A
variants (Fig. 2A). To exclude such effects, we trans
-comple-mented a DHBV plasmid defective for P protein translation
(pCD16-P-null) with P protein expression vectors encoding 3⫻
FLAG-tagged versions of the wt and the four mutant P proteins. Hence, regardless of the specific P protein, the pgRNA from the pCD16-P-null vector would always be packaged and reverse tran-scribed. Cytoplasmic lysates of the cotransfected cells were then analyzed by Southern blotting and by NAGE. The wt P protein
vector clearly rescued viral replication (Fig. 3C); the lower level of
viral DNA than that of the parental pCD16 vector (about 20%)
probably reflects the preferential cis-encapsidation of pgRNA
molecules from which the P protein is translated (4). Importantly,
all four mutant P proteins generated much weaker signals (5 to
FIG 3Box E nucleotide exchanges reduce replication at the P protein level but not at the pgRNA level. (A) Lack of a significant impact of box E mutations on steady-state levels of viral RNAs. LMH cells were cotransfected with wt DHBV expression vector pCD16 or its indicated derivatives plus a constant amount of a CMV promoter-based GFP expression vector. Mock indicates cells transfected with the GFP vector only. Cytoplasmic RNAs were analyzed by Northern blotting using probes specific for DHBV (top), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (middle), or GFP (bottom). Relative viral RNA levels were calculated by the normalization of the pgRNA plus preS/S RNA signals in one lane to the GAPDH (loading control) and GFP (transfection efficiency control) signals in the same lane, relative to the pCD16 wt signals, which were set at 100%. The range of relative signal intensities derived from 2 independent experiments is indicated. (B) Low levels of soluble P protein Y561A and I563A variants are due largely to insolubility rather than proteasomal degradation. LMH cells were transfected with pCD16 or its Y561A and I563A derivatives. At 3 days posttransfection, cells were split and cultured for another 24 h in the absence or presence of the proteasome inhibitor MG132. After cell lysis using NP-40 detergent, the soluble and insoluble fractions were separately analyzed by immunoblotting for DHBV P protein (Dpol), DHBV core protein (Dcore), and cellular tubulin (soluble) and lamin (insoluble), respectively. Note the strong Dpol signals for both variants in the insoluble fraction and their near absence in the soluble fraction; MG132 treatment did not change this distribution. (C and D) Poor replication rescue by box E-mutated P proteins intrans. LMH cells were cotransfected with a DHBV vector deficient for P protein production (pCD16-P-null, providing in each reaction mixture the same pgRNA as the encapsidation substrate) plus the indicated pCDNA-based expression vectors for 3⫻FLAG-tagged versions of the indicated P proteins or plus the empty pCDNA plasmid. Cells transfected with pCD16 served as the control. (C) Intracellular capsid-borne viral DNAs were analyzed by Southern blotting and quantified by phosphorimaging. Replication efficiencies, relative to those of pCD16-P-null complementation by the wt P protein expression vector, which were set at 100%, were calculated from three independent experiments. (D) Capsids and encapsidated DNA in aliquots from the same samples as those shown in panel C were subjected to NAGE. Capsids were monitored by immunoblotting, and encapsidated viral DNA was monitored by molecular hybridization with a32P-labeled probe.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.99.486.65.230.2]10% of those seen with the wt P protein vector); these data were confirmed by the equally small amounts of viral DNA per capsid
seen in the NAGE analysis (Fig. 3D). Hence, the reduced
replica-tion levels were attributable to the mutant P proteins and not to altered properties of the packaged pgRNA.
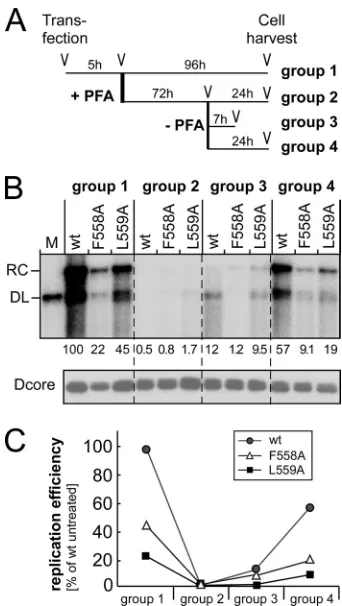
Reduced replication efficiency of box E F558A and L559A mutations is due to decreased polymerase activity.To directly confirm an impact of the F558A and L559A mutations on poly-merase activity, we monitored the kinetics of recovery of viral DNA formation after treatment with the pyrophosphate analog phosphonoformic acid (PFA). PFA traps HIV-1 RT in the
pre-translocated state (39), where the dNTP binding pocket is still
occupied by the primer 3=end and thus prevents the access of the
next incoming dNTP. PFA also inhibits the P protein’s polymerase
activity but not pgRNA encapsidation and protein priming (26).
Cells were transfected with pCD16 vectors encoding the two vari-ants or wt DHBV; at 5 h posttransfection, the cells were split and
subjected to four different treatments (Fig. 4A). Group 1 was
cul-tured without PFA for another 96 h to serve as untreated controls. The treatment groups received 0.4 mM PFA for 72 h; for group 2, treatment was continued until harvest at 96 h posttransfection; group 3 was cultured for another 7 h; and group 4 was cultured for another 24 h in the absence of PFA. The viral DNA in cytoplasmic
nucleocapsids was then analyzed by Southern blotting (Fig. 4B).
Untreated cells showed similar relative amounts of viral DNA as
before (Fig. 2), with the F558A variant reaching about 20% and
the L559A variant reaching about 40% of the level of the wt virus. Permanent PFA treatment reduced DNA levels to nearly unde-tectable amounts; notably, the signals for the two mutants were slightly stronger than those for wt DHBV, suggesting a low level of resistance to the drug, as confirmed below. In cells allowed to recover for 7 h, signals for wt DHBV already exceeded those for
both mutants (group 3) (Fig. 4B), and after 24 h, a similar ratio
of the DNA signals of wt DHBV versus the F558A variant versus the L559A variant was restored as in the untreated cells. A
graphic representation of the results is shown inFig. 4C. As
both variants encapsidated wt-like amounts of pgRNA (Fig.
2B), the reduced levels of viral DNA must result from reduced
polymerase activity.
This conclusion was corroborated by using the endogenous polymerase reaction, in which the P protein present in isolated
nucleocapsids extends immature replicative intermediatesin vitro
when provided with dNTPs; by employing at least one ␣-32
P-labeled dNTP, the extended products can be visualized directly by autoradiography. Here we subjected nucleocapsids from cells kept for 96 h in the presence of 0.4 mM PFA to this procedure and analyzed the levels of newly formed radiolabeled DNA 1 h, 3 h, 5 h, and 16 h after the addition of dNTP (see Fig. S2 in the
supplemen-tal material). As in cells (Fig. 4), thede novo synthesis of 32
P-labeled viral DNA in the mutant nucleocapsids proceeded 5 to 10 times slower and less efficiently than in wt nucleocapsids, corrob-orating a direct impact on polymerase activity.
To detect potentially strand-specific impairments in DNA syn-thesis activity, we analyzed viral DNAs from cells transfected with pCD16 vectors for the F558A and L559A variants by Southern blotting using strand-specific probes (data not shown). The levels of immature minus strands were affected similarly to the corre-sponding mature strands, and no accumulation of single-stranded minus strands (as expected from a defect in plus-strand synthesis)
was observed. Although not excluding minor impacts, these data gave no hints for strand-specific differences.
Box E in the DHBV P protein is important for protein prim-ing and⑀RNA binding.Forin vitropriming, the DHBV P protein
is provided with D⑀RNA as the template plus32P-labeled dNTPs;
the successful formation of the tyrosyl-DNA phosphodiester bond
results in the32P labeling of the protein. The simplest,
chaperone-independent system exploits severely truncated miniP proteins
that lack,inter alia, the C-terminal region, which in HIV-1 RT
forms the thumb (see Fig. S1 in the supplemental material), such
that the region encompassing box E is not stably folded (7). We
therefore used the full-length P proteinin vitrotranslated in RRL
to assess the impact of box E mutations on protein priming, using
[␣-32P]dGTP as the only exogenously added dNTP; dGMP,
spec-ified by the C residue at the last position of the D⑀bulge,
consti-tutes the natural first nucleotide that is added to the protein.
FIG 4Box E mutations F558A and L559A affect the P protein’s DNA poly-merase activity. (A) Treatment schemes for groups 1 to 4. Cells were trans-fected with pCD16 or its F558A and L559A variants. At 5 h posttransfection, cells received fresh media without PFA (group 1) or with 0.4 mM PFA (groups 2 to 4); 72 h later, PFA was removed for groups 3 and 4, and the cells were grown for another 7 h (group 3) or 24 h (group 4) in the absence of PFA. (B) Viral DNA production. Viral DNAs in cytoplasmic nucleocapsids were ana-lyzed by Southern blotting and quantitated by phosphorimaging. Values were normalized to the amount of DHBV core protein present in the same sample. Permanent PFA treatment reduced the DNA signals by 50- to 200-fold (group 2). A release of the PFA block caused a partial recovery of DNA levels at 7 h (group 3) and a more complete recovery at 24 h post-PFA removal (group 4). (C) Graphic evaluation of DNA levels upon different treatments relative to those of untreated group 1, which were set at 100%. Comparable results were obtained upon the monitoring ofde novoDNA synthesis in isolated nucleo-capsids by using the endogenous polymerase reaction (see Fig. S2 in the sup-plemental material).
on November 7, 2019 by guest
http://jvi.asm.org/
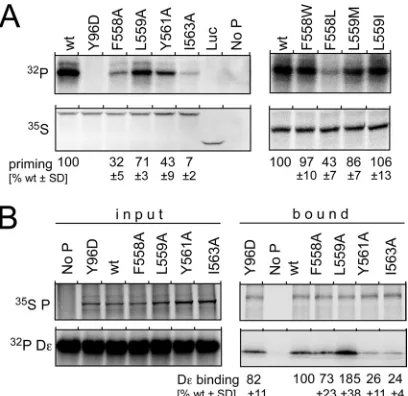
[image:7.585.335.506.64.367.2]The 35S-labeled in vitro translation products of the F558A, L559A, Y561A, and I563A variants plus the wt P protein as a pos-itive control and the Y96D variant as a negative control were first
subjected to centrifugation (at 16,000⫻gfor 20 min) to remove
insoluble molecules. SDS-PAGE followed by autoradiography showed similar amounts of P protein in all supernatants,
includ-ing for the Y561A and I563A variants (Fig. 5A). When subjected to
priming conditions, all reaction mixtures except those containing no P protein or the priming-deficient Y96D variant generated
specific signals but of markedly differing intensities (Fig. 5A).
The signal from the L559A variant was about 70% as intense as that from the wt protein, followed by the Y561A and F558A vari-ants (30% to 40% of the wt signal); the signal for the I563A variant was barely above background levels. These results correlated
largely with the results of the replication assays shown inFig. 2A,
except that the well-priming L559A variant poorly supported rep-lication (see below for a likely explanation). For the F558W,
F558L, L559M, and L559I variants (Fig. 5A, right), high-level
priming activity was seen when F558 was replaced by an aromatic but not an aliphatic residue, and L559 was tolerant to substitu-tions with other large hydrophobic (I and M) residues, as in the replication assays. This indicated that many of the box E muta-tions exert their effects already during the initiation of reverse transcription.
Finally, we directly assessed the D⑀RNA binding capacity of
the F558A, L559A, Y561A, and I563A variants; the wt P protein and the Y96D variant served as positive controls, and a reaction
mixture containing no P served as a negative control. Thein vitro
-translated,35S-labeled, His-tagged proteins were incubated with
32P-labeled D⑀RNA, the proteins were immobilized onto Ni-NTA
beads (6), and the protein and RNA remaining on the beads after
extensive washings were analyzed by SDS-PAGE and
autoradiog-raphy (Fig. 5B; the complete autoradiograms are shown in Fig. S3
in the supplemental material). For quantitation, the RNA signals
were normalized to the full-length P protein35S signals in the same
lane. Expectedly, the wt P protein and the Y96D variant bound
similar amounts of D⑀RNA, as did the F558A variant. Clearly, less
RNA was bound by the Y561A and I563A variants, which also had shown low (Y561A) and very low (I563A) levels of priming
activ-ity (Fig. 5A). Most notably, the L559A variant reproducibly
re-tained about twice as much RNA as the wt protein, indicating an
enhanced affinity for D⑀RNA. Nonetheless, it produced a weaker
priming signal (Fig. 5A), indicating a reduced capacity to use the
bound RNA as the template.
Box E, and even box C to box E, from HIV-1 RT can function-ally replace the corresponding sequences in the DHBV P pro-tein.Together, the results described above suggested that box E in the DHBV P protein is of a similar importance for functionality as the primer grip in HIV-1 RT, implying a similar structure. To substantiate this interpretation, we replaced the box E motif in the P protein (residues I556 to I563) with the primer grip sequence from HIV-1 RT (F227 to L234) (chimera 1), in which the GY
dipeptide is the only strictly identical feature (Fig. 1B). More
dar-ingly, in chimera 2, the entire⬃60-aa sequence, encompassing the
active-site YMDD motif in box C to box E, is derived from the HIV-1 enzyme (P protein N505 to I563 replaced by HIV-1 RT
D177 to L237) (Fig. 1A). Functional activity would be expected
only if both sequences, despite their divergent primary sequences (15 identical plus 11 similar residues within 59 residues total), could adopt similar 3D structures. To avoid impacts on pgRNA by the numerous nucleotide exchanges, functionality was assessed in the trans-complementation format. The cotransfection of pCD16-P-null with the wt P protein expression vector and the
empty pCDNA plasmid served as the control. As shown inFig. 6,
both chimeric P proteins supported the formation of the same
pattern of RC-DNA plus DL-DNA as thetrans-complemented wt
P protein albeit with a lower efficiency (around 30% of the wt). This was partly due to reduced levels of soluble chimeric P
pro-teins (Fig. 6), but additional contributions of the numerous
mu-tations to reduced polymerase activity are likely. Most impor-tantly, however, replacement by the short and even the long HIV-1 RT sequences still rendered the chimeric proteins capable of fulfilling all P protein functions, including pgRNA encapsida-tion and proper protein-primed reverse transcripencapsida-tion. Compared to the strong negative effects that even some single box E muta-tions exerted on DHBV P protein function, this high level of tol-erance toward multiple mutations derived from the homologous
FIG 5Differential impact of box E mutations onin vitropriming and D⑀RNA binding of the DHBV P protein. (A)In vitropriming. The wt P protein, the protein-priming-defective but D⑀RNA binding-proficient Y96D mutant, and the indicated box E variants werein vitrotranslated in rabbit reticulocyte lysates in the presence of [35S]Met; a reaction programmed for luciferase (Luc)
translation and nonprogrammed lysate (No P) served as the controls. For priming, the reaction mixtures were supplemented with D⑀RNA and [␣
-32P]dGTP in priming buffer, as detailed in Materials and Methods. Primed, 32P-labeled P proteins were detected by autoradiography after SDS-PAGE. The
amounts of soluble P protein present in each reaction mixture were deter-mined by SDS-PAGE and35S autoradiography of aliquots withdrawn from
each translation reaction mixture immediately before adjustment to priming conditions. Relative priming activities were assessed by normalizing the32P
signals to the35S-labeled P protein signals, with those for the wt P protein set at
100%. Mean values and SD are derived from data from three independent experiments. (B) D⑀RNA binding. His6-tagged P proteins werein vitro
trans-lated in the presence of [35S]Met and then mixed with32P-labeled D⑀RNA;
one aliquot each was analyzed directly by SDS-PAGE and autoradiography for protein and RNA (panel input). From the rest of each mixture, P protein and bound RNA were captured on Ni-NTA beads. The protein and RNA remain-ing bound after extensive washremain-ing were analyzed by SDS-PAGE/autoradiog-raphy as described above for panel A. D⑀RNA binding activity was assessed by normalizing the32P-labeled RNA signals to the35S-labeled P protein signal in
the same lane. Mean values relative to those produced by the wt P protein, set at 100%, and SD were derived from data from three independent experiments. Note the significantly enhanced RNA binding by the L559A variant. The com-plete autoradiograms are shown in Fig. S3 in the supplemental material.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.62.266.66.264.2]HIV-1 RT sequence strongly supports the formation of similar 3D structures by both protein sequences.
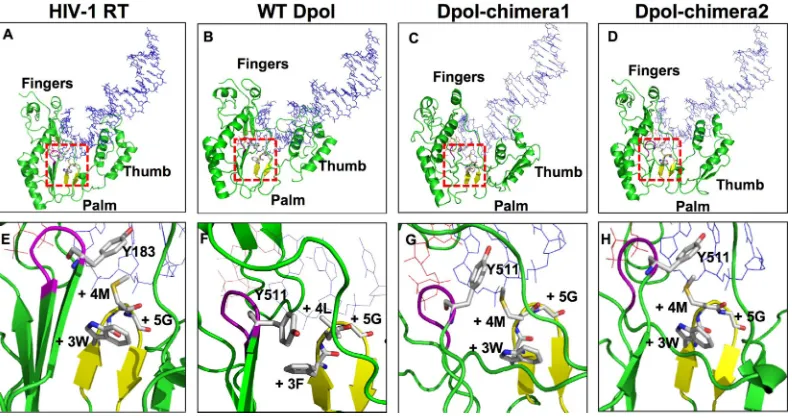
Molecular modeling is consistent with a primer grip-like structure of box E in the DHBV P protein.The modeled struc-tures of the RT domain of the HBV P protein have been derived by
using HIV-1 RT as the template (5,16,17). Here we took a similar
approach for the DHBV P protein and its two chimeric deriva-tives, as described in Materials and Methods. In brief, P protein residues 382 to 657, and the corresponding regions of chimeras 1 and 2, were modeled on residues 53 to 314 from chain A of the crystal structure of HIV-1 RT complexed with DNA and dTTP
(PDB accession number 1RTD [28]); that structure displays the
typical right-hand architecture with palm, fingers, and thumb (Fig. 7A). DNA and dTTP were then superimposed onto the en-ergy-minimized model structures.
The DHBV sequence adopted an architecture that was similar
overall to that of HIV-1 RT (Fig. 7B), and the box E residues
formed a hairpin structure (Fig. 7F), as in the HIV-1 RT primer
grip (Fig. 7E), around which the subdomains, including the
YMDD active-site motif, and the 3=-proximal primer strand
resi-dues organize. Resiresi-dues L559 and G560 were situated at the tip of
the primer grip hairpin (Fig. 7F), like HIV-1 RT residues M230
and G231 (residues⫹4 and⫹5) (Fig. 7E) and, thus, were similarly
able to contact the penultimate primer residue and properly
po-sition the terminal 3=-OH relative to the incoming dNTP. These
interactions would be perturbed by the replacement of either
res-idue with Ala. F558, at the⫹3 position, contacted Y511 in the
YMDD motif (highlighted in purple inFig. 7EtoH), an
arrange-ment that was overall similar although not identical to that of HIV-1 RT residues W229/M230 (in the primer grip) and Y183 (in the YMDD motif); there, M230, rather than W229, is closest to
FIG 6Replacement of box E, or box C to E, by the corresponding HIV-1 RT elements yields functional P proteins. Cells were cotransfected with pCD16-P-null plus pCDNA vectors encoding the 3⫻FLAG-tagged wt DHBV P pro-tein or its chimera 1 and 2 derivatives (Fig. 1A). Cotransfection with an empty pCDNA vector and transfection with the wt DHBV expression plasmid pCD16 served as controls. In chimera 1, box E residues 556 to 563 were replaced by HIV-1 RT residues 227 to 234; in chimera 2, residues 505 to 563 (box C to E) were replaced by HIV-1 RT residues 177 to 234. Viral DNA from cytoplasmic nucleocapsids was analyzed by Southern blotting, and levels of DHBV core protein and tubulin were monitored by Western blotting. The presence of soluble P proteins was addressed by using anti-FLAG and anti-DHBV P pro-tein antibodies. Replication capacities relative to those of thetrans -comple-mented wt P protein were calculated from the DNA signal intensities after normalization to tubulin and core protein levels in the same sample; means⫾ SD are from three independent experiments.
FIG 7Molecular modeling supports the formation of a primer grip-like structure by box E in the DHBV P protein. (A to D) Overall structures of the RT domains plus the bound template/primer duplex (in blue) and dTTP (in red) from HIV-1 RT (PDB accession number 1RTD) and modeled structures of the wt DHBV P protein (Dpol) and its chimeric derivatives in which box E (chimera 1) or box C to box E (chimera 2) were replaced by the corresponding HIV-1 RT elements. The primer grip hairpin is shown in yellow, template and primer strands are shown in blue, and bound dTTP is shown in red. The red squares indicate the parts of the structures that are enlarged in panels E to H. (E to H) Close-up views of the interactions between the active-site YMDD motif (in magenta, with Y183 in HIV-1 RT and Y511 in the P protein shown as stick models) and the primer grip loop. The residue numbering in the primer grip is described in the legend ofFig. 1B. As in the HIV-1 enzyme, primer grip residues⫹3,⫹4, and⫹5 are in close proximity to the penultimate primer residue and the Y residue in the YMDD motif; however, the major interaction with the latter is with the position⫹3 residue F558 rather than with the position⫹4 residue as in HIV-RT (M230); in the chimeric proteins, a more HIV-1 RT-like arrangement appears to be restored.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.95.232.62.276.2] [image:9.585.96.492.432.641.2]Y183. The replacement of F558 by smaller residues would weaken the interaction with the YMDD motif, consistent with the reduced replication capacity of the F558A variant, and would possibly af-fect resistance to NAs (a modest hypersensitivity toward LAM was indeed observed [see below]). The side chains of Y561 and I563 displayed extensive hydrophobic interactions with residues of the palm and thumb subdomains (L432, I570, and F609, and L432, I435, I556, and I570) (see Fig. S4 in the supplemental material), which would not be maintained by the smaller Ala side chain; the implied impact on overall folding is in line with partial misfolding
and the observed insolubility. The⫹7 position (Q562 in DHBV P)
shows substantial variability between different RTs (Fig. 1B), and
thus, the negligible impact of its replacement by Ala in the DHBV P protein is consistent with a nonessential role in folding and activity.
The modeling of the two chimeric DHBV P protein RT do-mains resulted in a similar overall architecture, with some
altera-tions in the thumb domain (Fig. 7C andD). The primer grip
hairpin was also formed, yet the contact to Y511 in the YMDD
motif was mediated largely by M559 (position⫹4) rather than
W558 (Fig. 7GandH), very similar to HIV-1 RT. Together, these
and other (7) modeling studies make a common structure of the
box E motifs in the DHBV P protein and HIV-1 RT highly plau-sible.
F558A and L559A in the DHBV P protein inversely affect sen-sitivity to lamivudine and PFA.As a final, independent line of evidence supporting such a similarity, we analyzed the impact of the box E mutations F558A and L559A in the DHBV P protein on sensitivity to lamivudine and PFA. In HIV-1 RT, the primer grip does not directly contact the incoming dNTP, but due to its scaf-folding function, mutations can indirectly affect susceptibility to nucleoside analogs. For instance, the box E mutation F227A ren-dered the enzyme 8-fold hypersensitive to azidothymidine (AZT), and various HIV-1 RT mutants selected for resistance to PFA
in-creased sensitivity to AZT (23,44). Both DHBV P protein
muta-tions induced an about 3-fold, statistically significant resistance to
PFA (mean IC50s of 189M and 143M, versus 55M for wt
DHBV), whereas sensitivity to lamivudine was about 2-fold
en-hanced (IC50s of 3.4 and 2.5M, versus 6.8M for wt DHBV)
(see Fig. S5 in the supplemental material for representative titra-tion experiments). For HBV, a similar cross talk between box E and the dNTP binding pocket in the HBV P protein was suggested by the changes in sensitivity to lamivudine and entecavir reported
previously for mutations of the box E⫹4 residue rtM250 (3,50);
for instance, rtM250V (in the absence of other mutations)
ren-dered the HBV enzyme 7-fold hypersensitive to LAM (50). Hence,
these data are again consistent with a common structural role for the box E motif in hepadnaviral P proteins and the DNA primer grip in HIV-1 RT.
Similar impacts on replication of box E mutations in human HBV and DHBV P proteins.DHBV and HBV are more closely related to each other than is either of them to HIV-1. Hence, if box E in the DHBV P protein can adopt a primer grip-like structure, this should also hold for the HBV protein, as also implied by
previously reported HBV RT models (5,16,17,50). Mutations of
box E in HBV P protein should then have functional consequences similar to those for DHBV. We therefore replaced residues F584, M585 (rtM250), Y587, and I589 in the HBV P protein (box E
positions⫹3, ⫹4, ⫹6, and ⫹8, corresponding to F558, L559,
Y561, and L563 in DHBV, respectively) with Ala and compared
the replication competence and pgRNA encapsidation capacity of the corresponding virus variants to those of wt HBV upon
trans-fection into the human hepatoma cell line Huh7 (Fig. 8A). Indeed,
all mutations reduced replication and RNA encapsidation
effi-ciency without detectably influencing capsid levels (Fig. 8Band
C). Relative reductions were similar to those of DHBV for the⫹6
position and were more pronounced for the⫹8 position and less
pronounced for the⫹3 and⫹4 positions than in DHBV (F558
and L559); a similarly minor effect of the rtM250A mutation at
position⫹3 was also seen previously in the context of a different
HBV genotype, where the corresponding residue is M598 (3).
Apart from the limitations in the accurate quantitation of South-ern blots and immunoblots (see Fig. S6 in the supplemental ma-terial for the DHBV mutants), the gradual differences in the mu-tational impact on HBV and DHBV likely reflect the substantial sequence divergence between the two proteins. However,
com-pared with the results reported for mutations at position⫹3 other
than M598A (3), the trend for DHBV L559 mutations was very
similar in that larger aliphatic amino acids were better tolerated than smaller ones. Hence, together, the data are fully compatible with a primer grip-like structure of the box E residues in HBV and DHBV P proteins.
DISCUSSION
Despite the importance of HBV as a pathogen, structural infor-mation on hepadnaviral replication is limited to homology-based
FIG 8Box E mutations in the HBV P protein generate replication phenotypes similar to those of the DHBV P protein. (A) Southern blot of HBV DNA from cytoplasmic nucleocapsids. Human Huh7 cells were transfected with the wild-type HBV expression vector pCH-9/3091 (pCH-wt) or derivatives carrying the indicated P protein mutations. For easier comparison, the box E positions of the mutated residues are indicated on the top; homologous residues in HBV versus DHBV are F584/F558, M585 (rtM250)/L559, Y587/Y561, and I589/ I563. Relative replication efficiencies were calculated from the DNA signals relative to the amounts of HBV core protein (Hcore) and tubulin present in the same samples; means⫾SD are from three independent experiments. ss, sin-gle-stranded DNA. (B) Relative viral DNA contents of cytoplasmic nucleocap-sids. Equal aliquots from the cytoplasmic lysates were analyzed by NAGE, followed by the immunodetection of HBV capsids and molecular hybridiza-tion of packaged DNA with a32P-labeled HBV probe. The relative DNA
con-tent per capsid was calculated from the intensity of the DNA signals normal-ized to the capsid signals from the same sample. (C) Relative viral RNA contents of cytoplasmic nucleocapsids. Huh7 cells transfected with the same vectors as those described above (A) were cultured in the presence of 250M LAM to prevent the conversion of packaged pgRNA into DNA. Aliquots from cytoplasmic lysates were subjected to NAGE and processed as described above (B). RNA contents per capsid relative to those of wild-type vector-transfected cells (100%) are derived from three experiments.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.302.543.65.214.2]models of the DNA polymerase (RT) domain of the HBV P
pro-tein (5,16,17,50). While consistent with NRTI resistance data,
HBV provides few experimental opportunities to directly validate the structural predictions. Using the related DHBV P protein as a
model, here we extended our previous functional analysis (11) to
the conserved box E, which forms the DNA primer grip in HIV-1 RT. All our experimental data plus molecular modeling strongly suggest that box E in the DHBV P protein as well as the HBV P protein adopts a structure similar to that of the primer grip in HIV-1 RT, with a similarly central role in coordinating the
inter-actions between RT subdomains and the primer 3=end. Most
compellingly, this is indicated by the replication competence of chimeric DHBV P proteins carrying box E, or the entire sequence from box C to box E, from HIV-1 RT. Our data provide new insights into the replication mechanism of hepadnaviruses that should also be useful for the design of new nonnucleosidic anti-HBV compounds.
Specific versus global effects of box E mutations of the DHBV P protein: evidence for a primer grip.Most box E mutations in the DHBV P protein, unless in chemically similar residues,
re-duced the overall replication efficiency (Fig. 2A); such negative
effects could be specific or may simply reflect global misfolding. For distinction, we tested the impacts of the mutations on P pro-tein solubility and stability, and we subjected the variants to mul-tiple functional assays, reflecting the consecutive steps of hepad-naviral reverse transcription.
The soluble fraction of the NP-40 lysates should contain P protein encapsidated in cytoplasmic nucleocapsids plus free, non-aggregated molecules; molecules that were non-aggregated and/or as-sociated with nuclei, large cellular complexes, or debris should accumulate in the insoluble fraction. In transfected cells, the frac-tion of nonencapsidated P protein exceeded that of encapsidated
molecules, and most of those apparently precipitated (59). We did
not differentiate between capsid-borne and free soluble P protein, yet clearly, the Y561A and I563A variants produced much less soluble protein than did the wt P protein and the other variants (Fig. 2Aand3B), suggesting a folding defect. In all viral RTs, the
corresponding primer grip positions⫹6 and⫹8 are occupied by
an aromatic residue and a large hydrophobic residue (Fig. 1B),
which undergo extensive hydrophobic interactions with the palm and thumb subdomains, and the same was predicted in our mod-eled DHBV RT domain (see Fig. S4 in the supplemental material). The lack of these interactions in the Y561A and I563A mutants could therefore easily promote misfolding and insolubility; nota-bly, an L234A exchange (equivalent to I563A) in HIV-1 RT also
affected proper folding (29). In turn, the lack of soluble P protein
largely explains the reduced replication capacities of these two
variants. Insolubility in the RRLin vitrotranslation system was less
pronounced, perhaps because only freshly translated protein was
analyzed. However, the low levels of D⑀RNA binding andin vitro
priming displayed by the two variants (Fig. 5) suggest that even the
RRL-soluble molecules are functionally impaired. In sum, an im-pact of the Y561A and I563A mutations on folding is compatible with the predicted primer grip structure, but more specific con-clusions can be drawn from the other variants, which did not display obvious folding problems.
Implications for the hepadnaviral replication mechanism.
While protein priming is unique to hepadnaviruses, the subse-quent chain elongation reactions must proceed as with other re-verse transcriptases. Mutations in the primer grip of HIV-1 RT,
depending on their nature and relative position, can severely
im-pair polymerase activity (14,21,22,29,34,56,57). Due to the
central position of the primer grip, such an impairment can occur by various mechanisms; these may be summarized as misalign-ments of the components involved in catalysis and/or a hindrance of the dynamic structure changes (“ratcheting”) that accompany the cycling of the enzyme through successive polymerization steps
(43). A comprehensive mechanistic analysis was beyond the scope
of this study, but the data still allow for several conclusions of relevance for the hepadnaviral replication mechanism.
Box E variants other than the Y561A and I563A variants dis-played low-replication phenotypes despite the absence of obvious
solubility or pgRNA packaging deficits (Fig. 2). We first confirmed
a direct effect on the P protein’s polymerase activity by monitoring the kinetics of DNA synthesis after the release of a block by PFA. In
both intact cells (Fig. 4) as well as isolated nucleocapsids (see Fig.
S2 in the supplemental material), the F558A and L559A variants generated less DNA from the encapsidated pgRNA than the did the wt protein, and they did so with slower kinetics. In HIV-1 RT,
the corresponding box E positions⫹3 and⫹4, W229 and M230,
and the universally conserved G at position⫹5 form the loop of
the12-13 primer grip hairpin and mediate the contacts to the Y
residue in the YMDD motif and the⫺2 residue in the primer (Fig.
1C). In our modeled DHBV P protein RT domain, F558 and L559
are located at equivalent positions (Fig. 7F). The tolerance of the
polymerase activity to replacement by the HIV-1-specific W and M residues (F558W and L559M) but not by Ala with its small side chain (F558A and L559A) is fully consistent with this model. Sim-ilarly, the replacement of the universally conserved glycine at
po-sition⫹5 by a more bulky amino acid would affect the interaction
with the second-last primer residue and the proper positioning of
the primer 3=end. Indeed, the G560A mutant displayed a
mod-estly reduced replication competence despite the maintained
pgRNA encapsidation efficiency (Fig. 2). Thus, all these data are in
line with box E of the P protein forming a primer grip structure with a function during polymerization homologous to that in other RTs.
Perhaps the most compelling evidence for a common catalytic core structure comes from the ability of the two chimeric P pro-teins to support DHBV replication, with the formation of the same replication products as those formed by the genuine DHBV
P protein (Fig. 6). Hence, despite the replacement of box E, or
even the entire sequence from box C containing the catalytic YMDD motif to box E, by the divergent sequence from the HIV-1 enzyme, the chimeric proteins were able to specifically package pgRNA and to initiate and complete reverse transcription,
includ-ing RNase H-mediated pgRNA degradation. Thus, the unique⑀
RNA and TP-dependent steps of hepadnaviral replication also proceeded normally although with decreased efficiency. Notably, the reduction in the overall replication level was not more pro-nounced than that caused by the single F558A and L559A muta-tions in the genuine P protein context.
Together with the importance of the YMDD motif in P pro-teins and our previous demonstration that DHBV P protein F451 in box A fulfills a role in dNTP versus NTP discrimination analo-gous to that of Y115 in HIV-1 RT, our new data firmly establish a common catalytic core structure in the RT domains of P proteins and retroviral reverse transcriptases; in turn, this strongly sup-ports the plausibility of the modeled P protein RT domain struc-tures.