Enfuvirtide (T20)-Based Lipopeptide Is a
Potent HIV-1 Cell Fusion Inhibitor:
Implications for Viral Entry and
Inhibition
Xiaohui Ding,a,bXiujuan Zhang,cHuihui Chong,a,bYuanmei Zhu,a,b Huamian Wei,a,bXiyuan Wu,a,bJinsheng He,cXinquan Wang,dYuxian Hea,b MOH Key Laboratory of Systems Biology of Pathogens, Institute of Pathogen Biology, Chinese Academy of Medical Sciences, and Peking Union Medical College, Beijing, Chinaa; Center for AIDS Research, Chinese Academy of Medical Sciences, and Peking Union Medical College, Beijing, Chinab; College of Life Sciences and Bioengineering, School of Science, Beijing Jiaotong University, Beijing, Chinac; Ministry of Education Key Laboratory of Protein Science, Center for Structural Biology, School of Life Sciences, Tsinghua University, Beijing, Chinad
ABSTRACT The peptide drug enfuvirtide (T20) is the only viral fusion inhibitor used in
combination therapy for HIV-1 infection, but it has relatively low antiviral activity and easily induces drug resistance. Emerging studies demonstrate that lipopeptide-based fu-sion inhibitors, such as LP-11 and LP-19, which mainly target the gp41 pocket site, have greatly improved antiviral potency andin vivostability. In this study, we focused on de-veloping a T20-based lipopeptide inhibitor that lacks pocket-binding sequence and tar-gets a different site. First, the C-terminal tryptophan-rich motif (TRM) of T20 was veri-fied to be essential for its target binding and inhibition; then, a novel lipopeptide, termed LP-40, was created by replacing the TRM with a fatty acid group. LP-40 showed markedly enhanced binding affinity for the target site and dramatically in-creased inhibitory activity on HIV-1 membrane fusion, entry, and infection. Unlike LP-11 and LP-19, which required a flexible linker between the peptide sequence and the lipid moiety, addition of a linker to LP-40 sharply reduced its potency, implying different binding modes with the extended N-terminal helices of gp41. Also, inter-estingly, LP-40 showed more potent activity than LP-11 in inhibiting HIV-1 Env-mediated cell-cell fusion while it was less active than LP-11 in inhibiting pseudovirus entry, and the two inhibitors displayed synergistic antiviral effects. The crystal struc-ture of LP-40 in complex with a target peptide revealed their key binding residues and motifs. Combined, our studies have not only provided a potent HIV-1 fusion in-hibitor, but also revealed new insights into the mechanisms of viral inhibition.
IMPORTANCE T20 is the only membrane fusion inhibitor available for treatment of
viral infection; however, T20 requires high doses and has a low genetic barrier for resistance, and its inhibitory mechanism and structural basis remain unclear. Here, we report the design of LP-40, a T20-based lipopeptide inhibitor that has greatly im-proved anti-HIV activity and is a more potent inhibitor of cell fusion than of cell-free virus infection. The binding modes of two classes of membrane-anchoring lipo-peptides (LP-40 and LP-11) verify the current fusion model in which an extended prehairpin structure bridges the viral and cellular membranes, and their complemen-tary effects suggest a vital strategy for combination therapy of HIV-1 infection. More-over, our understanding of the mechanism of action of T20 and its derivatives bene-fits from the crystal structure of LP-40.
KEYWORDS HIV-1, gp41, fusion inhibitor, T20, lipopeptide
Received20 May 2017Accepted14 June 2017
Accepted manuscript posted online28 June 2017
CitationDing X, Zhang X, Chong H, Zhu Y, Wei H, Wu X, He J, Wang X, He Y. 2017. Enfuvirtide
共T20兲-based lipopeptide is a potent HIV-1 cell
fusion inhibitor: implications for viral entry and
inhibition. J Virol 91:e00831-17.https://doi.org/
10.1128/JVI.00831-17.
EditorFrank Kirchhoff, Ulm University Medical Center
Copyright© 2017 American Society for
Microbiology.All Rights Reserved.
Address correspondence to Xinquan Wang, xinquanwang@mail.tsinghua.edu.cn, or Yuxian He, yhe@ipbcams.ac.cn.
X.D., X.Z., and H.C. contributed equally to this work.
VACCINES AND ANTIVIRAL AGENTS
crossm
on November 7, 2019 by guest
http://jvi.asm.org/
H
uman immunodeficiency virus type 1 (HIV-1) infection requires fusion between viral and cellular membranes, which is mediated by the trimeric viral envelope (Env) glycoproteins gp120 and gp41 (1, 2). Binding of the surface subunit of gp120 to the cell receptor CD4 and a coreceptor, CCR5 or CXCR4, triggers a cascade of confor-mational changes in the gp120/gp41 complex, which activate the fusogenic function of gp41. The fusion peptide (FP) at the N terminus of gp41 is exposed, enabling its insertion into the cell membrane, thus generating a prehairpin intermediate (PHI) that bridges the viral and target cell membranes. Next, the C-terminal heptad repeat (CHR) of gp41 collapses in an antiparallel manner into the hydrophobic grooves created by the trimeric N-terminal heptad repeat (NHR) coiled coil, resulting in a stable six-helix bundle (6-HB) that drives two membranes in close apposition for a fusion reaction (3–5). A prominent feature of 6-HB structures is the deep pocket on the C-terminal portion of NHR helices, which is penetrated by three hydrophobic residues (Trp628, Trp631, and Ile635) from the pocket-binding domain (PBD) of the CHR helix (3–6). Recently, a subpocket, located immediately downstream of the deep pocket, has been identified (7–9). Both pockets play critical roles in the stability of the 6-HB core and offer ideal target sites for anti-HIV agents (6, 7, 9–11).Peptides derived from the NHR or CHR sequence of gp41 can bind to the prehairpin intermediate to form a heterogeneous 6-HB and prevent viral 6-HB formation, thereby inhibiting membrane fusion in a dominant-negative fashion (10, 12, 13). T20 (enfu-virtide) is currently the only approved viral fusion inhibitor used in combination therapy of HIV-1 infections; however, T20 has several defects that limit its clinical use, including a low genetic barrier for drug resistance and a shortin vivohalf-life (14–17). Regardless, the success of T20 has stimulated considerable efforts to explore the mechanisms of viral fusion and inhibition and to design next-generation inhibitors with improved pharmaceutical profiles (18–20). The CHR peptide C34 (34 residues) has been widely used as a template for inhibitor design, as it contains the pocket-binding sequence and exhibits high antiviral activity, but most of C34-based inhibitors usually inherit a longer sequence, such as sifuvirtide (36 residues) (18), SC35EK (35 residues) (19), and T2635 (36 residues) (20). The discovery of the M-T hook structure, which can markedly enhance the binding affinity and antiviral activity of CHR peptides, provides an additional strategy for designing or optimizing inhibitors (21–24). On the basis of the M-T hook, we designed highly potent short-peptide inhibitors that mainly target the deep-pocket site of gp41 (25–27). Two such peptides, HP23 and 2P23, showed the highest binding and anti-HIV activities compared to a panel of C34-based inhibitors, and they exhibited dramatically improved inhibition of T20-resistant HIV-1 mutants and high genetic barriers to developing resistance (25, 26).
Emerging studies have demonstrated that lipid-modified peptides (lipopeptides) possess improved antiviral activity andin vivostability, which have been considered to interact preferentially with the cell membranes where viral fusion occurs, thus raising the local concentration of the peptide at the target site (28–35). We modified HP23 with different classes of lipids (fatty acid, cholesterol, and sphingolipids), resulting in a panel of lipopeptides with increased binding and antiviral activities (36). One of the lipopep-tides, LP-11, showed highly potent and long-lasting activity in inhibiting diverse subtypes of HIV-1 strains and T20-resistant HIV-1 mutants, possibly due to its enhanced
␣-helical stability and membrane-anchoring property. Very recently, by conjugating 2P23 with lipids, we developed the lipopeptide LP-19, which showed robust inhibition of HIV-1, HIV-2, and simian immunodeficiency virus (SIV) and a highly potent thera-peutic effect on simian-human immunodeficiency virus (SHIV)-infected rhesus ma-caques (37).
Compared to C34-based inhibitors, the drug T20 lacks the pocket-binding sequence, but it has a C-terminal tryptophan-rich motif (TRM) (WASLWNWF) that can bind the target cell membrane. It has been shown that this 8-residue lipid-binding domain is important for T20’s inhibitory activity (38, 39). In this study, we further verified the essential roles of TRM for both T20’s binding and inhibitory functions. Then, we created a T20-based lipopeptide, named LP-40, by replacing its TRM with a C16 fatty acid
on November 7, 2019 by guest
http://jvi.asm.org/
(palmitic acid). Amazingly, LP-40 showed increased activity in inhibiting HIV-1 fusion, entry, and infection, but unlike HP23-based lipopeptides, which require a longer linker between the peptide sequence and lipid moieties, introducing a flexible linker into LP-40 sharply reduced its inhibitory activity, suggesting different binding modes with the target site on the extended N-terminal helices of gp41. While LP-40 was less active than LP-11 in inhibiting HIV-1 entry, it showed more potent activity in blocking Env-mediated cell-cell fusion, and the two inhibitors had synergistic inhibitory effects on diverse subtypes of HIV-1 isolates. We determined the crystal structure of LP-40 in complex with a target mimic peptide, which revealed the key binding residues and motifs. Taken together, we believe that our studies have provided new insights into mechanisms of viral fusion and inhibition, not only for HIV-1, but also for other enveloped viruses.
RESULTS
The TRM of T20 plays essential roles in binding and inhibitory activities. To characterize the function of the TRM in T20, we synthesized a truncated peptide, termed T20-TRM, by deleting the TRM from T20 (Fig. 1). First, the antiviral activities of the two peptides were compared by three experimental systems. As shown in Table 1, T20-TRM fully lost its inhibitory activity against HIV-1HXB2Env-mediated cell-cell fusion,
HIV-1NL4-3Env-mediated pseudovirus entry, and replicative HIV-1JRCSFinfection,
verify-ing the essential role of TRM in T20’s anti-HIV potency. Next, we sought to determine whether the TRM affects the interaction of T20 with its NHR target. To this end, the NHR peptide N39, which has a sequence corresponding to the T20-binding site on the N-terminal helices (Fig. 1), was synthesized as a target surrogate, and the␣-helicity and thermostability of 6-HBs formed between N39 and T20 or T20-TRM were measured by
FIG 1Schematic illustration of HIV-1 gp41 protein and its NHR- and CHR-derived peptides. The gp41 numbering of HIV-1HXB2was used. TM, transmembrane domain. The positions and sequences correspond-ing to the T20-resistant site and pocket-formcorrespond-ing site in the NHR are marked in blue. The positions and sequences corresponding to the M-T hook structure, PBD, and tryptophan-rich motif in the CHR are marked in green, red, and purple, respectively. “C16” in inhibitors represents a fatty acid group.
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.40.368.69.368.2]circular dichroism (CD) spectroscopy. As shown in Fig. 2A, an equimolar mixture of N39 and T20 at 10M concentrations displayed typical double minima at 208 and 222 nm, which indicated the formation of a secondary structure with an␣-helical content of 48.6%. The thermal-unfolding transition (melting temperature [Tm]) value was
deter-mined to be 43.9°C (Fig. 2B). However, the mixture of N39 and T20-TRM did not show appreciable ␣-helical folding, suggesting that the TRM plays an essential role in the interaction of T20 with the NHR site.
A T20-based lipopeptide has dramatically improved antiviral activity.To further investigate the roles of the TRM and to develop a more potent fusion inhibitor, we generated the lipopeptide LP-40 by substituting a C16fatty acid for the TRM of T20 and
then assessed its anti-HIV activity in different experiments. Surprisingly, LP-40 showed dramatically increased anti-HIV activity relative to T20 and the previously reported lipopeptide DP-C16 (39) (Table 1). In the cell fusion assay, LP-40 showed a mean 50% inhibitory concentration (IC50) of 0.41 nM, whereas T20 showed a mean IC50of 24.17
nM, which indicated a 58.95-fold increase of potency for LP-40. Compared to DP-C16, which inhibited cell fusion with a mean IC50of 4.72 nM, the inhibitory activity of LP-40
also increased about 11.51-fold. In the single-cycle entry assay, LP-40 showed a mean IC50 of 0.44 nM, whereas T20 and DP-C16 had mean IC50s of 9.41 and 8.9 nM,
respectively, indicating increases of 21.38- and 20.23-fold, respectively. For a replicative strain, LP-40 inhibited HIV-1JRCSFwith a mean IC50of 0.28 nM, which was 10.54-fold
more potent than T20 (5.19 nM) and 39.82-fold more potent than DP-C16 (11.15 nM). These results demonstrated that LP-40 is a potent inhibitor of HIV-1-mediated cell fusion, viral entry, and infection. Meanwhile, we also determined the cytotoxicities of T20 and LP-40 (data not shown). It was found that both inhibitors had a 50% cytotoxic concentration (CC50) of greater than 100 M, which suggested an extremely high
therapeutic selectivity index (CC50/IC50ratio).
[image:4.585.41.548.83.304.2]A T20-based lipopeptide has greatly increased binding affinity.To understand the mechanism underlying the increased antiviral potency of LP-40, we applied CD spectroscopy to determine its interaction with the NHR peptide N39 in comparison to DP-C16 (Fig. 2C and D). Interestingly, the N39/LP-40 complex displayed a helical content of 60.9% and aTmvalue of 51.3°C, which were much higher than those of the
TABLE 1Inhibitory activities of inhibitors on HIV-1 fusion, entry, and infectiona
Inhibitor Sequenceb
IC50(nM)c
Fusion (HXB2) Entry (NL4-3)d Infection (JRCSF)
T20 based
T20 YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF 24.17⫾2.65 9.41⫾1.19 5.19⫾0.31 T20-TRM YTSLIHSLIEESQNQQEKNEQELLELDK ⬎2,000 ⬎2,000 ⬎2,000 LP-40 YTSLIHSLIEESQNQQEKNEQELLELDK(C16) 0.41⫾0.1 0.44⫾0.01 0.28⫾0.04 DP-C16 YTSLIHSLIEESQNQQEKNEQELLEK(C16) 4.72⫾1.71 8.9⫾1.23 11.15⫾1.94 LP-41 YTSLIHSLIEESQNQQEKNEQELLELD-AHX-K(C16) 6.8⫾0.3 1.57⫾0.69 4.89⫾0.52 LP-42 YTSLIHSLIEESQNQQEKNEQELLELD-PEG 2-K(C16) 8.72⫾1.1 2.29⫾0.91 6.62⫾0.33 LP-43 YTSLIHSLIEESQNQQEKNEQELLELD-PEG 4-K(C16) 11.57⫾2.17 14.14⫾1.04 5.06⫾0.81 LP-44 YTSLIHSLIEESQNQQEKNEQELLELD-PEG 8-K(C16) 15.22⫾0.73 95.36⫾31.69 30.17⫾4.26 LP-45 YTSLIHSLIEESQNQQEKNEQELLELD-PEG 12-K(C16) 35.09⫾3.56 185.26⫾56.78 45.59⫾8.91
HP23 based
HP23 EMTWEEWEKKIEEYTKKIEEILK 0.82⫾0.15 0.63⫾0.03 0.94⫾0.01 LP-1 EMTWEEWEKKIEEYTKKIEEILK(C16) 569.7⫾26.24 235.39⫾27.81 149.96⫾5.16 LP-2 EMTWEEWEKKIEEYTKKIEEILK-AHX-K(C16) 9.43⫾0.51 4.65⫾0.54 0.98⫾0.12 LP-4 EMTWEEWEKKIEEYTKKIEEILK-PEG 2-K(C16) 8.4⫾0.85 5.72⫾1.2 8.61⫾1.17 LP-6 EMTWEEWEKKIEEYTKKIEEILK-PEG 4-K(C16) 4.72⫾0.21 0.93⫾0.05 0.85⫾0.32 LP-8 EMTWEEWEKKIEEYTKKIEEILK-PEG 8-K(C16) 1.12⫾0.09 0.26⫾0.01 0.37⫾0.06 LP-10 EMTWEEWEKKIEEYTKKIEEILK-PEG 12-K(C16) 0.65⫾0.3 0.2⫾0.02 0.27⫾0.01 LP-11 ELTWEEWEKKIEEYTKKIEEILK-PEG 8-K(C16) 1.36⫾0.04 0.18⫾0.04 0.39⫾0.05
aThe assay was performed in triplicate and repeated 3 times. b“C
16” represents a fatty acid group; AHX is 6-aminohexanoic acid.
cThe data are expressed as means⫾standard deviations. dAn NL4-3 pseudovirus with a D36G mutation in gp41 was used.
on November 7, 2019 by guest
http://jvi.asm.org/
N39/T20 complex; the N39/DP-C16 complex had a helical content of 53.9% and aTm
value of 43°C, similar to those of the N39/T20 complex. Taken together, the CD results indicated that a fatty acid substitution for the TRM confers highly enhanced binding affinity and inhibitory activity for T20 and that three C-terminal amino acids in LP-40 (LDK) critically determine such capacities.
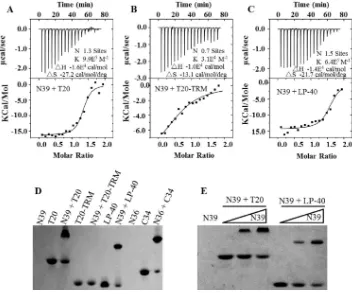
We further performed an isothermal titration calorimetry (ITC) experiment to deter-mine the thermodynamic profiles of the molecular interaction between N39 and LP-40, T20, and T20-TRM. The heat released or absorbed during the interaction allowed an accurate measurement of the binding constant (K), reaction stoichiometry (N), enthalpy (ΔH), and entropy (ΔS). As shown in Fig. 3A to C, three inhibitors interacted with N39
FIG 2Biophysical properties of T20-based inhibitors determined by CD spectroscopy. The␣-helicity (A) and thermostability (B) of T20- and T20-TRM-based 6-HBs, the␣-helicity (C) and thermostability (D) of LP-40- and DP-C16-based 6-HBs, and the␣-helicity (E) and thermo-stability (F) of LP-40 with linker-based 6-HBs were determined, with the final concentration of each peptide in PBS at 10M. The␣-helicity andTmvalues are shown in parentheses. NA, not applicable for calculation. The experiments were repeated at least two times, and
representative data are shown.
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.41.477.69.548.2]in a typical enthalpy-driven reaction in which a large amount of heat was released. T20 showed a K value of 9.9 ⫻ 105 M⫺1, but the deletion of its TRM resulted in a
dramatically decreasedKvalue (3.1⫻104M⫺1). LP-40 displayed aKvalue of 6.4⫻105
M⫺1, again confirming that addition of a lipid tail could largely recover the lost binding
affinity.
We also used a native polyacrylamide gel electrophoresis (N-PAGE)-based method to visualize the interactions between N39 and inhibitors. As shown in Fig. 3D, the NHR peptide N39 showed no band in the native gel because it could migrate up and off the gel due to its net positive charges, similar to the NHR peptide N36. The negatively charged inhibitors T20, T20-TRM, and LP-40 showed specific bands, as did the control peptide, C34. While T20 and its lipid derivative LP-40 were mixed with N39, the specific bands corresponding to the 6-HB appeared similar to that of the N36/C34 complex. Unexpectedly, T20-TRM could not form a complex with N39 in the gel, verifying its weak binding stability. Figure 3E demonstrates that both T20 and LP-40 interact with N39 in a dose-dependent manner.
Introduction of a linker sharply reduces the antiviral activity of LP-40. We previously demonstrated that a flexible linker was required to conjugate a C16group to
the C terminus of the short-peptide HP23-based inhibitors, which might facilitate the peptides overcoming the steric hindrance for binding (36, 37). The lipopeptides LP-11 and LP-19, with a relatively long linker (polyethylene glycol 8 [PEG 8]), showed potent and long-acting anti-HIV activity, and they were physically stable under high temper-ature and humidity. Inspired by this, we expected to further improve LP-40 by intro-ducing a linker between the peptide sequence and the lipid moiety. Thus, a panel of new T20-based lipopeptides with diverse linkers (LP-41 to LP-45) were synthesized, and
FIG 3Interaction of LP-40 and control peptides with an NHR-derived peptide. (A to C) The thermodynamic profiles of the molecular interactions between T20 (A), T20-TRM (B), or LP-40 (C) and N39 were determined using ITC technology. The titration traces are shown at the top, and the binding affinities when the N39 solution was injected into an T20, T20-TRM, or LP-40 solution are shown at the bottom. (D) Visualization of the binding of LP-40 and control peptides to the NHR peptide N39 or N36 by native PAGE; each of the peptides was used at a final concentration of 40M. (E) Dose-dependent binding of T20 and LP-40 (40M) to N39 (0, 20, and 40M).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.43.400.72.366.2]their anti-HIV activities were measured. Surprisingly, the potency of inhibitors sharply decreased with the increased lengths of linkers (Table 1), totally contrary to the HP23-based lipopeptides, which repeatedly achieved improvement along with the extension of the linkers. In particular, a direct C-terminal conjugation of the truncated T20 generated the most active lipopeptide, LP-40, while such conjugation in HP23 (LP-1) sharply damaged potency. The␣-helicity and thermostability of 6-HBs formed by the lipopeptides with linkers were also measured by CD spectroscopy (Fig. 2E and F). It was found that introducing a linker partially reduced the␣-helical contents and that the lipopeptide LP-45, which has the longest linker, PEG 12, exhibited a significantly decreasedTmvalue (46.6°C), consistent with the dramatic loss of anti-HIV activity.
Binding modes of lipopeptide-based fusion inhibitors. The above-mentioned data suggest that steric hindrance might not be a problem for such lipopeptide-based inhibitors and that LP-11 and LP-40 possess different binding modes, which correlate with the mechanisms of viral fusion and inhibition; in other words, the antiviral activities of LP-11 and LP-40 are primarily determined by their docking sites on the NHR helices of gp41. As depicted in Fig. 4, while the short peptide HP23 is attached to the target cell membrane, it needs a long linker to reach the deep pocket located at the extreme C termini of the NHR helices (binding mode I), and thus, a direct lipid conjugation (LP-1) would place the peptide in the wrong position, resulting in sharply decreased antifusion activity. On the other hand, T20 targets the membrane-proximal NHR site, and its anchoring via a lipid molecule does not affect its binding (mode II binding), but conversely, an introduced linker can impede the docking of inhibitors.
LP-40 has greatly improved activity against diverse HIV-1 subtypes.HIV-1 evolves through great genetic diversity, raising concerns about the peptide inhibitors that target the variable Env protein. To investigate this, we constructed two large panels of pseudoviruses with diverse subtypes of viral Envs. The panel 1 Envs were derived from the primary subtype A, B, and C viruses and the recombinant-form CRF01_BC and CRF01_AE viruses that were routinely used in our laboratory; the panel 2 Envs, the so-called “global panel,” were recently selected based on the genetic and antigenic variability of the viral Envs that are representative of the global AIDS epidemic (40). As shown in Table 2, LP-40 demonstrated greatly improved activity over T20 in
FIG 4Binding models of membrane-anchoring lipopeptides. Both classes of lipopeptide inhibitors can bind to the extended PHI during the early stage of viral membrane fusion. LP-11 mainly targets the pocket site located at the extreme C terminus of the NHR helices, and thus, it needs a long linker to reach the position (binding mode I). LP-40 targets the membrane-proximal NHR site, and its anchoring via a lipid molecule does not affect its binding (binding mode II). Red represents peptide sequences, purple represents fatty acid molecules, and the black line represents a PEG 8 linker.
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.87.326.69.279.2]inhibiting diverse subtypes of HIV-1 isolates, with mean IC50s of 4.29 nM and 29.45 nM,
respectively. As a control, the lipopeptide LP-11 inhibited the viruses with a mean IC50
of 0.83 nM and was the most potent inhibitor compared to T20 and LP-40.
LP-40 is a more potent inhibitor of HIV-1 Env-mediated cell-cell fusion. Obvi-ously, the above-mentioned data demonstrated that LP-40 had much improved activity over the parent peptide, T20, in the inhibition of diverse subtypes of HIV-1 Envs, but its inhibitory activity was about 5-fold lower than that of LP-11. However, our previous and present data indicate that LP-11 was less sensitive to Env-mediated cell-cell fusion relative to its inhibition of both the wild-type (WT) and pseudotyped viruses (Table 1). More interestingly, the initial results shown in Table 1 also revealed that LP-40 had more potent activity than LP-11 in inhibiting HIV-1HXB2Env-mediated cell fusion (0.41
nM versus 1.36 nM), which was contrary to their potencies against pseudovirus. To address whether this phenomenon was caused by a specific Env or revealed a general phenotype, we used a dual-split-protein (DSP) assay to determine the activity of inhibitors on cell-cell fusion mediated by a panel of Envs. As shown in Table 2, LP-40 had dramatically increased potency over LP-11 (8.25-fold) and T20 (56.55-fold), with their mean IC50s at 0.2, 1.68, and 11.31 nM, respectively.
[image:8.585.45.545.81.376.2]The combination of LP-40 and LP-11 exhibits a synergistic anti-HIV effect.We were interested in knowing whether LP-40 and LP-11 had synergistic roles in terms of their different binding modes and inhibitory activities. To investigate this, we initially tested the cooperative effects of two lipopeptides on three representative HIV-1 pseudoviruses. As shown in Fig. 5, the combination of LP-40 and LP-11 resulted in synergism against NL4-3, JRFL, and SF162, with combination index (CI) values of 0.49, 0.27, and 0.55, respectively. We further determined the synergistic effects with diverse subtypes of HIV-1 isolates, which showed a mean CI of 0.47. The mean dose reductions were 5.54-fold for LP-40 and 5.02-fold for LP-11, confirming synergism between LP-40 TABLE 2Inhibition by LP-40 and control peptides of HIV-1 Env-mediated cell entry and fusiona
Primary Env Subtype
Inhibitionc(IC
50[nM]) of:
Viral entry Cell fusion
T20 LP-40 LP-11 T20 LP-40 LP-11
92UG037.8 A 5.36⫾0.53 2.47⫾0.12 0.92⫾0.05 4.7⫾0.62 0.14⫾0.04 0.98⫾0.26 92RW020 A 1.215⫾0.28 1.32⫾0.13 0.84⫾0.04 0.81⫾0.17 0.01⫾0 0.28⫾0.06 398-F1_F6_20b A 12.69⫾1.3 3.58⫾0.27 0.5⫾0.02 7.2⫾0.08 0.12⫾0.04 3.25⫾0.49
AC10.0.29 B 3.55⫾0.26 1.94⫾0.53 0.47⫾0.02 2.59⫾0.51 0.06⫾0.02 0.66⫾0.06 SC422661.8 B 20.65⫾4.44 18.68⫾4.24 0.28⫾0 2.32⫾0.29 0.1⫾0.01 0.53⫾0.08 TRO.11b B 8.18⫾0.62 3.49⫾0.48 0.99⫾0.01 24.12⫾7.12 0.54⫾0.15 2.53⫾0.6
BJOX002000.03.2b B 34.38⫾3.6 3.35⫾0.88 0.96⫾0.02 1.66⫾0.59 0.06⫾0.01 0.4⫾0.1
X2278_C2_B6b B 6.42⫾0.2 1.82⫾0.8 0.16⫾0 4.57⫾1.11 0.17⫾0.09 1.94⫾0.35
B02 B’ 7.7⫾0.18 0.91⫾0.16 1.26⫾0.11 4.89⫾0.4 0.06⫾0.02 0.9⫾0.10 B04 B’ 5.01⫾0.68 1.14⫾0.14 0.79⫾0.03 8⫾2.11 0.15⫾0.01 2.09⫾0.81 CAP45.2.00.G3 C 170.9⫾2.46 21.74⫾2.46 0.98⫾0.01 25.37⫾5.42 0.68⫾0.09 2.7⫾0.14 ZM53 M.PB12 C 34.00⫾5.57 4.63⫾0.27 1.04⫾0.02 38.79⫾8.12 0.22⫾0.06 2.17⫾0.91 HIV_25710-2.43b C 14.73⫾0.74 4.35⫾0.53 0.4⫾0 13.75⫾2.89 0.08⫾0.01 0.59⫾0.09
CE1176_A3b C 4.66⫾0.24 1.58⫾0.43 0.81⫾0.01 5.73⫾1.58 0.09⫾0.01 1.57⫾0.08
CE703010217_B6b C 43.11⫾9.81 3.52⫾0.53 0.65⫾0.01 6.23⫾0.47 0.09⫾0 0.31⫾0.05
X1632-S2-B10b G 15.49⫾0.98 2.27⫾0.13 1.08⫾0.11 6.78⫾1 0.22⫾0.09 1.02⫾0.11
246_F3_C10_2b A/C 41.24⫾4.17 3.89⫾0.21 0.84⫾0.02 4.1⫾0.91 0.11⫾0.01 2.46⫾0.28
AE03 A/E 8.66⫾0.56 3.62⫾0.63 0.75⫾0.04 17.45⫾1.95 0.16⫾0.01 2.19⫾0.34 AE04 A/E 11.72⫾0.67 0.81⫾0.07 1.38⫾0.12 24.57⫾3.59 0.21⫾0.05 3.01⫾0.33 CNE8b A/E 35.24⫾4.8 3.46⫾0.55 0.76⫾0.01 33.96⫾7.12 0.23⫾0.09 3.97⫾0.23
CNE55b A/E 23⫾1.45 2.68⫾0.44 0.45⫾0.01 9.77⫾1.12 0.06⫾0.01 1.62⫾0.85
CH64.20 B/C 50.69⫾8.86 4.98⫾0.1 0.5⫾0.02 3.15⫾0.28 0.18⫾0.06 0.62⫾0.12 CH070.1 B/C 80.8⫾3.89 3.83⫾0.08 1.33⫾0.08 13.08⫾2.45 1.08⫾0.06 3.89⫾0.86 CH119.10b B/C 22.84⫾1.6 2.22⫾0.19 0.79⫾0.02 2.63⫾0.52 0.04⫾0.01 0.81⫾0.13
CH120.6 B/C 74.06⫾3.63 4.95⫾0.56 1.93⫾0.12 9.89⫾0.52 0.18⫾0.04 1.49⫾0.42
Mean 29.45 4.29 0.83 11.31 0.20 1.68
aThe assay was performed in triplicate and repeated 3 times.
bA global panel of HIV-1 isolates representing the genetic and antigenic diversities worldwide.
cThe data are expressed as means⫾standard deviations.
on November 7, 2019 by guest
http://jvi.asm.org/
and LP-11 (Table 3). However, we were very surprised by these results, as previous studies had suggested highly potent synergism between unmodified T20 and C34-based inhibitors (T1249, T1144, and SFT) (41, 42). Thus, we back-checked the synergistic effects of the combinations between T20 and HP23 or T20 and T1249. Unexpectedly, two pairs of free peptides had no or slight synergistic effects in inhibiting NL4-3, JRFL, and SF162 (Fig. 6).
Inhibitory activity of LP-40 against drug-resistant HIV-1 mutants. We also determined the inhibitory activity of LP-40 against a panel of pseudoviruses that possessed T20-resistant mutations (Table 4). Here, a D36G mutant of HIV-1 isolate NL4-3 was referred to as a T20-sensitive virus because its WT virus had a naturally occurring aspartic acid at position 36 of gp41 that could confer high-level resistance to T20. Compared to T20, LP-40 showed increased activity in inhibiting only three viruses (WT, D36G, and I37T), but it showed no or minor improvement against the majority of the T20-resistant mutants. In contrast, the short-peptide inhibitors HP23 and LP-11 main-tained their high inhibitory potencies, consistent with our previous results (26, 36).
We previously showed that a single L57R mutation in the gp41 pocket mediated high-level resistance to short-peptide inhibitors targeting the pocket site and that a secondary mutation in the CHR site (E136G) could significantly boost the resistance phenotype (43). It was also noticed that the L57R-based mutants were more sensitive to the inhibition of T20. Here, we sought to determine the effects of L57R and L57R/E136G mutations on the anti-HIV activity of LP-40. As shown in Table 4, the
FIG 5Synergistic anti-HIV effects of LP-40 and LP-11. The synergistic inhibition by inhibitors of diverse HIV-1 pseudoviruses was determined by single-cycle infection assay. Peptides were tested individually and in combination at a fixed molar ratio achieving equivalent IC50s. LP-40/LP-11 ratios, 3:1 (A), 1:2 (B), and 2:1 (C). Each sample was tested in triplicate, and the experiment was repeated 3 times. An NL4-3 pseudovirus with a D36G mutation in gp41 was used. The error bars indicate standard deviations (SD).
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.121.291.74.407.2]pseudoviruses with L57R or L57R/E136G mutations displayed increased sensitivity to T20 and conferred high-level resistance to HP23 and LP-11. In sharp contrast, LP-40 exhibited markedly increased potency over T20, HP23, and LP-11 in inhibiting two HIV-1 mutants. More interestingly, while LP-40 inhibited the WT virus with an IC50of
39.42 nM, it inhibited the L57R and L57R/E136G mutants with IC50s of 1.12 and 1.41 nM,
respectively, which indicated an⬃30-fold-increased potency for LP-40 against HP23-and LP-11-resistant viruses. These results further highlighted the fact that a combina-tion of LP-40 and LP-11 provided complementary effects for the treatment of HIV-1 infection.
Structural basis of LP-40.To elucidate the structural basis of the efficacy of LP-40, we performed crystallographic studies of its complex with the NHR-derived peptide N44. The crystal of the N44/LP-40 complex belonged to the space groupP21, contained
three pairs of N44/LP-40 peptides (one complete 6-HB) per asymmetric unit, and diffracted X rays to a resolution of 1.7 Å. Unfortunately, although the crystal structure had relatively high quality and resolution, the fatty acid (C16) could not be observed in
the electron density map, probably due to its flexible character. Other than that, we could build most of the residues of the N44/LP-40 peptides, with a refined model having good refinement statistics and stereochemistry qualities (Table 5).
As anticipated, the crystal structure of N44/LP-40 formed a typical 6-HB structure similar to other HIV gp41 core structures. From an overall perspective, the structure was a 6-HB analogous to three helical hairpins, each composed of an N44 helix paired with an antiparallel LP-40 helix. A number of intrahelical and interhelical salt bridges and hydrogen bonds formed binding networks and contributed to the stabilization of the helical conformation of the 6-HB. As shown in Fig. 7, there were four hydrogen bonds on LP-40 peptides increasing the␣-helix stability: the OH group of Glu-151 formed a hydrogen bond with the NH group of Gln-147, the OH group of Glu-146 accepted a hydrogen bond from the NH group of Gln-142, the OH group of Glu-143 accepted a hydrogen bond from the NH group of Gln-139, and the NH group of Gln-141 donated a hydrogen bond to the OH group of Asn-145.
[image:10.585.43.544.83.286.2]The complicated interactions of interhelical hydrogen bonds were mainly gathered together in the region of residues Glu-137 to Glu-146 on LP-40 and residues Gly-36 to Glu-49 on N44, except that the OH group of Leu-152 on LP-40 accepted a hydrogen bond from the NH group of Arg-31 on N44 (Fig. 8). In particular, the OH group of Glu-146 on LP-40 accepted a hydrogen bond from the NH group of N-42 on the left N44 peptide. The NH group of Gln-142 on LP-40 donated a hydrogen bond to the OH group TABLE 3Synergistic inhibition by LP-40 and LP-11 of diverse HIV-1 isolatesa
Virus Subtype CI
LP-40 inhibition (IC50 [nM])
Dose reduction (n-fold)
LP-11 inhibition (IC50 [nM])
Dose reduction (n-fold)
Alone In mixture Alone In mixture
92UG037.8 A 0.37 2.36 0.52 4.54 0.85 0.13 6.54
NL4-3D36G B 0.49 0.89 0.25 3.56 0.37 0.08 4.63
JRCSF B 0.31 5.69 0.22 25.86 0.82 0.22 3.73
R3A B 0.39 0.44 0.11 4.00 0.77 0.11 7.00
AC10.0.29 B 0.28 1.62 0.24 6.75 0.46 0.06 7.67
TRO.11 B 0.50 4.93 1.08 4.56 0.95 0.27 3.52
JRFL B 0.27 1.76 0.22 8.00 3.10 0.44 7.05
SF162 B 0.55 3.97 1.15 3.45 1.87 0.58 3.22
B02 B’ 0.42 0.93 0.25 3.72 1.66 0.25 6.64
CE1176_A3 C 0.64 1.32 0.64 2.06 1.04 0.16 6.50
X1632-S2-B10 G 0.48 2.50 0.70 3.57 0.86 0.17 5.06
AE03 A/E 0.46 3.29 0.71 4.63 0.75 0.18 4.17
CNE8 A/E 0.51 4.51 1.25 3.61 1.37 0.32 4.28
CNE55 A/E 0.77 3.46 1.03 3.36 0.55 0.26 2.12
CH64.20 B/C 0.52 4.14 1.09 3.78 0.43 0.11 3.91
CH119.10 B/C 0.55 2.22 0.69 3.22 0.72 0.17 4.24
Mean 0.47 2.75 0.63 5.54 1.04 0.22 5.02
aThe assay was performed in triplicate and repeated 3 times, and representative data are shown.
on November 7, 2019 by guest
http://jvi.asm.org/
of the backbone of Val-38 on the N44 trimer. Interestingly, the OH group of Gln-139 on the inhibitor formed a hydrogen bond with the NH group of Arg-46 and the OH group of Glu-49 on the N44 trimer. The NH group of Asn-145 and Gln-141 on LP-40 simulta-neously donated a hydrogen bond to the OH group of the backbone of Gly-36 on the N44 trimer. The OH group of Gln-142 and the OH group of the backbone of Ser-138 on LP-40 accepted a hydrogen bond from the NH group of Gln-40 on the N44 trimer, and simultaneously, the OH group of the side chain of Ser-138 formed a hydrogen bond with the OH group of Gln-40. Coincidently, Glu-137 on the inhibitor also formed hydrogen bonds with Gln-39 and Asn-43. In addition to the complicated hydrogen bond network, the negatively charged residue Glu-137 on LP-40 formed a salt bridge with the positively charged residue Arg-46 on the N44 trimer (distance, 3.71 Å), and extremely C-terminal of the inhibitor, the negatively charged residue Asp-153 formed a salt bridge with Arg-31 on the N44 trimer (distance, 3.05 Å), which greatly enhanced the interaction between the N44 trimer and LP-40. Notably, the single residue Gln-142 was able simultaneously to link two residues (Val-38 and Gln-40) on both sides of the NHR helices, which undoubtedly enhanced the stability of the 6-HB structure. Surpris-ingly, Gln-139 on inhibitors could form hydrogen bonds with two residues located at two adjacent turns of the␣-helix, forming a stable “triangle.” It happened that there
FIG 6Synergistic anti-HIV effects of unconjugated peptide inhibitors. The synergistic inhibition by inhibitors of HIV-1 pseudoviruses was determined by single-cycle infection assay. Peptides were tested individually and in combination at a fixed molar ratio achieving equivalent IC50s. T20/HP23 ratios, 5:1 (A), 1:1 (B), and 1:2 (C); T20/T1249 ratios, 3:1 (D), 1:1 (E), and 3:1 (F). Each sample was tested in triplicate, and the experiment was repeated 3 times. The error bars indicate SD.
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.42.430.70.457.2]was a similar case, where Glu-137 also formed hydrogen bonds with Gln-39 and Asn-43 located at neighboring turns of the␣-helix, and more surprisingly, it also formed a salt bridge with Arg-46, simultaneously increasing the binding force between the NHR and the inhibitors. In summary, the LP-40 helix had plenty of interactions of salt bridges and hydrogen bonds with the neighboring two NHR helices, simultaneously forming a network structure and enhancing the stability of the 6-HB structure.
Hydrophobic amino acids clustered together at both termini of the sequence of LP-40 had abundant interactions with the paired N44 peptides, contributing to the stability of the 6-HB structure (Fig. 9). At the N termini of the inhibitors, the C2 atom and C␦2 atom of Tyr-127 had hydrophobic interactions with the adjacent C␦1 atom of Leu-57, which belonged to the residues of the pocket domain, a well-known drug target on the NHR trimer. In addition, the C␥1 atom of Ile-131 on inhibitors also had hydrophobic interactions with the C␦2 atom of Leu-54 on the N44 trimer, another key residue of the pocket domain. Notably, the C␥1 atom of Ile-131, together with the C␦2 atom of Leu-130 on LP-40 inhibitors, also had numerous hydrophobic interactions with Ala-50 on the N44 trimer simultaneously. In addition, Leu-134 on LP-40 inhibitors had interactions with Ala-47 on the N44 trimer. At the C termini of the LP-40 inhibitors, the hydrophobic side chain of Leu-149 had hydrophobic interactions with the side chain of Val-38 on the N44 trimer. Notably, the C␦1 atom of Leu-152 of LP-40 had interactions with the C␦2 atom of Leu-33 on the N44 trimer, contributing the binding force of 6-HB. In short, the crystal structure of N44/LP-40 has plenty of molecular interactions evenly scattered in the structure to stabilize the binding of the inhibitors and the NHR peptides. First, hydrophobic interactions mainly exist at two ends of the inhibitors, stabilizing the binding force of both terminals. Second, abundant hydrogen bonds mainly appear in the middle part of the inhibitors, forming a chemical bond network to stabilize the structure of 6-HB. Third, two salt bridges located on two sides of the LP-40 inhibitors further increase the stability of 6-HB. Our crystal structure provides a molecular basis for the mechanism of action of LP-40.
DISCUSSION
In this study, we first verified the essential roles of the C-terminal TRM in T20 and then created a T20-based lipopeptide (LP-40) by replacing the TRM with a C16fatty acid.
[image:12.585.41.369.83.285.2]Compared to T20 and a previously reported T20-based lipopeptide (DP-C16), LP-40 had greatly improved binding affinity and inhibitory activity. Interestingly, addition of a flexible linker between the peptide sequence and the lipid moiety of LP-40 greatly TABLE 4Inhibition by LP-40 and control peptides of drug-resistant HIV-1 mutantsa
NL4-3
Inhibition (IC50[nM]) (meanⴞSD)
T20 LP-40 HP23 LP-11
T20 sensitive
D36G 10.6⫾3.07 0.72⫾0.2 0.85⫾0.04 0.36⫾0.06
T20 resistant
WT 109.7⫾39.73 39.42⫾7.02 0.69⫾0.2 0.29⫾0.1 I37T 932.12⫾98.89 334.42⫾89.39 0.79⫾0.03 0.45⫾0.19 V38A 2,318.17⫾167.32 1,644.42⫾212.88 0.65⫾0.17 0.26⫾0.09 V38 M 1,097.47⫾194.59 732.49⫾231.62 1.03⫾0.08 0.43⫾0.07 Q40H 1,152.53⫾241.02 1,366.83⫾415.38 0.85⫾0.16 0.24⫾0.06 N43K 621.85⫾200.95 409.17⫾142.05 0.68⫾0.18 0.32⫾0.13 N126K 181.41⫾46.43 381.97⫾15.93 1.89⫾0.24 0.75⫾0.17 D36S/V38 M 444.93⫾204.65 405.86⫾212.05 1.11⫾0.57 0.63⫾0.22 I37T/N43K ⬎2,000 ⬎1,500 0.82⫾0.03 0.33⫾0.1 V38A/N42T ⬎2,000 ⬎1,500 0.44⫾0.04 0.12⫾0.04
HP23 resistant
L57R 19.57⫾2.83 1.12⫾0.39 243.5⫾36.73 8.74⫾5.15 L57R/E136G 12.65⫾2.09 1.41⫾0.15 ⬎430 20.13⫾6.54
aThe assay was performed in triplicate and repeated 3 times.
on November 7, 2019 by guest
http://jvi.asm.org/
reduced its anti-HIV potency, suggesting a different binding mode than LP-11, a lipopeptide inhibitor specifically targeting the gp41 pocket. We found that LP-40 was much more active in inhibiting HIV-1 Env-mediated cell-cell fusion than in inhibiting pseudovirus-mediated entry, which was also opposite to LP-11, implying that the two membrane-anchoring inhibitors have distinct antiviral effects during the viral entry process. Consistent with this, LP-40 and LP-11 displayed a synergistic inhibitory efficacy against diverse subtypes of HIV-1 isolates, which suggested a combination strategy for treating HIV-1 infection. To further understand the mechanism of action of LP-40, we determined its crystal structure in complex with a target mimic NHR peptide, which revealed the critical binding residues and motifs. Combined, the data presented have provided multiple implications for mechanisms of viral entry and inhibition and have offered a novel lipopeptide HIV-1 fusion inhibitor for further development.
Discovery of T20 (originally termed DP178) from the CHR of HIV-1 gp41 opened a promising avenue for exploring the mechanism of viral entry and for developing antiviral drugs (10, 44). Subsequently, a more potent CHR peptide, C34 (Fig. 1), was identified (45); the fusogenic gp41 core structure (6-HB) was determined (3, 5), and T20 was approved for clinical use in 2003 (46, 47). The inhibitory model of T20 was initially thought to target one site within the viral fusion protein gp41 (44); however, a body of evidence suggested a multifaceted mechanism (48). First, T20 targets the NHR helices, thus preventing the formation of the 6-HB structure, as predicted in its initial model and similar to other CHR-derived peptides (12, 44). Indeed, T20-resistant mutations are largely mapped to the NHR, with the36GIV38motif being a hot spot (14, 16). Second,
[image:13.585.40.373.85.404.2]several studies suggested that T20 may inhibit the self-interaction of the N terminus of TABLE 5Crystallographic data collection and refinement statisticsa
Parameter Valueb
Data collection
Beamline SSRF BL17U
Wavelength (Å) 0.9796
Resolution range 50.0–1.76 (1.80–1.76)
Space group P21
Cell dimensions
a,b,c(Å) 50.83, 36.14, 53.44
a,b,g(°) 90, 99.87, 90
Redundancy 3.8
Total no. of reflections 65,449
No. of unique reflections 17,147
Rmerge(%) 4.2 (19.3)
I/I 16.0 (4.1)
Completeness (%) 90.4 (88.6)
Refinement
Resolution (Å) 24.3–1.76 (1.85–1.76)
No. of reflections 17,133 (2,432)
Rwork/Rfree(%) 20.7/25.1
No. of atoms
Protein 1,720
Water 122
B factors (Å2)
Protein 34.3
Water 43.5
RMS deviations
Bond lengths (Å) 0.006
Bond angles (°) 0.66
Ramachandran plot (%)
Favored 100
Allowed 0
Disallowed 0
aR
workandRfreeare defined as follows:R⫽ ⌺hkl||Fobs|⫺|Fcalc||/⌺hkl|Fobs|, whereh,k, andlare the indices of the reflections (used in refinement forRwork; 5%, not used in refinement forRfree) andFobsandFcalcare the structure factors deduced from intensities and calculated from the model, respectively.
bValues in parentheses are for the highest-resolution shell.
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
gp41 while it is extended in the early fusion steps; alternatively, T20 may prevent the interaction of the membrane-proximal external region (MPER) with the N terminus of gp41 in the late fusion steps (49–51). Third, it was proposed that T20 has two binding sites with different binding affinities; while the lower-affinity site is the NHR, the higher-affinity site is still unknown (52–54). It was speculated that T20 may bind to a parallel endogenous region, thus preventing aggregation of several Env trimers needed to enlarge the fusion pore (52). The transmembrane domain (TMD) of gp41 may also be targeted by T20, because its inhibitory activity could be blocked by TMD-derived peptides (55). Fourth, several lines of evidence supported the involvement of gp120 in sensitivity to T20 (56–59). Also, T20 was able to block the interaction of gp120-CD4 complexes with the CXCR4 coreceptor, and peptides derived from the coreceptor binding site of gp120 could competitively block the inhibitory activity of T20 (55, 60, 61). Therefore, the coreceptor binding site on gp120 has been considered a molecular target for T20. Fifth, it is well established that T20 can bind to the target cell membrane through its C-terminal TRM, which critically determines its inhibitory activity (38, 39, 62, 63). All of these studies, by using T20 as a probe, shed light from different angles on the mechanisms of viral fusion and inhibition. Taken together, we think that more efforts should be made to understand the mechanism of action of T20 and to develop T20-based fusion inhibitors, while the PBD-based CHR peptides are widely focused.
As an inhibitor for drug development, LP-40 has multiple advantages over its parental peptide, T20. First, it has greatly increased binding and antiviral activities; second, the lipid peptides have been validated, with a dramatically improvedin vivo half-life; third, it has complementary effects with PBD-containing inhibitors; and fourth, its small size contributes to cost-effective production. However, it is more valuable as a new tool to investigate the mechanisms of viral fusion and inhibition. On the basis of the 6-HB structure, it is hypothesized that gp41 adopts an extended PHI that bridges the viral and target cell membranes and that peptide fusion inhibitors bind to the PHI and block the formation of viral 6-HB structure. By comparing two membrane-anchoring lipopeptides that target different sites on the NHR helices of gp41 (LP-40 and LP-11), the present study provides convincing experimental data to support the PHI
FIG 7Intrahelical hydrogen bonds of LP-40 in the 6-HB structure. (A) Ribbon model of 6-HB structure formed by the N44/LP-40 complex (PyMol). The N44 trimer is colored gray. LP-40 inhibitors are colored green. Residues related to hydrogen bonds on LP-40 are shown as stick models colored green with labels. The C terminus of LP-40 and the N terminus of N44 are labeled. (B) Amino acid sequence of LP-40. The glutamine residues are colored red, the glutamic acid residues are blue, and the asparagine residue is green. The solid black lines indicate hydrogen bonds formed between residues on LP-40.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:14.585.122.289.69.296.2]conformation. The effects of introduced linkers in lipopeptides reveal two binding models for two classes of inhibitors; while a lipopeptide mainly targeting the pocket site needs a relatively long linker, conversely, a T20-based lipopeptide mainly targeting the membrane-proximal NHR site does not.
Previous studies found that T20 was a more potent inhibitor of cell-cell fusion than of infection by cell-free virus, and a fusion pore dilation model was proposed to explain
FIG 8Interhelical salt bridges and hydrogen bonds in the 6-HB structure of N44/LP-40. (A) Portion of a ribbon model of the 6-HB structure formed by N44/LP-40 (PyMol). The N44 trimer is colored gray. Residues related to hydrogen bonds and salt bridges on the N44 trimer are shown as stick models colored gray with labels. The LP-40 inhibitors are colored green. Residues related to hydrogen bonds and salt bridges on LP-40 are shown as stick models colored green with labels. The dashed lines indicate salt bridges and hydrogen bonds between residues on the N44 trimer and the inhibitor. The N terminus of N44 and the C terminus of LP-40 are labeled. (B) View of the crystal structure in panel A rotated 90° to the left. The same representation and color scheme as in the crystal structure in panel A are used. (C) A single LP-40 interacting with two NHR helices shown in a sequence map. The positively charged residues related to salt bridges are red, and the negatively charged residues related to salt bridges are green. The dashed black lines indicate salt bridges and hydrogen bonds between the N44 trimer and LP-40. The C and N termini of the sequences are labeled.
FIG 9Interactions of N and C termini of LP-40 in the 6-HB crystal structure. Hydrophobic interactions of LP-40 are presented horizontally in a ribbon model (PyMol). The N44 trimer is colored gray. Residues related to hydrogen bonds are shown as stick models in gray. Hydrophobic amino acids related to hydrophobic interaction on the N44 trimer are shown as stick models and colored in corresponding colors. LP-40 inhibitors are green. Hydrophobic amino acids related to hydrophobic interaction on LP-40 are shown as stick models and colored in corresponding colors. The N and C termini of LP-40 and the N terminus of N44 are labeled.
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
[image:15.585.54.353.72.350.2] [image:15.585.121.291.582.671.2]the difference (44, 53). In our recent work, we repeatedly observed this phenomenon with different HIV-1 and SIV isolates (37). In this study, in which a panel of 25 Envs derived from diverse subtypes of HIV-1 isolates was used (Table 2), T20 again showed higher activity in inhibiting cell-cell fusion than in inhibiting pseudovirus. However, this unusual phenomenon was even more pronounced in the inhibitory activity of LP-40 (0.2 nM versus 4.29 nM). In sharp contrast, the pocket-targeting lipopeptide LP-11 was a more efficient inhibitor of cell-free virus than of Env-mediated cell-cell fusion, as evidenced by our previous and present studies (36). Although, the reason for the reversal in potency of T20, LP-40, and LP-11 in Env-mediated cell fusion and virus-mediated infection remains unclear, it may reflect differences in the two modes of infection (e.g., cell-cell transmission versus cell-free virus transmission) and correlate with the binding modes of two classes of lipopeptide inhibitors. Considering that the cell-cell transmission may be a predominant pathwayin vivo, T20-based inhibitors such as LP-40 would provide additional advantages for the treatment of HIV-1 infection. Our data also demonstrate that LP-40 and LP-11 have synergistic effects in inhibiting diverse HIV-1 isolates, which may depend on their different binding sites and/or their complementary inhibitions of transmission pathways. Regardless, they may be used in combination to treat HIV-1 infection. In this regard, our previous studies also showed that LP-11 is a highly potent inhibitor of diverse T20-resistant mutants, while T20 had improved inhibitory activity even against HIV-1 variants that were resistant to short-peptide inhibitors targeting the gp41 pocket, such as HP23 and LP-11.
The invention of T20 initiated a new era in exploring the nature of viral infection and developing antiviral drugs; however, the precise mechanism of action of T20 remains a puzzle, and so far, its structure has not been determined. In this study, the crystal structure of LP-40 revealed the details of the interactions between the inhibitor and its NHR target, which provides valuable information to understand the mechanisms of T20 and its derivatives. For example, it had been thought that T20 does not target the deep NHR pocket site because its N terminus lacks the pocket-binding sequence; however, the structure of LP-40 bound to an NHR peptide (N44) shows the huge interactions of the N-terminal segment of the inhibitor with the pocket-forming residues, such as Leu-54 and Leu-57, which critically determine the binding stability of the 6-HB struc-ture. Thus, it is possible to understand why HIV-1 mutants carrying an L57R substitution display markedly increased susceptibility to T20 and LP-40, which may change the binding method of the inhibitors. Moreover, the structure shows that the residue Leu-152 in the C terminus of LP-40 has interactions with the residue Leu-33 on the NHR trimer, and its carbonyl group also accepts a hydrogen bond from the NH group of Arg-31, together greatly stabilizing the binding force of the inhibitors. The lipopeptide DP-C16 lacks Leu-152 in its C terminus, which explains its dramatically decreased binding and inhibitory activities compared to LP-40.
Finally, it would be interesting to know whether LP-40, when it is bound to the cell membrane, has multifaceted actions, as does T20. Namely, how can T20, and possibly its derivatives, interact with different gp41 sites (e.g., N-terminal sequence, NHR, CHR, and TRM), the gp120 coreceptor-binding region, and the target cell membrane, simul-taneously or independently? We have finished the current project, but we will continue our efforts to elucidate the inhibitory mechanism of LP-40 and its template, T20, and we also hope to evaluate the therapeutic efficacy of LP-40 in nonhuman primate models.
MATERIALS AND METHODS
Cell lines and plasmids.The following reagents were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH: HL2/3 cells, which contain stably integrated copies of the HIV-1 molecular clone HXB2/3gpt that express high levels of HIV Gag, Env, Tat, Rev, and Nef proteins, from Barbara K. Felber and George N. Pavlakis; TZM-bl indicator cells, which stably express large amounts of CD4 and CCR5, along with endogenously expressed CXCR4, from John C. Kappes and Xiaoyun Wu; U87 CD4⫹ CXCR4⫹cells stably transduced to express CD4 and CXCR4, from Hongkui Deng and Dan R. Littman; the Panel of Global HIV-1 Env Clones, which contains 12 envelope clones as reference strains representing the global epidemic, from David Montefiori; and the plasmid pYK-JRCSF encoding an infectious molec-ular clone of HIV-1JRCSF, from Irvin S. Y. Chen and Yoshio Koyanagi. The plasmids DSP1–7and DSP8 –11for
on November 7, 2019 by guest
http://jvi.asm.org/
the DSP-based fusion assay were provided by Zene Matsuda at the Institute of Medical Science of the University of Tokyo (Tokyo, Japan).
Peptide synthesis and lipid conjugation. Peptides were synthesized on Rink amide 4-methylbenzhydrylamine (MBHA) resin by using a standard solid-phase 9-fluorenylmethoxy carbonyl (FMOC) method as described previously (21). All peptides were acetylated at the N terminus and amidated at the C terminus. The template peptides contain a lysine residue at their C termini with a Dde side chain-protecting group, enabling the conjugation of a fatty acid that requires a special deprotection step in a solution of 2% hydrazinehydrate/N,N-dimethylformamide (DMF), as described by Shai and collaborators (30, 39). All the peptides were purified by reverse-phase high-performance liquid chroma-tography (HPLC) to⬎95% homogeneity and were identified by mass spectrometry. The concentrations of the peptides were determined by UV absorbance and a theoretically calculated molar-extinction coefficient based on the tryptophan and tyrosine residues.
Cell-cell fusion assays.Inhibition of peptides on HIV-1 Env-mediated cell-cell fusion was measured using a reporter gene assay based on the activation of an HIV long terminal repeat (LTR)-driven luciferase cassette in TZM-bl (target) cells by HIV-1 tat from HL2/3 (effector) cells (27). Briefly, TZM-bl cells were plated in 96-well clusters (1 ⫻ 104/well) and incubated at 37°C overnight. The target cells were cocultured with HL2/3 cells (3⫻104/well) for 6 h at 37°C in the presence or absence of a test peptide at graded concentrations. Luciferase activity was measured using luciferase assay reagents and a luminescence counter (Promega, Madison, WI, USA). Background luminescence in TZM-bl cells was determined without the addition of HL2/3 cells. The percent inhibition of fusion by the peptides and IC50 values of fusion were calculated using Graph-Pad Prism software (GraphPad, San Diego, CA, USA).
A DSP-based fusion assay was also performed to determine the inhibitory activity of peptides, as described previously (64, 65). Briefly, a total of 1.5⫻104HEK293T cells (effector cells) were seeded on a 96-well plate, and a total of 8⫻104U87 CD4⫹CCR5⫹cells (target cells) were seeded on a 24-well plate. On the following day, the effector cells were transfected with a mixture of an Env-expressing plasmid and a DSP1–7plasmid, and the target cells were transfected with a DSP8 –11plasmid. Forty-eight hours posttransfection, the target cells were resuspended in 300l prewarmed culture medium, and to each well, 0.05l EnduRen live-cell substrate (Promega) was added. Then, aliquots of 75l of the target cell suspension were transferred over each well of the effector cells in the presence or absence of serially 3-fold-diluted inhibitors. The cells were then spun down to maximize cell-cell contact and incubated for 1 h at 37°C. Luciferase activity was measured, and the percent inhibition of fusion and IC50s were calculated using GraphPad Prism software.
Single-cycle infection assay.Inhibition of peptides on HIV-1 entry was determined by single-cycle infection assay as described previously (66). Briefly, HIV-1 pseudovirus was generated via cotransfection of 293T cells with an Env-expressing plasmid and a backbone plasmid, pSG3Δenv, that carried an Env-defective, luciferase-expressing HIV-1 genome. Supernatants were harvested 48 h after transfection and the 50% tissue culture infectious dose (TCID50) was determined in TZM-bl cells. Peptides were prepared in 3-fold dilutions and mixed with 100 TCID50viruses and then incubated for 1 h at room temperature. The mixture was added to TZM-bl cells (104/well) in triplicate and incubated for 48 h at 37°C, and luciferase activity was measured. The percent inhibition and IC50s were calculated as described for the cell fusion assay.
Inhibition of a replicative HIV-1 isolate.Inhibition of peptides on a replication-competent HIV-1 isolate was determined using a molecular clone of JRCSF (subtype B, R5). Briefly, the plasmid pYK-JRCSF was transfected into 239T cells, and virus stock was harvested and quantified 48 h posttransfection. A total of 100 TCID50viruses was used to infect TZM-bl cells in the presence or absence of serially diluted peptides. Cells were harvested 2 days postinfection and lysed in cell lysis buffer, and luciferase activity was measured. The percent inhibition and IC50s were calculated as described above.
Cytotoxicity assay.The cytotoxicity of T20 and LP-40 on TZM-bl cells was measured using a CellTiter 96 AQueous One Solution cell proliferation assay (Promega). Briefly, 50l of peptides (in Dulbecco’s modified Eagle’s medium [DMEM]) at graded concentrations was added to TZM-bl cells, which were seeded on a 96-well tissue culture plate (1⫻104cells per well). After incubation at 37°C for 2 days, 20
l of CellTiter 96 AQueous One Solution reagent was pipetted into each well and further incubated at 37°C for 2 h. The absorbance was measured at 490 nm using a SpectraMax M5 microplate reader.
CD spectroscopy. CD spectroscopy was performed as described previously (67). Briefly, a CHR peptide was incubated with an equal molar concentration of the NHR peptide N39 at 37°C for 30 min in phosphate-buffered saline (PBS) (pH 7.2). CD spectra were acquired on a Jasco spectropolarimeter (model J-815) using a 1-nm bandwidth with a 1-nm step resolution from 195 to 260 nm at room temperature. Spectra were corrected by subtraction of a solvent blank. The␣-helical content was calculated from the CD signal by dividing the mean residue ellipticity ([]) at 222 nm by the value expected for 100% helix formation (⫺33,000° · cm2· dmol⫺1). Thermal denaturation was performed by monitoring the ellipticity change at 222 nm from 20°C to 98°C at a rate of 1.2°C/min, andTmwas defined as the midpoint of the
thermal-unfolding transition.
N-PAGE.N-PAGE was carried out to determine the interaction between the N and C peptides, as described previously (68, 69). Briefly, an N peptide (N39 or N36) was incubated with a C peptide (T20, T20-TRM, LP-40, or C34) (the final concentration of each peptide was 40M) at 37°C for 30 min. The sample was mixed with Tris-glycine native sample buffer at a ratio of 1:1 and then loaded onto a 10- by 1.0-mm Tris-glycine gel (20%) at 25l/per well. Gel electrophoresis was carried out with 100 V constant voltage at 4°C for 3 h. The gel was then stained with Coomassie blue and imaged with a Bio-Rad imaging system.
T20-Based Lipopeptide HIV-1 Fusion Inhibitor Journal of Virology
on November 7, 2019 by guest
http://jvi.asm.org/
ITC.The ITC assay was performed using an ITC200 microcalorimeter instrument (MicroCal, North-ampton. MA, USA) as described previously (18, 26). Briefly, 1 mM N39 peptide dissolved in double-distilled H2O (ddH2O) was injected into a chamber containing 100M T20, T20-TRM, or LP-40. The experiments were performed at 25°C. The time between injections was 240 s, and the stirring speed was 500 rpm. Data acquisition and analysis were performed using MicroCal Origin software (version 7.0).
Synergy analysis.The synergistic effects of inhibitors on diverse HIV-1 pseudoviruses were deter-mined by single-cycle infection assays as described above. Peptides were tested individually and in combination at a fixed molar ratio, which was based on the IC50 of single drugs that were tested separately over a range of serial dilutions. The inhibition data were analyzed for cooperative effects by using the method of Chou and Talalay (70, 71). The analysis was conducted in a stepwise fashion by calculating IC50s based on the dose-response curves of single drugs that were tested separately and two drugs tested in combination. Then, the CI was calculated by using the median-effect equation with the CalcuSyn program to assess the synergistic effect of combinations. A CI of⬍1 indicates synergism (CI values were interpreted as follows: 0.1, very strong synergism; 0.1 to 0.3, strong synergism; 0.3 to 0.7, synergism; 0.7 to 0.85, moderate synergism; and 0.85 to 0.90, slight synergism), a CI of 1 or close to 1 indicates additive effects, and a CI of⬎1 indicates antagonism. Dose reduction was calculated by dividing the IC50of a peptide when it was tested alone by that of the same peptide tested in combination with another peptide(s).
Crystallization of the N44/LP-40 complex.Equal amounts of N44 and LP-40 were dissolved in denaturing buffer (100 mM NaH2PO4, 10 mM Tris-HCl, pH 8.0, 8 M urea). To refold the peptides, the mixture was dialyzed against buffer containing 50 mM Tris-HCl, pH 7.5, 100 mM NaCl at 4°C overnight. The dialyzed sample was concentrated by centrifugation and subsequently loaded on a size exclusion column (Superdex 75 10/300 GL; GE Healthcare). An elution mixture corresponding to the molecular weight of a 6-HB was collected and concentrated prior to the crystallization trials. The N44/LP-40 complex was crystallized by mixing equal volumes (0.2l) of purified peptide complex (⬃10 mg/ml) and reservoir solution (0.1 M Tris-HCl, pH 8.5, 10% [wt/vol] PEG 4000) in a sitting-drop vapor diffusion system at 18°C. Cryocooling of the crystals was achieved by soaking the crystals for 5 s in reservoir solution containing 30% (vol/vol) glycerol followed by flash freezing to 100 K in liquid nitrogen. All data sets were collected on beamline BL17U at the Shanghai Synchrotron Research Facility (SSRF) and processed with HKL2000. All data collection and processing statistics are listed in Table 5.
Structural determination and refinement.The structure was determined by molecular replace-ment with the crystallographic software PHASER (72). The search model was the gp41 core structure (Protein Data Bank [PDB] accession no.1ENV), and iterative refinement with the program PHENIX and model building with the program COOT were performed to complete the structure refinement (73, 74). Structure validation was performed with the program PROCHECK, and all structural figures were generated with PyMol.
ACKNOWLEDGMENTS
We thank Zene Matsuda at the Institute of Medical Science of the University of Tokyo (Tokyo, Japan) for providing the DSP plasmids.
This work was supported by grants from the National Natural Science Foundation of China (81630061, 81473255, 81673484, and 81271830) and the CAMS Innovation Fund for Medical Sciences (2017-I2M-1).
REFERENCES
1. Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol 4:309 –319.https://doi.org/10 .1038/nrm1076.
2. Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem 70:777– 810.https://doi.org/10.1146/ annurev.biochem.70.1.777.
3. Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273.https://doi.org/10.1016/ S0092-8674(00)80205-6.
4. Tan K, Liu J, Wang J, Shen S, Lu M. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A 94:12303–12308.
https://doi.org/10.1073/pnas.94.23.12303.
5. Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426 – 430.
https://doi.org/10.1038/387426a0.
6. Chan DC, Chutkowski CT, Kim PS. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc Natl Acad Sci U S A 95:15613–15617.https://doi.org/10.1073/ pnas.95.26.15613.
7. Qiu Z, Chong H, Yao X, Su Y, Cui S, He Y. 2015. Identification and charac-terization of a subpocket on the N-trimer of HIV-1 Gp41: implication for viral entry and drug target. AIDS 29:1015–1024.https://doi.org/10.1097/QAD .0000000000000683.
8. Crespillo S, Camara-Artigas A, Casares S, Morel B, Cobos ES, Mateo PL, Mouz N, Martin CE, Roger MG, El Habib R, Su B, Moog C, Conejero-Lara F. 2014. Single-chain protein mimetics of the N-terminal heptad-repeat region of gp41 with potential as anti-HIV-1 drugs. Proc Natl Acad Sci U S A 111:18207–18212.https://doi.org/10.1073/pnas.1413592112. 9. Chu S, Gochin M. 2013. Identification of fragments targeting an
alterna-tive pocket on HIV-1 gp41 by NMR screening and similarity searching. Bioorg Med Chem Lett 23:5114 –5118. https://doi.org/10.1016/j.bmcl .2013.07.026.
10. Chan DC, Kim PS. 1998. HIV entry and its inhibition. Cell 93:681– 684.
https://doi.org/10.1016/S0092-8674(00)81430-0.
11. Weng Y, Weiss CD. 1998. Mutational analysis of residues in the coiled-coil domain of human immunodeficiency virus type 1 transmembrane protein gp41. J Virol 72:9676 –9682.
12. He Y. 2013. Synthesized peptide inhibitors of HIV-1 gp41-dependent mem-brane fusion. Curr Pharm Des 19:1800 –1809. https://doi.org/10.2174/ 1381612811319100004.
13. Eggink D, Berkhout B, Sanders RW. 2010. Inhibition of HIV-1 by fusion inhibitors. Curr Pharm Des 16:3716 –3728. https://doi.org/10.2174/ 138161210794079218.
14. Rimsky LT, Shugars DC, Matthews TJ. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J Virol 72:986 –993.