Cross-Coupling Chemistry as a Tool for the
Synthesis of Diverse Heterocyclic Systems and
Natural Products
A Thesis Submitted for the Degree of Doctor of
Philosophy of the Australian National University
by
Michael Elvis Dlugosch
Research School of Chemistry
Canberra, Australia
Acknowledgements
First I would like to thank my supervisor Professor Martin Banwell for having given me the
opportunity and the privilege to undertake my PhD studies in his research group. Like a captain, he
would guide me through the rough seas of my research and make sure that I would never lose track
of where I was going. He would encourage me and he would inspire me to never give up and always
keep going, even and especially at times of doubt and uncertainty. If it was not for Martin Banwell I
never would have made it to the point where I am now. Thank you very much for your guidance and
for your patience with me.
Thank you to Dr Brett Schwartz for being both my lab mate and also for his role as a member of my
supervisory panel. On countless occasions he has given me pivotal ideas which would push forward
my research into completely new directions. I never would have finished my work on the
epi-kirkamide if it hadn’t been for Dr Schwartz sharing his expertise and profound knowledge with me
and providing me with the relevant ideas to bring my research to a successful conclusion. I would
also like to thank Associate Professor Malcolm McLeod for his role as a member of my supervisory
panel. His feedback and his ideas which he shared with me on numerous occasions during our
discussions have sharpened my perspective on how and where to direct my research. I thank the
technical staff in the Research School of Chemistry for all their support. Thank you to Anitha
Jeyasingham, Joe Boileau, Adam Carroll and Thy Truong for all the mass specs you ran for me. Also, a
big thank you to Daniel Bartkus for all your work you invested into setting up the bioreactor and
helping me with the actual biotransformation. Thank you also to Drs Paul Carr and Jas Ward for all
the X-ray structures you ran for me. Thank you also to Drs Hideki Onagi and Nicholas Kanizaj for not
only being helpful in the lab, but also for being two very good friends.
Also I would like to thank the various project and visiting students I had the pleasure of supervising
and working with. Thank you to Alfred Fung, Michael Clark and Yun Qiu for your hard work.
A great thank you to my beautiful housemates Jenny and Nick for always making me feel at home,
and also to Daniel Smith for your dark humour and sarcasm, that helped me through so many
difficult situations. Thanks also to Moira, Zoë and Brad for tolerating my existence. I would also like
to thank my very good friend Junna Hayashi for all the beautiful and vibrant times we had together.
Without you my time here at the RSC and in Australia would have been far less enjoyable. I will
always cherish the memories of the expeditions we went on, but also the dinner and movie nights
we had. Thank you also all the members of the HDR Student Representative Committee. It was a
pleasure working with you towards an even better RSC.
Table of Contents
Declaration
i
Acknowledgements
ii
Table of Contents
iv
Publications
v
Relative Contributions to Publications
vii
Abstract
1
Synopsis
2
Thesis Overview
3
Statement of Contribution
20
Publication One
24
Publication Two
36
Publication Three
157
Publication Four
238
Publication Five
264
Publications
The following list details the publications that have resulted from the author’s research work
performed during his candidature for the Degree of Doctor of Philosophy
Publications:
1.
The Palladium-catalysed Ullmann Cross
-
coupling Reaction:
A Modern Variant on a Time-honored Process
Faiyaz Khan, Michael Dlugosch, Xin Liu and Martin G. Banwell
Accounts of Chemical Research, 2018, 51, 1784-1795.
2.
Palladium-Catalyzed Ullmann Cross-Coupling of β-Iodoenones and β-Iodoacrylates with
o-Halonitroarenes or o-Iodobenzonitriles and Reductive Cyclization of the Resulting Products
To Give Diverse Heterocyclic Systems
Faiyaz Khan, Michael Dlugosch, Xin Liu, Marium Khan, Martin G. Banwell, Jas S. Ward, and
Paul D. Carr, Organic Letters 2018, 20, 2770–2773.
3.
Reductive Cyclization of o-Nitroarylated-α,β-Unsaturated Aldehydes and Ketones with
TiCl
3/HCl or Fe/HCl Leading to 1,2,3,9-Tetrahydro-4H-carbazol-4-ones and Related
Heterocycles
Yun Qiu, Michael Dlugosch, Xin Liu, Faiyaz Khan, Jas S Ward, Ping Lan, and Martin G Banwell
J. Org. Chem., 2018, 83, 12023–12033.
4.
Synthesis of a Highly Functionalised and Homochiral 2-Iodocyclohexenone Related to the
C-Ring of the Polycyclic, Indole Alkaloids Aspidophytine and Haplophytine
Michael Dlugosch and Martin Banwell
Australian Journal of Chemistry, 2018, 71, 573-579.
Article featured on the cover of the journal
5.
Syntheses of Structurally and Stereochemically Varied Forms of C
7N Aminocyclitol
Derivatives from Enzymatically-derived and Homochiral cis-1,2-Dihydrocatechols
6.
Chemical Syntheses of the Cochliomycins and Certain Related Resorcylic
Acid Lactones
Commentary on the Contributions of Mr Michael Dlugosch to the
Six Papers Included in this PhD Thesis by Publication
Publication 1. This is a review article that was written by Professor Martin Banwell. It incorporates
descriptions of research on palladium-catalyzed Ullmann cross-coupling reactions conducted by the
co-authors including the author of this thesis.
Publication 2. Professor Martin Banwell proposed the research work reported in this article. The
author carried out 40 % of the reported laboratory work. In addition he collated and formatted 40 %
of the reported spectral data presented in the Supporting Information. The author also wrote 40 %
of the Experimental Section and conducted relevant literature surveys. Professor Martin Banwell
wrote the body of the paper.
Publication 3. The initial idea for this project came from Professor Martin Banwell. The author
carried out 65 % of the laboratory work reported in this article. In addition, he collated and
formatted 60 % of the reported spectral data presented in the Supporting Information. The author
also wrote 65 % of the Experimental Section and conducted relevant literature surveys. Professor
Martin Banwell wrote the body of the paper.
Publication 4. The initial idea for this project came from Dr Lorenzo White. The author carried out
the entirety of the laboratory work reported in this article. In addition, he collated and formatted
the entirety of the reported spectral data presented in the Supporting Information. The author also
wrote the entirety of the Experimental Section and conducted relevant literature surveys. Professor
Martin Banwell wrote the body of the paper.
Publication 5. The initial idea for this project came from Professor Martin Banwell. The author
carried out the entirety of the laboratory work associated with the reported synthesis of
epi-kirkamide (Schemes 1 and 2) and the enantiomeric switching regime (Scheme 6). In addition, he
collated and formatted the entirety of the reported spectral data sets presented in the Supporting
Information. He also wrote the entirety of the corresponding portion of the Experimental Section
and conducted relevant literature surveys on epi-kirkamide. He also conducted extensive research of
the relevant literature pertaining to the synthesis of various kirkamide analogues. Professor Martin
Banwell wrote the body of the paper.
Publication 6. This is a review article that was written by Professor Martin Banwell. It incorporates
Abstract
Publication 1 comprises a review article concerned with palladium-catalyzed Ullmann cross-
coupling reactions. Specifically, it details modern variants of these type of reactions and their
extensive use, most notably by the Banwell group, in the synthesis of various heterocyclic systems,
including ones encountered in natural products. Publication 1 contextualizes the research described
in Publications 2-4.
Publication 2 is concerned with the palladium-catalyzed Ullmann cross-coupling reactions of
β-iodoenones or β-iodoacrylates with o-iodonitrobenzenes or o-iodobenzonitriles, as well as the
reductive cyclization of the resulting products to give various heterocyclic systems. Thus, this
publication is concerned with a two-step approach to the synthesis of structurally diverse and
biologically active heterocycles, including quinolones and benzomorphans which are normally only
accessible via multistep-syntheses.
Publication 3 outlines research on palladium-catalyzed Ullmann cross-coupling reactions of
α-iodoenones with o-iodonitrobenzenes and the reductive cyclization of the ensuing coupling products.
Specifically, it details the exploration of two distinct modes of reductive cyclization that allow for the
synthesis of structurally “complementary” heterocyclic ring systems from a common precursor.
Publication 4 describes a chemoenzymatic synthesis of a highly functionalized and homochiral
-iodocyclohexenone that it is expected will serve as a precursor, through the application of
palladium-catalyzed Ullmann cross-coupling and reductive cyclization reactions, to the complex
indole alkaloids aspidophytine and haplophytine. The synthesis starts with an enantiomerically pure
cis-1,2-dihydrocatechol that is itself obtained through the whole-cell biotransformation of
bromobenzene.
Publication 5 is concerned with the developing syntheses of certain C
7N aminocyclitols, a significant
group of biologically active natural products. In particular, this paper details chemoenzymatic total
syntheses of several novel compounds within the class, including analogues of the recently isolated
natural product kirkamide. The syntheses exploit, as starting materials, enzymatically-derived and
homochiral cis-1,2-dihydrocatechols obtained from either iodo- or bromo-benzene. Methods for
obtaining the enantiomers of the reported C
7N aminocyclitol derivatives have been identified. Once
again, palladium-catalyzed cross-coupling chemistries were employed as key steps in these
syntheses.
Publication 6 is concerned with developing chemical syntheses of cochliomycins and related,
synthesis of an important subset of the large class of structurally distinct and biologically significant
natural products known as resorcylic acid lactones.
Synopsis
Cross-coupling reactions provide a particularly effective means for the formation of carbon-carbon
bonds. Many, well-established methods for forming such bonds now exist, perhaps the most
noteworthy being palladium-catalyzed cross-couplings that involve the linking of a halide or
pseudo-halide with an organometallic or metalloid species. While these reactions often give good yields of a
single product, one drawback is the need to form the requisite organometallic species, usually from
the corresponding organo-halide. While the classical Ullmann cross-coupling reaction has the
advantage that it can affect the direct coupling of two distinct organo-halides, harsh reaction
conditions are usually involved (temperatures in excess of 250 °C are frequently required) and often
homo-coupling is the predominant, if not the only process observed.
1The palladium-catalyzed
Ullmann cross-coupling affords many of the advantages of the standard palladium-catalyzed
processes as well as those associated with the original Ullmann reaction. As such, it is now possible
to carry out hetero-couplings of two distinct organo-halides under mild conditions.
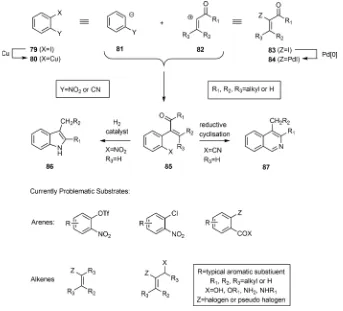
The focus of the research described in this thesis is the deployment of palladium-catalyzed Ullmann
cross-coupling reactions in the synthesis of biologically active heterocyclic systems, as well as the
total synthesis of natural products. A common theme is the cross-coupling of iodoenones with
substituted o-iodo- or o-bromo-benzenes as shown, for example, in Scheme 1. The substituents (R’)
associated with the haloarenes are usually strongly electron-withdrawing ones, such as nitrile or
nitro groups. The proposed reaction mechanism for this type of couplings is based on a previously
reported reaction mechanism by Shimizu and co-workers.
2In the first step the Pd(0) catalyst
Scheme 1: An Example of the Palladium-Catalyzed Ullmann Cross-Coupling Reaction and the
Proposed Reaction Mechanism
Such palladium-catalyzed Ullmann cross-couplings can be conducted using a range of iodinated
enones, as described in Publications 2 and 3, and so providing ready access to diverse heterocyclic
systems in just one or two steps. As described in Publication 4, it is anticipated that the reaction can
also be deployed for the assembly of more elaborate systems. The application of related
cross-coupling reactions to syntheses of natural products and natural product analogues are described in
Publications 5 and 6.
Thesis Overview
Publication 1: The Palladium-catalysed Ullmann Cross-coupling Reaction:
A Modern Variant on a Time-honored Process
reaction sequence shown in Scheme 2 is also one direct example of the author’s immediate
contributions to this publication.
Scheme 2: Synthesis of Indole 4
Another example is the synthesis of a class of compounds called carbolines. There are four isomeric
carbolines, as shown in Figure 1, and each of these is encountered in natural products and/or within
pharmacologically significant compounds.
Figure 1: The Structures of the Four Isomeric Carbolines
The synthesis of the carboline-type natural product harman (13)
3(possessing anti-HIV activity
4)
shown in Scheme 3 further exemplifies the utility of the protocols developed by the Banwell group.
Scheme 3: Synthesis of the Natural Product Harman
Publication 2. Palladium-Catalyzed Ullmann Cross-Coupling of β-Iodoenones and β-Iodoacrylates
with o-Halonitroarenes or o-Iodobenzonitriles and Reductive Cyclization of the Resulting Products
to Give Diverse Heterocyclic Systems
Previous work conducted by the Banwell group on the palladium-catalyzed Ullmann cross-coupling
reaction was largely focussed on the coupling of α-iodoenones with o-halonitrobenzenes.
Transformations of this type provide access to different types of natural products including the
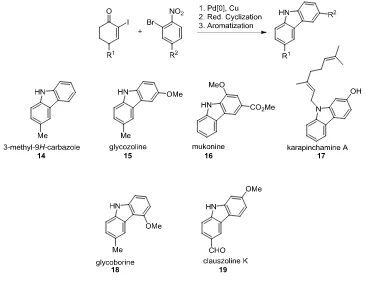
carbazoles shown in Figure 2.
5Figure 2: Structures of Diverse Carbazoles
[image:13.595.115.481.160.443.2]The successful syntheses of the members of the the uleine alkaloid family shown in Figure 3 further
highlight the power of such methodologies.
6Figure 3: Members of the Uleine Alkaloid Family Accessible Using the Palladium-Catalyzed Ullmann
The research reported in Publication 2 is complementary to such earlier work. The focus in this
publication is on the participation of β-iodoenones and β-iodoacrylates (rather than, say,
α-iodoenones) in palladium-catalyzed Ullmann cross-coupling reactions. Depending on the precise
nature of such coupling partners, as well as the methods used for the reductive cyclization, then
various heterocyclic systems including quinolones, dihydroquinolones, benzomorphanes and
naphthydrines can be obtained. A number of these have now become much more readily accessible
as a result. The benzomorphans reported in this publication are of particular interest, as many of
them have a number of notable medicinal properties including by serving as analgesics.
7Scheme 4
highlights some such transformations.
Scheme 4: Palladium-Catalyzed Ullmann Cross-Coupling of β-iodoenones with o-Halonitrobenzenes
Giving Access to Heterocyclic Systems such as Azacoumarins and Benzomorphans
In this study it was also shown that depending upon the mode of reduction of the initial Ullmann
cross-coupling product different cyclization products can be obtained. For instance, if compound 37
(Scheme 5) is treated with Fe in AcOH/HCl then quinolone 38 is obtained while exposing the same
substrate to standard hydrogenation conditions (H
2/Pd on C) affords the fully reduced product,
namely dihydroquinolone 36.
On the basis of the studies reported in Publication 2, it is clear that palladium-catalyzed Ullmann
cross-couplings involving β-iodoenones and β-iodoacrylates provide a versatile means for obtaining
hitherto unknown or less readily accessible heterocyclic frameworks.
Publication 3: Reductive Cyclization of o-Nitroarylated-α,β-Unsaturated Aldehydes and Ketones
with TiCl
3/HCl or Fe/HCl Leading to 1,2,3,9-Tetrahydro-4H-carbazol-4-ones and Related
Heterocycles
Publication 3 further articulates the utility of the palladium-catalyzed Ullmann cross-coupling
/reductive cyclization sequence. In particular, it focuses on different methods for effecting the
reductive cyclizations of the coupling products. Significantly, depending upon the mode of the
reductive cyclization a given coupling product may afford distinct heterocyclic products. This further
extends the range of heterocycles that can be obtained. In particular, Publication 3 focuses on the
complementary behaviours of the H
2/Pd on C and the TiCl
3/HCl reduction systems. So, as shown in
Scheme 6, if coupling product 40 is treated with H
2in presence of Pd on C then it reacts to give
product 39 while on treatment with TiCl
3/HCl then congener 41 is obtained.
Scheme 6: Divergent Reductive Cyclization Pathways for Compound 40
Similarly, as shown in Scheme 7, while subjection of cross-coupling products 46 or 47 to reductive
cyclization using H
2and Pd on C provides indole 45, on treating the same substrates with TiCl
3in HCl
Scheme 7: Synthesis of Reductive Cyclization Products 45 and 48
In contrast, the readily available cinnamaldehyde derivative 49 shown in Scheme 8 affords the
known anti-proliferative agent 50
8on treatment with TiCl
3
/HCl in acetone.
Scheme 8: TiCl
3-Mediated Cyclization of Compound 49
In a further example of the utility of such cyclization reactions, levoglucosenone derivative 51
provides, as depicted in Scheme 9, the tetracyclic product 52 upon treatment with TiCl
3in HCl.
Scheme 9: TiCl
3-Mediated Cyclization of Compound 51 to Give the Tetracyclic
Levoglucosenone-Based Compound 52
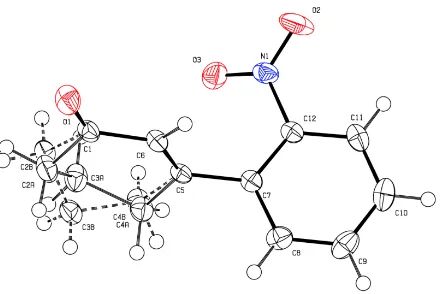
With this method it is also possible to form novel dihydroquinolines. Thus, ketones such as acetone
can be incorporated into the reductive cyclization product. As illustrated in Scheme 10, this
presumably occurs via Schiff base condensation, electrocyclic ring-closure then a prototropic shift
and accompanying re-aromatization. If, for instance, compound 31 is reacted with TiCl
3/HCl in
53. The amine residue so-formed then undergoes a Schiff base condensation with the added ketone
to yield intermediate 55 that undergoes electrocyclic ring closure to give intermediate 56. A
prototropic shift and accompanying re-aromatization then leads to the final product 54, the
structure of which was confirmed by single-crystal X-ray analysis.
Scheme 10: Formation of Dihydroquinoline 54 from Cross-coupling Product 31 and Acetone in the
Presence of TiCl
3/HCl
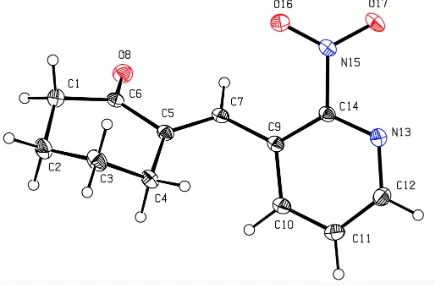
As shown in Scheme 11 a range of other dihydroquinolines was prepared by simply reacting
compound 53 with aldehydes or ketones, usually in presence of aqueous HCl. For instance, when
this substrate was reacted with benzaldehyde, then product 57 was obtained. Similarly, the reaction
between compound 53 and butanone afforded compound 58, while the reaction involving
cyclohexanone gave the spirocyclic product 59.
Scheme 11: Acid-Promoted Reactions of Compound 53 with Aldehydes or Ketones to Give
Overall, then, Publication 3 builds, in a distinctly complementary way, on the work reported in
Publication 2.
Publication 4: Synthesis of a Highly Functionalised and Homochiral 2-Iodocyclohexenone Related
to the C-ring of the Polycyclic, Indole Alkaloids Aspidophytine and Haplophytine
Aspidophytine is a constituent of the heterodimeric compound (+)-haplophytine (Figure 4) that
occurs in the Mexican cockroach plant Haplophyton cimididum.
9Dried leaves of this plant have been
used for their insecticidal properties since at least the Aztec era. A range of synthetic studies has
been conducted on the total synthesis of aspidophytine, the first synthesis of the
(–)-enantiomeric form having been reported by Corey and his co-workers in 1999.
10This was
followed by the reports of Fukuyama (2003)
11,12, Padwa (2006)
13,14, Marino (2006)
15, Nicolaou
(2008)
16, Tokuyama (2013)
17and Qiu (2013).
18Figure 4: Structures of the Alkaloids Haplophytine and Aspidophytine
A nine step synthesis of a highly functionalized and homochiral 2-iodocyclohexenone that is related
to the C-Ring of aspidophytine and haplophytine is reported in Publication 4. Unlike all previously
reported asymmetric total syntheses of these compounds, a chemo-enzymatic approach is used to
establish the required functionality and stereochemistry.
Scheme 12: Synthesis of the Homochiral 2-Iodocyclohexenone 70
The aminoalkyl chain associated with the target 2-iodocyclohexenone was introduced via Suzuki–
Miyaura cross-coupling of compounds 63 and 64. The quaternary stereocenter of the target was
then established using the allylic alcohol moiety embedded within compound 66 by treating it with
dimethylformamide dimethyl acetal (DMADMA) and thus effecting an Eschenmoser–Claisen
rearrangement. Deprotection of the newly generated allylic alcohol, followed by oxidation and
Johnson
-iodination then gave α-iodoenone 70.
Scheme 13: Possible Elaboration of the Palladium-Catalyzed Ullmann Cross-Coupling Product 70 to
Tetracyclic Compound 73
Scheme 14 illustrates a series of possible further reactions, including a Corey–Winter olefination,
that could be applied to compound 73 and so yielding ent-aspidophytine. Compound 73 may be
reacted with 2-bromoethanol to yield amino-alcohol 74, which can then be mesylated. Treating the
indole moiety with a strong base should then lead to deprotonation and subsequently to an
intramolecular displacement of the mesylate and ring-formation. The unprotected diol in compound
76 would then be converted to the corresponding olefin via Corey–Winter olefination. Hydrolysis of
the dimethyl amide and esterification should then give ent-aspidophytine.
Publication 5: Syntheses of Structurally and Stereochemically Varied Forms of C
7N Aminocyclitol
Derivatives from Enzymatically-derived and Homochiral cis-1,2-Dihydrocatechols
In 2015 the isolation of the new C
7N aminocyclitol kirkamide as well as an eleven step synthesis of it
from methyl N-acetyl-D-glucosamine was reported by Gademann and co-workers (Scheme 15).
19Scheme 15: Gademann’s Synthesis of Kirkamide
Kirkamide is found in leaf nodules of the plant Psychotria kirkii and likely produced by Candidatus
Burkholderia kirkii, a leaf symbiont of this plant.
20,21Since kirkamide has shown to be toxic to aquatic
arthropods and insects it might be acting as an anti-feedant and thus protecting the leaves of the
host plant.
In Publication 5 total syntheses of several derivatives of kirkamide, including an epimer, are
reported. In contrast to the previously reported synthesis, the approach taken in the author’s work is
a chemo-enzymatic one. The starting material for this purpose is the cis-dihydrocatechol 62, which
also served as the starting material for the synthesis described in Publication 4.
In Scheme 16 the total synthesis of epi-kirkamide is shown. Thus, treatment of previously reported
acetamide derivative, 87, with KHMDS gave, via the intermediate aziridine 88, the isomeric oxazoline
89. The introduction of the hydroxymethyl group associated with the C
7N aminocyclitols was
iodoalkene 91 in presence of N,O-dimethylhydroxylamine under a carbon monoxide atmosphere
gave the α,β-unsaturated Weinreb amide 92. A two-step reduction of this amide (using lithium
aluminium hydride for the formation of the corresponding aldehyde and subsequent Luche
reduction) yielded compound 95 and global deprotection of this using aqueous acetic acid then gave
epi-kirkamide (96).
Scheme 16: A Total Synthesis of epi-Kirkamide (96)
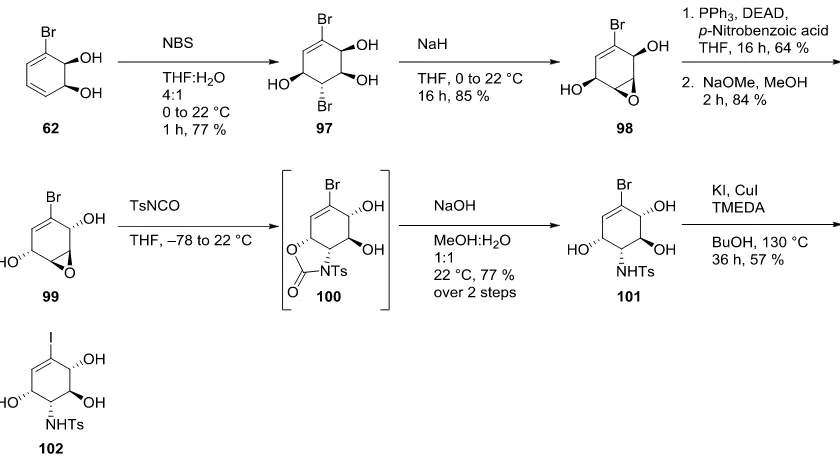
Scheme 17: Synthesis of Alkenyl Iodide 102 from cis-Dihydrocatechol 62
[image:23.595.88.508.71.305.2]Compound 102 was also synthesized in a more direct manner from the cis-dihydrocatechol 103
(Figure 5). However, while compound ent-62 is known, congener ent-103 is not. As such, only
synthetic sequences starting from the bromo-compound 62 can be used to produce the
enantiomeric forms of the targeted kirkamide analogues.
Figure 5: cis-Dihydrocatechol 103
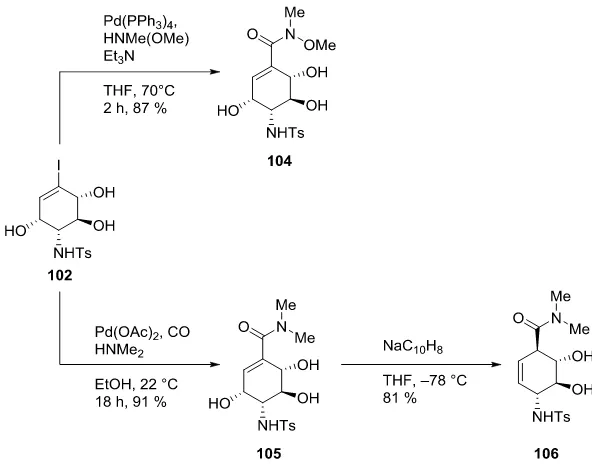
Compound 102 was engaged in two carbopalladation reactions (Scheme 18) and thereby affording
the C
7N-aminocyclitols 104 and 105. Furthermore, upon treating compound 105 with freshly
Scheme 18: Synthesis of New C
7N-Aminocyclitol Derivatives 104, 105 and 106 from Iodoalkenyl 102
[image:24.595.148.444.71.305.2]The three new aminocyclitols 104, 105 and 106 thus obtained have the potential to serve as
precursors to a diverse range of aminocarbasugars. Compounds 104 and 105 can also be regarded as
precursors to ent-kirkamide (ent-86) (Figure 6).
Figure 6: ent-Kirkamide (ent-86)
Publication 6. Chemical syntheses of the cochliomycins and certain related resorcylic acid lactones
Small molecule natural products (SMNPs) are often utilized as therapeutic agents, as precursors to
these or as an inspiration for them.
22Among SMNPs the resorcylic acid lactones (RALs) are notable
for the diversity of their biological activities, their unique structural features and their frequent
occurrence among fungal metabolites.
23This article reviews synthetic studies on and the biological
and is particularly interesting from a medicinal point of view because it shows good binding affinity
for human opioid receptors.
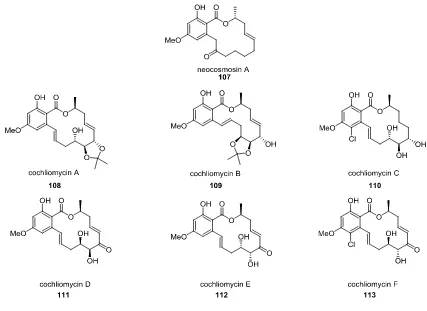
Figure 7: Structures of Various Cochliomycins
Scheme 19 shows the total synthesis of cochliomycin C as reported by the Banwell group. This
synthesis featuring a Stille cross-coupling as a crucial step is another example highlighting the broad
applicability of cross-coupling reactions for the total synthesis of natural products.
References
1
Ullmann F., Bielecki J. , Chem. Ber., 1901, 34, 2174–2185
2
Shimizu N., Kitamura T., Watanabe K., Yamaguchi T, Shigyo H., Ohta T., Tetrahedron Lett., 1993, 34,
3421-3424
3
Yan, Q.; Gin, E.; Banwell, M. G.; Willis, A. C.; Carr, P. D., J. Org. Chem., 2017, 82, 4328-4335
4
Ishida J., Wang H.K., Oyama M., Cosentino M.L., Hu C.Q., Lee K.H., J. Nat. Prod., 2001, 64, 958-960
5
Yan Q., Gin E., Wasinska-Kalwa M., Banwell M. G., J. Org. Chem. 2017, 82, 4148−4159
6
Tang F., Banwell M. G., Willis A., J. Org. Chem. 2016, 81, 2950-2957
7
Cittern, P.A., Kapoor V.K., Parfitt R.T., J. Med. Chem., 1986, 29, 1929–1933
8
Miao B., Zheng Y., Wu P., Li S., Ma S. Adv. Synth. Catal. 2017, 359, 1691
9
Snyder H.R., Fischer R.F., Walker J.F., Els H.E., Nussberger G.A., J. Am. Chem. Soc. 1954, 76, 4601-4605
10
He F., Bo Y., Altom J.D., Corey E.J., J. Am. Chem. Soc. 1999, 121, 6771-6772
11
Sumi S., Matsumoto K., Tokuyama H., Fukuyama T., Org. Lett., 2003, 5, 1891-1893
12
Sumi S., Matsumoto K., Tokuyama H., Fukuyama T., Tetrahedron Lett., 2003, 59, 8571-8587
13
Mejía-Oneto J.M., Padwa A., Org. Lett., 2006, 8, 3275-3278
14
Mejía-Oneto J.M., Padwa A., Helv. Chim. Acta, 2008, 91, 285-302
15
Marino J.P., Cao G., Tetrahedron Lett., 2006, 47, 7711-7713
16
Nicolaou K.C., Dalby S.M., Majumder U., J. Am. Chem. Soc., 2008, 130, 14942-14943
17
Satoh H., Ueda H., Tokuyama H., Tetrahedron, 2013, 69, 89-95
18
Yang R., Qiu F.G., Angew. Chem. Int. Ed., 2013, 52, 6015-6018
19
Sieber S., Cerlier A., Neuburger M., Grabenweger G., Eberl L., Gademann K., Angew. Chem. Int. Ed. 2015,
54, 7968-7970
20
VanOevelen S., DeWachter R., Vandamme P., Robbrecht E., Prinsen E., Int. J. Syst. Evol. Microbiol. 2002,
52, 2023 –2027
21
Carlier A.L., Eberl L., Environ. Microbiol. 2012, 14, 2757- 2769; Carlier A.L., Omasits U., Ahrens C.H.,
Eberl L., Mol. Plant-Microbe Interact. 2013, 26, 1325 –1333
22
Dias, D.A. Urban, S. Roessner, U. Metabolites 2012, 2, 303-336
23
Winssinger N. Barluenga S. Chemical Communications 2007, 22-36
24
Zhang Y. Dlugosch M. Jübermann M. Banwell M.G. Ward J.S. J. Org. Chem., 2015, 80, 4828-4833
25
Gao J., Radwan M.M., Leon F., Dale O.R., Husni A.S., Wu Y., Lupien S., Wang X., Manly S.P., Hill R.A,
The Palladium-Catalyzed Ullmann Cross-Coupling Reaction:
A Modern Variant on a Time-Honored Process
Faiyaz Khan, Michael Dlugosch, Xin Liu, and Martin G. Banwell
*
Research School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, ACT 2601, Australia
CONSPECTUS:
Cross-coupling reactions, especially those that are catalyzed by palladium, have revolutionized the way in
which carbon
−
carbon bonds can be formed. The most commonly deployed variants of such processes are the Suzuki
−
Miyaura,
Mizoroki
−
Heck, Stille, and Negishi cross-coupling reactions, and these normally involve the linking of an organohalide or
pseudohalide (such as a tri
fl
ate or nona
fl
ate) with an organo-metallic or -metalloid such as an organo-boron, -magnesium, -tin,
or -zinc species. Since the latter type of coupling partner is often prepared from the corresponding halide, methods that allow for
the direct cross-coupling of two distinct halogen-containing compounds would provide valuable and more atom-economical
capacities for the formation of carbon
−
carbon bonds. While the venerable Ullmann reaction can in principle achieve this, it has
a number of drawbacks, the most signi
fi
cant of which is that homocoupling of the reaction partners is a competitive, if not the
dominant, process. Furthermore, such reactions normally occur only under forcing conditions (viz., often at temperatures in
excess of 250
°
C). As such, the Ullmann reaction has seen only limited application in this regard, especially as a mid- to
late-stage feature of complex natural product synthesis. This Account details the development of the palladium-catalyzed Ullmann
cross-coupling reaction as a useful method for the assembly of a range of heterocyclic systems relevant to medicinal and/or
natural products chemistry. These couplings normally proceed under relatively mild conditions (<100
°
C) over short periods of
time and, usually, to the exclusion of (unwanted) homocoupling events. The keys to success are the appropriate choice of
coupling partners, the form of the copper metal employed, and the choice of reaction solvent.
At the present time, the cross-coupling partners capable of engaging in the title reaction are con
fi
ned to halogenated and
otherwise electron-de
fi
cient arenes and, as complementary reactants,
α
- or
β
-halogenated,
α
,
β
-unsaturated aldehydes, ketones,
esters, lactones, lactams, and cycloimides. Nitro-substituted (and halogenated) arenes, in particular, serve as e
ff
ective
participants in these reactions, and the products of their coupling with the above-mentioned carbonyl-containing systems can be
manipulated in a number of di
ff
erent ways. Depending on the positional relationship between the nitro and carbonyl groups in
the cross-coupling product, the reduction of the former group, which can be achieved under a range of di
ff
erent conditions,
provides, through intramolecular nucleophilic addition reactions, including Schi
ff
base condensations, access to a diverse range
of heterocyclic systems. These include indoles, quinolines, quinolones, isoquinolines, carbazoles, and carbolines. Tandem
variants of such cyclization processes, in which Raney cobalt is used as a catalyst for the chemoselective reduction (by
dihydrogen) of nitro and nitrile groups (but not ole
fi
ns), allow for the assembly of a range of structurally challenging natural
products, including marinoquinoline A, (
±
)-1-acetylaspidoalbidine, and (
±
)-gilbertine.
1. INTRODUCTION
Arguably, carbon
−
carbon bond formation is the most
important process in organic chemistry, and the development
of means for doing so has been a source of conscious e
ff
ort for
almost two centuries.
1In modern times, cross-coupling
reactions, perhaps most especially those catalyzed by
palladium, nickel, copper, and iron species, have revolutionized
the way in which more complex organic compounds are
assembled from simpler ones.
2Named reactions such as the
Suzuki
−
Miyaura, Mizoroki
−
Heck, Stille, Sonogashira, and
Negishi cross-couplings immediately spring to mind in
considering such matters.
2aThe coupling partners involved
in these processes are normally an organohalide or
metallic species that is, more often than not, obtained from a
halide precursor. In view of this and the frequently unstable/
sensitive nature of the organometallic species, there have been
many e
ff
orts directed at e
ff
ecting the reductive cross-coupling
of two structurally distinct organohalides, the most
con-spicuous examples of which involve adaptations of the
venerable Wurtz
1and Ullmann
3reactions. In their traditional
forms, however, these processes have not found extensive
application because of competition from homocoupling
reactions and/or the need to use rather aggressive reaction
conditions that are incompatible with other functionalities
Received: April 13, 2018
Article
pubs.acs.org/accounts
Cite This:Acc. Chem. Res.2018, 51, 1784−1795
Downloaded via AUSTRALIAN NATL UNIV on September 18, 2018 at 05:05:02 (UTC).
present in the substrates. In recent times, so-called
cross-electrophile couplings (XECs), especially ones carried out in
reductive mode and often involving multimetallic catalysts,
have come to the fore, with notable contributions having been
reported in the past few years by various groups.
4The
versatility of such processes is quickly becoming apparent.
Herein we detail the outcomes of our own ongoing work
concerned with the development of the palladium-catalyzed
Ullmann cross-coupling reaction of structurally distinct, sp
2-hybridized, halogen-associated electrophiles with one
anoth-er.
5,6These reactions enable the construction of products that
are useful in their own right and/or can participate in reductive
cyclization reactions and thus a
ff
ording various heterocyclic
motifs encountered in a range of interesting natural products.
2. THE CLASSICAL ULLMANN REACTION
The Ullmann reaction (
Scheme 1
) was
fi
rst reported in 1901
3aand in the intervening period has found extensive application
in chemical synthesis, most notably in the reductive coupling
of aryl halides (e.g.,
1
) to form the corresponding symmetrical
biaryls (e.g.,
2
).
3,6Its limitations also became evident rather
quickly. These include the need to use high reaction
temperatures (>200
°
C), the attendant functional group
incompatibilities, an inability to cleanly generate
unsym-metrical biaryls from two structurally distinct aryl halide
precursors (because of competing homocoupling processes),
and the frequently erratic yields obtained. Manifold e
ff
orts to
redress such de
fi
ciencies have been undertaken over the years,
including through the application of on-surface processes,
3gthe
introduction of metal-chelating species,
3eand the use of
varying forms of copper as well as other metal species.
3,6These
have had useful impacts, as summarized in a range of recent
review articles.
33. DISCOVERY OF THE PALLADIUM-CATAYLZED
ULLMANN CROSS-COUPLING REACTION
Some years ago, in connection with work directed toward
establishing a total synthesis of the alkaloid rhazinal,
7a potent
spindle toxin, we required access to an arylated pyrrole. We
initially attempted to prepare this key intermediate through
conventional Ullmann cross-coupling of commercially available
o
-bromonitrobenzene (
1
) with the known iodinated pyrrole
3
(see
Scheme 2
), but only traces of target
4
were obtained.
Upon undertaking an extensive literature survey, we came
across the work of Thompson
8and Shimizu,
9both of whom
reported that the synthesis of certain arylated pyridines
through the Ullmann cross-coupling of the relevant aryl halide
and halogenated pyridine is greatly facilitated by the addition
of a palladium catalyst. Upon applying such observations to
our system,
7using DMF as solvent and three equivalents of
compound
1
, we were able to obtain, under ultrasonication
conditions, target
4
in 88% yield (based on recovered starting
material (brsm)), with the major byproduct being 2,2
′
-dinitrobiphenyl (
2
) (55%) (
Scheme 2
).
These observations triggered extensive studies of the title
process that continue in our group to this day. These studies
have provided, through the reductive cyclization of the initially
formed cross-coupling products, useful new means for the
construction of a wide range of heterocyclic compounds,
including ones embodying previously unreported frameworks.
Details of these processes are presented in the following
sections and categorized according to the heterocyclic
frameworks that are generated.
4. APPLICATION TO THE SYNTHESIS OF
HETEROCYCLES
4.1. Indoles
Our
fi
rst e
ff
orts to comprehensively develop the title reaction
involved the cross-coupling of readily available
α
-halo-enones
and -enals with
o
-halonitroarenes and the reductive cyclization
of the ensuing
α
-arylated-enones and -enals to give indoles,
including annulated variants.
10The simple reaction sequences
shown in
Scheme 3
serve to highlight the possibilities for the
assembly of such heterocycles, and others have since exploited
these processes in the total synthesis of a range of natural
products.
11Some of our own e
ff
orts in this regard are detailed
in the next section.
In the course of optimizing these sorts of cross-coupling
processes, we established that a range of di
ff
erent sources of
Pd[0] can be used, that DMSO appears to be the optimal
solvent, that electron-de
fi
cient, halogenated arenes are
required, and that a lower reaction temperature leads to a
better ratio of cross-coupling to homocoupling products.
Indeed, in favorable circumstances the cross-coupling reactions
can be conducted at near ambient temperatures and essentially
to the exclusion of the homocoupling process. Thus, a close to
1:1 ratio of coupling partners could often be employed, an
important consideration in exploiting these processes in
complex natural product synthesis, where such transformations
are exploited at a late stage. Mechanistically speaking, we
believe that these couplings proceed as suggested by Shimizu
9Scheme 1. Original (1901) Ullmann Reaction Resulting in
the Reductive Coupling of
o
-Bromonitrobenzene (1) To
A
ff
ord
o
,
o
′
-Dinitrobiphenyl (2)
Scheme 2. Palladium-Catalyzed Ullmann 1 and 3 Leading to Arylated Pyrrole 4
(see the penultimate section for details), wherein palladium[0]
oxidatively adds to the
α
-iodo-enone or -enal and the resulting
palladium[II] complex reacts with the ortho-cuprated
nitro-arene arising from the other coupling partner, thereby
producing a palladated intermediate that undergoes reductive
elimination to deliver the observed product (and, of course,
regenerates the Pd[0] catalyst). The nature of the copper used
in these reactions has some impact on the e
ffi
ciency of the
process, with freshly prepared activated copper
12being
particularly e
ff
ective though somewhat tedious to prepare.
The simple expedient of adding some sand to the reaction
mixture containing normal copper powder (copper bronze),
and thus continuously generating a fresh metal surface through
abrasion, is an operationally simple means of achieving often
the desired process,
11balthough the precise origins of this
bene
fi
t remain to be fully understood.
Highly functionalized indolic substructures are encountered
in therapeutically signi
fi
cant alkaloids such as vincristine
(
Scheme 4
), and we sought to establish methods for
assembling these using our protocols.
13In a representative
process,
α
′
-carbomethoxylated cycloheptenone
12
was
sub-jected to Pinhey arylation with plumbated indole
13
, thereby
a
ff
ording compound
14
, which was itself engaged in a
Johnson-type
α
-iodination
14reaction to a
ff
ord iodide
15
. The
palladium-catalyzed Ullmann cross-coupling of this last
compound with
o
-iodonitrobenzene (
5
) gave product
16
,
which upon reductive cyclization a
ff
orded bis(indole)
17
Scheme 3. Palladium-Catalyzed Ullmann Cross-Coupling/Reductive Cyclization Sequences Leading to Indoles 8 and 11
Scheme 4. Synthetic Sequence Leading to Bis(indole) 17 Resembling the Southern Hemisphere of Vincristine
Scheme 5. Palladium-Catalyzed Ullmann Cross-Coupling/Reductive Cyclization Sequence Leading to Oxindole 20
4.2. Oxindoles
Oxindoles, which represent privileged structures in medicinal
chemistry and motifs encountered in biologically active natural
products,
15are readily obtained using analogous processes
wherein an
α
-brominated
α
,
β
-unsaturated cycloimide, lactam,
or lactone is used as the coupling partner in a reaction with an
o
-halonitroarene and the product of this process then subjected
to reductive cyclization.
15The e
ffi
cient
5
and
18
(
Scheme 5
)
to produce arylated
N
-methylmaleimide
19
followed by its
reductive cyclization under standard conditions to give
oxindole
20
is illustrative of these types of processes.
4.3. Quinolines and Related Heterocycles
A further extension of our original processes, as shown in
Scheme 3
, has allowed the formation of quinolones and related
coupling of arene
2
with aldehyde
21
a
ff
ords arylated enal
22
,
which, upon reductive cyclization, produces
cyclopenta-annulated quinolone
23
. In a related but less e
ffi
cient manner,
cross-coupling of compounds
2
and
24
a
ff
ords ester
25
which
upon reductive cyclization delivers the 2-quinolone
26
. By
similar means a range of alternately substituted/annulated
quinolones, phenanthridines, and 6(5
H
)-phenanthridinones
can be obtained. The capacity to generate electrophiles such as
21
directly from the corresponding ketone (in this case
cyclopentenone) through a Vilsmeier
−
Haack haloformylation
reaction is likely to enhance the utility of these processes.
164.4. Carbazoles
When
α
-iodocyclohex-2-en-1-ones are cross-coupled with
halogenated nitroarenes such as
1
and
5
using the protocols
Scheme 6. Palladium-Catalyzed Ullmann Cross-Coupling/Reductive Cyclization Sequences Leading to Quinoline 23 and
Quinolone 26
Scheme 7. Synthetic Routes to the Carbazole Natural Products Clauszoline K (32) and Karapinchamine A (34)
that their fully aromatic counterparts (viz., carbazoles) are
encountered in a wide range of biologically active natural
products, we sought to produce such heterocycles using
variations of our earlier protocols. The routes to clauszoline K
and karapinchamine A shown in
Scheme 7
are illustrative of
the possibilities the title reaction o
ff
ers in this regard.
17Thus,
reductive cross-coupling of electrophiles
27
and
28
under our
now standard conditions a
ff
orded product
29
(80%), which
upon reductive cyclization using dihydrogen in the presence of
Raney nickel a
ff
orded tetrahydrocarbazole
30
(65%). This
could then be oxidized to its fully aromatic counterpart,
namely carbazole
31
(88%), upon exposure to 10% Pd on C in
diphenyl ether at 210
°
C (various attempts to e
ff
ect the
conversion
29
→
31
in a direct manner, or at least in a
one-pot-process, have been unsuccessful to date). Upon exposure
of compound
31
to
2,3-dichloro-5,6-dicyano-1,4-benzoqui-none (DDQ) it was oxidized, in 66% yield, to the natural
product clauszoline K (
32
). On the other hand, treatment of
compound
31
with BBr
3e
ff
ected cleavage of the associated
ether residue and, thereby, the formation of the anticipated
phenolic product
33
(84%). Deprotonation of the latter
compound with
n
-butyllithium and reaction of the ensuing
anion with geranyl bromide resulted in alkylation at nitrogen
and the formation of the carbazole-containing natural product
karapinchamine A (
34
), which was obtained in 50% yield.
4.5. Carbolines
There are four isomeric carbolines, namely, the
α
,
β
,
γ
, and
δ
forms (
35
−
38
, respectively;
Figure 1
), and each of these
frameworks is encountered in both natural products and
pharmacologically active agents.
18While various methods have
been developed for their synthesis, a uni
fi
ed approach to them
had remained elusive until our recent deployment of the title
cross-coupling reaction for this purpose.
18An illustrative example of our approach is presented in
Scheme 8
. It starts with the palladium-catalyzed Ullmann
cross-coupling of bromonitropyridine
39
with readily available
α
-iodinated cyclohexenone
40
. Engaging the ensuing product
41
(80%) in a reductive cyclization reaction gives
tetrahy-drocarboline
42
(83%), which is then dehydrogenated to give
the fully aromatic compound
43
(83%) representing the
structure of the natural product harman.
18The challenge associated with deploying this type of
approach to the carbolines is the need to construct the
requisite polysubstituted pyridine-based coupling partner.
Thus, for example, the nitration reaction associated with the
synthetic sequence leading to compound
39
also produced a
regioisomer, and these could only be separated from one
another by HPLC techniques.
184.6.β-Haloenones and Related Compounds as Cross-Coupling Partners
Recently we have established that
β
-haloenones such as
44
couple particularly e
ff
ectively with electrophiles including
5
(
Scheme 9
) to form the anticipated cross-coupling product
45
(91%), a compound that upon exposure to standard reductive
cyclization conditions using methanol as the solvent a
ff
ords
3,4-benzomorphan
46
in 73% yield.
19In a further illustration
of the extensive utility of these types of processes, the coupling
of brominated pyridine
47
with the
β
-iodinated crontonate
48
proceeded with retention of con
fi
guration and a
ff
orded the
anticipated product
49
(84%). Reductive cyclization of this last
compound using iron
fi
lings in an acidic medium then gave the
1,8-naphthyridin-2(1
H
)-one
50
(76%). Interestingly,
o
-iodo-benzonitriles can be engaged in related couplings,
19although
these are less e
ffi
cient than those involving iodinated
nitroarenes, presumably because of the weaker
electron-withdrawing properties of the cyano group.
4.7. Formation of Unsymmetrical Biaryls
An obvious application of the title reaction is in the production
of unsymmetrical biaryls. While we have yet to explore such
processes in any comprehensive fashion, early indications have
been very positive. Thus, as shown in
Scheme 10
for example,
the cross-coupling of aryl iodide
51
with bromide
52
under our
by now standard conditions provided the desired biaryl
53
(60%).
20This last compound was readily elaborated to the
alkaloid zephycandidine III, a natural product reported to
possess acetylcholinesterase (AChE) inhibitory properties,
20which were not evident in the synthetically derived material
despite the spectroscopic equivalence of the natural and
synthetic materials. More pertinent to the present discussion is
that all our attempts to prepare compound
53
and related
systems using Suzuki
−
Miyaura cross-coupling reactions were
unsuccessful.
205. APPLICATION TO THE TOTAL SYNTHESIS OF
NATURAL PRODUCTS
As our understanding of the palladium-catalyzed Ullmann
cross-coupling reaction has developed, we have been exploiting
it on an increasingly frequent basis in developing syntheses of
various natural products. Such is our con
fi
dence in the
Figure 1.The four isomeric carbolines.
Scheme 8. Synthetic Route to the Carboline Natural
Product Harman (43)
conjunction with reductive cyclization reactions that enable the
conversion of the cross-coupling products into various
heterocyclic frameworks. Speci
fi
c examples are given in the
following sections.
5.1. Synthesis of Marinoquinoline A
As part of an ongoing interest in the cross-coupling chemistries
of pyrroles,
21we were attracted to the development of a
synthesis of marinoquinoline A, an alkaloid isolated from a
marine gliding bacterium that displays AChE inhibitory
22and
antimalarial activities. The route that we ultimately established
in obtaining this compound is shown in
Scheme 11
. It starts
with the palladium-catalyzed Ullmann cross-coupling of
o
-bromonitrobenzene (
1
) with iodinated pyrrole
54
to a
ff
ord the
target
55
in 74% yield. Signi
fi
cantly, all of our attempts to
e
ff
ect the Suzuki
−
Miyaura cross-coupling of compound
54
with
o
-nitrophenylboronic acid failed.
22In a related vein, when
o
-iodonitrobenzene (
5
) was used as a coupling partner in this
process, its homocoupling (to give 2,2
′
-dinitrobiphenyl)
became the dominant process. Such outcomes highlight the
capacity to facilitate cross-coupling processes by attenuating
the reactivity of one substrate through changing the associated
halogen.
The elaboration of coupling product
55
to the target alkaloid
was straightforward and involved the initial addition of
methyllithium to the associated aldehyde residue and oxidation
of the resulting alcohol,
56
, to the corresponding methyl
ketone
57
using the Dess
−
Martin periodinane (DMP).
Reductive cyclization of this last compound to the target
framework was e
ff
ected using magnesium in methanol, and this
was accompanied by cleavage of the tosyl group, thus a
ff
ording
marinoquinoline A (
58
) in 85% yield.
5.2. Total Syntheses of the Aspidosperma Alkaloids Aspidospermidine, Limaspermidine, and
1-Acetylaspidoalbidine and Approaches to Vindoline
In a more elaborate reaction sequence and as part of an
ongoing campaign to develop a synthesis of the binary indole
−
indoline alkaloid vincristine (see
Scheme 4
), we
fi
rst developed
a route to the alkaloid aspidospermidine.
23This entailed, as
one of two key steps, the cross-coupling of
α
-iodinated
cyclohexenone
59
with arene
5
to a
ff
ord product
60
in 75%
yield (
Scheme 12
). Compound
60
was readily elaborated to
azide
61
that upon heating engaged in an intramolecular [3 +
2] cycloaddition reaction followed by nitrogen extrusion to
a
ff
ord aziridine
62
, thereby establishing the piperidine ring
Scheme 9. Palladium-Catalyzed Ullmann Cross-Coupling/Reductive Cyclization Sequences Leading to Heterocycles 46 and 50
Scheme 10. Palladium-Catalyzed Ullmann Cross-Coupling of Halogenated Arenes 51 and 52 Leading to Biaryl 53, a Precursor
the Alkaloid Zephycandidine III
Scheme 11. Total Synthesis of Marinoquinoline A
then deployed in elaborating compound
62
to
aspidospermi-dine.
A related but more convergent protocol was employed in
obtaining the alkaloid limaspermidine.
24As shown in
Scheme
13
, compounds
5
and
63
were cross-coupled to give the
α
-arylated enone
64
(85%). When this was subjected to
reductive cyclization using dihydrogen in the presence of
Raney cobalt, the indole-annulated and cis ring-fused
octahydroquinoline
65
was obtained in 85% yield. This
conversion involves the selective reduction of the nitro and
cyano groups within substrate
64
while the enone moiety
remains intact. As a result, the associated ketone carbonyl
engages in an intramolecular Schi
ff
base condensation reaction
with the aniline or
N
-hydroxyaniline arising from reduction of
the nitro group while the 1
°
amine arising from the cyano
residue undergoes a hetero-Michael addition reaction, thus
forming both the indole and piperidine rings in a one-pot
operation. The use of properly prepared Raney cobalt
25is
critical to the success of this transformation because of the
chemoselectivities it allows for. If the more active Raney nickel
is used as the catalyst, then reduction of the carbon
−
carbon
double bond of the enone residue also occurs, with the result
that piperidine ring formation does not take place.
24Elaboration of compound
65
to (
±
)-limaspermidine was
achieved over four additional steps, including several closely
related to those employed in the conversion of compound
62
into (
±
)-apsidospermidine (
Scheme 12
). Two additional
steps, including an oxidative cyclization reaction employing
mercuric acetate, were required to convert (
±
)-limaspermidine
into (
±
)-1-acetylaspidoalbidine.
24The extension of the protocols de
fi
ned above in an
enantioselective approach to the alkaloid vindoline
(represent-ing a crucial substructure of vincristine) is shown in
Scheme
14
.
26Cross-coupling of iodinated nitroarene
66
with
homochiral
α
-iodinated cyclohexenone
67
(a compound
obtained from an enzymatically derived
cis
-1,2-dihydrocate-chol
27) gave the anticipated product
68
in 92% yield.
Reduction of this last compound using dihydrogen in the
presence of Raney cobalt resulted in the formation of the
tandem reductive cyclization product
69
(85%) embodying a
cis ring-fused octahydroquinoline. Over a further four steps
this could be elaborated to the hexacyclic compound
70
embodying many of the features of vindoline, which we are
seeking to convert into that alkaloid.
5.3. Formal Total Synthesis of the Cage-like Alkaloid Kopsihainanine A
The tandem reductive cyclizations of the palladium-catalyzed
Ullmann cross-coupling products
64
and
68
are presumed to
proceed under kinetic control, thus a
ff
ording cis ring-fused
products. Given that trans ring-fused perhydroquinolines are
encountered in a range of natural products, we sought methods
to access such systems. Despite extensive investigations of the
cyclization reactions, including examination of a range of
modi
fi
cations to the conditions employed, the cis ring-fused
products were invariably formed on an exclusive basis.
Scheme 12. Total Synthesis of (
±
)-Aspidospermidine
Scheme 13. Total Syntheses of (
±
)-Limaspermidine and (
±
)-1-Acetylaspidoalbidine
Therefore, we sought ways to e
ff
ect epimerization at the
ring-junction carbon center bearing the piperidine nitrogen. This
turned out to be a straightforward process, as illustrated in our
formal total synthesis of the cage-like alkaloid kopsihainanine A
(
Scheme 15
).
28The reductive cyclization product
65
could be
converted, over
fi
ve steps, into the angularly allylated congener
71
, which upon exposure to iodosobenzene in
dichloro-methane at ambient temperatures was oxidized to the
corresponding imine
72
. Upon reduction of compound
72
with sodium borohydride, the epimeric octahydroquinoline
73
was obtained. Since this last compound has previously been
converted into kopsihainanine A, the illustrated synthetic
sequence constitutes a formal total synthesis of the racemic
modi
fi
cation of this alkaloid.
5.4. Syntheses of the Uleine Alkaloids and Approaches to the Strychnos Alkaloids
members of the uleine family of alkaloids.
29Cross-coupling of
compounds
5
and
74
under the usual conditions a
ff
orded the
anticipated product
75
(88%), and the reductive cyclization of
this with dihydrogen in the presence of Raney cobalt a
ff
orded
the tetracyclic product
76
(60%) as a result of the same type of
tandem processes as shown in
Schemes 13
and
14
. Selective
Boc protection of the piperidine nitrogen within compound
76
a
ff
orded carbamate
77
, and this could be elaborated over two
steps, including a pyridinium chlorochromate-mediated
oxidation reaction to introduce a carbonyl moiety at the
methylene adjacent to the indole ring, to hydroxyketone
78
.
Reaction of this last compound with methyllithium proceeded
smoothly, and the resulting tertiary alcohol engaged in a
cycloetheri
fi
cation reaction upon treatment with protic acid.
Cleavage of the Boc group also occurred under these
conditions, and the resulting 2
°
amine was subjected to
reductive N-methylation to a
ff
ord (
±
)-gilbertine.
29aScheme 14. Synthesis of Compound 70, an Analogue of the Alkaloid Vindoline
Scheme 15. Conversion of cis Ring-Fused Octahydroquinoline 65 into Its trans-Con
fi
gured Congener 73, an Advanced
Precursor to the Alkaloid Kopsihainanine A
stereoselective manner,
29bwhile the ABCDE ring system of the
Strychnos
alkaloids proved accessible by similar means.
306. MECHANISTIC AND SYNTHETIC OVERVIEW
Our current thinking about the title process is dominated by
the original mechanistic proposals of Shimizu.
9Thus, as shown
in
Scheme 17
, aryl iodide
79
is presumed to react with the
added copper through an oxidative addition/reductive
deiodination process to give arylcopper(I)
80
, representing
the aryl anion synthon
81
. This reacts with the aryl cation
synthon
82
, which is produced through oxidative addition of
Pd[0] to the carbonyl-containing coupling partner
83
, thus
a
ff
ording intermediate
84
. The coupling event presumably
[image:40.625.143.486.454.765.2]Scheme 16. Total Synthesis of the Alkaloid (
±
)-Gilbertine
Figure 2.Structures of the simpler uleine alkaloids.