0022-538X/05/$08.00⫹0 doi:10.1128/JVI.79.11.7239–7247.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Loss of Interferon Regulatory Factor 3 in Cells Infected with Classical
Swine Fever Virus Involves the N-Terminal Protease, N
pro
S. Anna La Rocca,
1Rebecca J. Herbert,
1Helen Crooke,
2Trevor W. Drew,
2Thomas E. Wileman,
1and Penny P. Powell
1*
Department of Immunology, BBSRC Institute for Animal Health, Ash Road, Pirbright, Surrey GU24 0NF,1and Department of
Virology, Veterinary Laboratories Agency, Weybridge, Surrey KT15 3NB,2United Kingdom
Received 22 September 2004/Accepted 5 January 2005
We show that cells infected with the pestivirus classical swine fever virus (CSFV) fail to produce alpha/beta interferon not only following treatment with double-stranded RNA but also after superinfection with a heterologous virus, the alphavirus Sindbis virus, a virus shown to normally induce interferon. We investigated whether the inhibition of interferon synthesis by CSFV involved a block in interferon regulatory factor 3 (IRF3) activity. Cells infected with CSFV exhibited a lack of translocation of green fluorescent protein-IRF3 to the nucleus; however, constitutive shuttling of IRF3 was not blocked, since it could still accumulate in the nucleus in the presence of leptomycin B. Interestingly subcellular fractionation analysis showed that IRF3 was lost from the cytoplasm of infected cells from 18 h postinfection onwards. Using IRF3 promoter-luciferase reporter constructs, we demonstrate that loss of IRF3 was due to an inhibition of transcription of the IRF3 gene in CSFV-infected cells. Further, we investigated which viral protein may be responsible for the inhibition of interferon and loss of IRF3. We used cell lines expressing the CSFV N-terminal protease (Npro
) to show that this single viral protein, unique to pestiviruses, inhibited interferon production in response to Sindbis virus. In addition to being lost from CSFV-infected cells, IRF3 was lost from Npro
-expressing cells. The results demonstrate a novel viral evasion of innate host defenses, where interferon synthesis is prevented by inhibiting transcription of IRF3 in CSFV-infected cells.
Classical swine fever virus (CSFV) belongs to thePestivirus
genus in the Flaviviridae family, together with bovine viral diarrhea virus (BVDV) and border disease virus (26). The CSFV genome is a single positive-stranded RNA about 12.5 kb in length, with a single large open reading frame encoding a polyprotein that is processed into 12 known proteins (38). CSFV causes a severe disease of pigs characterized by fever, leukopenia, and hemorrhage (40), and there is widespread apoptosis of uninfected lymphocytes (35, 36). The disease causes significant economic loss worldwide and is an Office International des Epizooties list A pathogen (25). Pestiviruses can also cross the placenta, resulting in the birth of persistently infected animals due to failure of the dam to raise an innate immune response capable of preventing infection of the fetus (8). This interesting property of pestiviruses may partly be due to their ability to inhibit interferon production in cells they infect. CSFV causes no visible cytopathic effect in cells in culture, it induces the production of proinflammatory cyto-kines, yet does not stimulate interferon secretion (5). More-over, both CSFV and BVDV can inhibit the induction of in-terferon and apoptosis induced by double-stranded RNA (dsRNA) (5, 29). Production of interferon during a viral infec-tion is dependent on the activainfec-tion of several transcripinfec-tion factors, including interferon regulatory factor 3 (IRF3), NF-B, and ATF2 (reviewed in references 3 and 13). IRF3 is central for induction of antiviral genes, such as alpha
interferon, RANTES, ISG-15, ISG-54, ISG-56, and inducible nitric oxide (14). IRF3 is expressed constitutively as two forms, one of which is phosphorylated at its N terminus (27). In unstimulated cells, IRF3 shuttles between the nucleus and the cytoplasm, with cytoplasmic localization predominating (18). During viral infection, dsRNA produced during viral replica-tion activates the latent IRF3 via phosphorylareplica-tion on C-termi-nal serine residues (20, 30, 32). It is thought that there are multiple pathways leading to activation of IRF3 following virus infection (31), with a number of kinases involved, including among others, DNA-dependent protein kinase (16), IKK ep-silon, and TBK1 (10, 33). Recent work has described an im-portant pathway distinct from the dsRNA-dependent protein kinase R in the activation of IRF3 by viral RNA (34). The activated IRF3 then dimerizes and translocates to the nucleus, where it can bind one of the histone acetylases, CREB binding protein (CBP) or p300 (44). This causes IRF3 localization to become predominantly nuclear. IRF3 and CBP/p300 form a virally activated factor as part of the enhanceosome which binds to the beta interferon promoter and stimulates inter-feron production (41).
A number of small RNA viruses encode proteins that block interferon induction through inhibition of IRF3 activity; for the flaviviruses, several studies have looked for a mechanism of blocking IRF3. The serine protease complex NS3/4A of hep-atitis C virus expressed from subgenomic replicons inhibits IRF3 phosphorylation and translocation to the nucleus (11). Also, cells infected with both noncytopathic and cytopathic strains of the pestivirus BVDV prevent IRF3 binding to DNA, although the transcription factor does translocate to the nu-cleus in response to infection with a heterologous virus (1, 2).
* Corresponding author. Mailing address: Department of Immunol-ogy, Institute for Animal Health, Ash Road, Pirbright, Surrey GU24 0NF, United Kingdom. Phone: 44 1483 231090. Fax: 44 1483 232448. E-mail: penny.powell@bbsrc.ac.uk.
7239
on November 8, 2019 by guest
http://jvi.asm.org/
A variety of other virus families have the ability to block in-terferon through IRF3, including the paramyxoviruses (7), Ebola virus (4), and Bunyamwera virus (17).
In the present study, we show that the pestivirus CSFV inhibits interferon induced both by a heterologous virus, the alphavirus Sindbis virus, and by dsRNA. In contrast to findings for BVDV, CSFV infection did not induce IRF3 translocation to the nucleus. However, there was no inhibition of the con-stitutive shuttling of IRF3 between the cytoplasm and nucleus, shown by its accumulation in the nucleus following blockage of nuclear export by leptomycin B treatment. Interestingly, the cytoplasmic form of IRF3 was lost from cells during CSFV infection, becoming undetectable after 24 h postinfection (hpi). The loss of IRF3 was not affected by proteolysis inhib-itors and was concomitant with viral protein expression. Using constructs containing the IRF3 promoter upstream of a lucif-erase reporter gene, we present evidence that the loss of IRF3 is due to inhibition of transcription in CSFV-infected cells.
Recently, the importance of the N-terminal protease (Npro)
of CSFV in virulence and in the inhibition of interferon pro-duction has been demonstrated (23, 27). Npro is a cysteine
autoprotease which cleaves itself from the core protein (28); it is also nonessential, as it is not required for virus replication in culture (23). In animals, viruses with a deletion in Nprowere
attenuated and protected the animal from lethal doses of highly virulent CSFV (23). Importantly, mutant virus with a deletion in the N-terminal protease Nproabrogated the
inhi-bition of interferon production observed in wild-type virus (27). In our study, we have extended these findings by express-ing CSFV Nproin cells, and we have found that this single viral
protein not only inhibits interferon production induced by Sindbis virus but Nproexpression also results in a loss of IRF3
protein which is similar to that observed in cells infected with virus.
MATERIALS AND METHODS
Reagents. The rabbit anti-IRF3 antibody was a gift from Michael David (UCSD, California) and was raised against amino acids 107 to 208 of human IRF3 fused to glutathioneS-transferase. The antibody recognizing Npro
was raised in rabbits by immunizing them with peptides with the sequence KTNKQ KPMGVEEPVYDATGKPLFGDPS, which corresponds to amino acids 11 to 37 of the Brescia strain of CSFV. Alpha and gamma tubulin antibodies were from Sigma. CSFV was stained with WH303 anti-E2 monoclonal antibody (9). Sindbis virus was stained with a rabbit anti-E2 polyclonal antibody provided by Sondra Schessinger (Washington University Medical School, St. Louis, Missouri). The plasmid encoding green fluorescent protein (GFP)-IRF3 was a kind gift from John Hiscott (McGill University, Montreal, Canada). The IRF3 promoter plas-mids pIRF3-79Luc and pIRF3-80Luc, containing fragments of the 5⬘upstream region of the IRF3 gene (from KpnI to HindIII and BamHI to HindIII, respec-tively) cloned upstream of the luciferase cDNA in the pGL3 basic vector (21) were a gift from Paula Pitha-Rowe (Johns Hopkins School of Medicine, Balti-more, Maryland).
Cell culture and viruses.Cells were maintained at 37°C in 5% CO2. PK15
cells, derived from a pig kidney, were grown in Dulbecco’s modified Eagle’s medium, 10% fetal calf serum (demonstrated to be BVDV free), 100 units/ml glutamine, and penicillin-streptomycin. The Max cell line is from an inbred NIH minipig major histocompatibility complexd/dhaplotype, kindly provided by A. Saalmuller, and was grown in Iscove’s modified Dulbecco’s medium, 10% BVDV-free fetal calf serum, 100 units/ml glutamine, and penicillin-streptomycin. MDBK cells, stably transfected with a construct in which the human interferon-induced MxA promoter drives transcription of a chloramphenicol acetyltrans-ferase (CAT) cDNA (MDBK T2 cells) were provided by Bryan Charleston (12). The virulent isolate of CSFV Brescia, used for all infections, was kindly provided by the Institute for Animal Science and Health, Lelystad, The Netherlands (43),
and adapted for cell culture by serial passage on Max cells and PK15 cells as described by Van Gennip et al. (39). Virus was isolated by freeze-thaw lysis, titrated by immunostaining with anti-E2 antibody WH303 (5), and used in ex-periments at a multiplicity of infection (MOI) of 2 50% tissue culture infective doses (TCID50) per cell. Sindbis virus was generated from an infectious cDNA
clone provided by Sondra Schessinger (SinTOTO1101). Briefly, cDNA encoding the entire Sindbis virus genome was linearized with XbaI. RNA (⬎10 kb) was transcribed in vitro with SP6 RNA polymerase using the Message Machine kit (Ambion). RNA was electroporated into BHK cells, and cells were grown for 24 h. Virions were harvested from the supernatant and titrated by enzyme-linked immunosorbent assay using an anti-Sindbis virus antibody (a gift from S. Schess-inger). Sindbis virus was adapted to PK15 cells and Max cells by serial passages and used to infect cells at an MOI of 2 TCID50per cell. In some experiments,
Sindbis virus strain Edgar 339 obtained from the national collection of patho-genic viruses (CAMR) was used to stimulate interferon from PK15 cells.
IRF3 promoter transfections and luciferase assay.PK15 cells were infected with CSFV for 24 h and then transfected with IRF3 promoter plasmids, either pIRF3-79luc or pIRF3-80luc (21), using Fugene 6 (Roche). Cell extracts were collected 48 h after transfection, and the luciferase assay was performed by following the manufacturer’s instructions (Promega).
Interferon bioassay. Interferon bioactivity was measured using a sensitive reporter gene assay consisting of MDBK cells stably transfected with Mx-CAT (MDBK-t2 cells) (12). Interferon was induced in PK15 cells by infection with Sindbis virus or by transfection with pIpC (100g/ml; Sigma) using Fugene 6 (Roche). Cells were washed thoroughly 1 hour after treatment, and media were removed at the time point specific for each experiment. Supernatants were heated to 56°C for 1 hour to inactivate viruses or treated with RNase to remove residual pIpC. MDBK-t2 cells, maintained in 10g/ml blasticidin, were seeded into six-well plates. Cells were incubated with sample supernatants overnight. Cell extracts were prepared by repeated freezing-thawing, and the protein con-centration was measured by bicinchoninic acid (Pierce). Lysates (30g) were assayed for CAT using an enzyme-linked immunosorbent assay (Roche).
Western blots.Total cell extracts were prepared by lysis of cells in boiling sodium dodecyl sulfate (SDS) sample buffer. Protein extracts were quantified with the bicinchoninic acid kit (Pierce). Cell lysates, containing equal amounts of protein, were separated on SDS-polyacrylamide gel electrophoresis gels and transferred to a Hybond-C membrane (Amersham). Blocking and subsequent incubation with primary and secondary antibodies were performed in 5% dry skim milk dissolved in phosphate-buffered saline (PBS) and 0.02% Tween 20. Filters were probed overnight at 4°C with the primary antibodies. After extensive washing, the immune complexes were detected with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antiserum as appropriate (Bio-Rad Laboratories) followed by an enhanced chemiluminescence reaction (Pierce).
Subcellular fractionation. PK15 cells, either uninfected or infected with CSFV, were washed in PBS, scraped, and pelleted. Nuclear and cytoplasmic extracts were prepared using the ReadyPrep protein extraction kit according to the manufacturer’s instructions. Briefly, the cells were resuspended in CPEB buffer, incubated 30 min on ice, and then passed through a syringe needle. The lysates were centrifuged, and the supernatant was kept as a cytoplasmic fraction. The nuclear pellet was resuspended in PSB buffer and then centrifuged at maximum speed. This last step was repeated twice. The last supernatant is used as a nuclear fraction extract. Equivalent amounts of cytoplasmic and nuclear protein were separated by 10% SDS-polyacrylamide gel electrophoresis. Proteins were transferred to a Hybond-C membrane (Amersham) and probed with the relevant antibodies.
Cloning and expression of the N-terminal protease, Npro.PK15 cells were infected with CSFV at an MOI of 2.0 TCID50/cell for 0, 8, 18, and 24 hpi. The
cells were washed and lysed in Trizol (Invitrogen). Total RNA was extracted, resuspended in water, and reverse transcribed using avian myeloblastosis virus reverse transcriptase and random hexanucleotide primers at 42°C for 1 hour. Amplification of the Nproopen reading frame withTaqpolymerase (Promega)
was carried out by PCR with 15 cycles of 94°C for 1 min, 55°C for 1 min, and 72°C for 1 min using forward primer 5⬘-ATGGAGTTGAATCATTTTGAACTTTTA TAC-3⬘and reverse primer 5⬘-GCAACTGGTAACCCACAATGGACA-3⬘. The PCR product was cloned into pcDNA3.1 and transfected into PK15 cells using Fugene 6 (Roche). For stable expression in all cells, either cultures were grown in G418 (1 mg/ml; Invitrogen) for several weeks or, where indicated, cells were cotransfected with pBabe-puro (0.5g/ml; obtained from Silvia Soddu, Regina Elena Cancer Institute) and selected for 3 days in puromycin (1.5g/ml; Sigma). Nproexpression was enhanced by infecting cells with vaccinia virus modified
on November 8, 2019 by guest
http://jvi.asm.org/
vaccinia Ankara (MVA) T7 carrying the T7 RNA polymerase cDNA for 24 h before cell lysis.
Immunohistochemistry and fluorescence microscopy. Porcine kidney cells (Max) were grow on 13-mm glass coverslips to 50% confluence and infected with CSFV for 24 h. Cells were then transiently transfected using Lipofectamine (Invitrogen) with a plasmid containing IRF3-GFP and left overnight before fixing in 100% methanol, permeabilizing in 0.1% Triton X-100, and blocking in PBS containing 30% normal goat serum and 0.2% gelatin. CSFV-infected cells were stained with anti-CSFV E2 antibody (WH303). Sindbis virus was stained using an anti-Sindbis virus E2 antibody. In some experiments, leptomycin B was added at 10 nM for 2 hours before fixing. Cells were incubated with Alexa Fluor 488- or Alexa 594-conjugated secondary antibody for 60 min before being stained for 5 min with 4⬘, 6⬘-diamidino-2-phenylindole (DAPI) (Sigma). Cells were examined using a Nikon E800 microscope or with a Leica TCS NT confocal microscope.
RESULTS
CSFV inhibits interferon production by a heterologous virus and by dsRNA.Interferon secreted from PK15 cells was mea-sured using a sensitive bioassay in which supernatants were assayed on cells expressing the Mx-CAT reporter gene, and CAT expression was measured by enzyme-linked immunosor-bent assay. We induced interferon from PK15 cells using a heterologous virus, the alphavirus Sindbis virus. The charac-teristics of Sindbis virus infection of PK15 cells are shown in Fig. 1a. Although there were lower viral titers and longer cell survival seen in these cells than in BHK cells, there was a substantial amount of interferon produced even by 2 hpi (Fig.
FIG. 1. (a) Sindbis virus replication in PK15 cells and kinetics of interferon production. SV induced similar levels of interferon secre-tion from PK15 cells after 2, 4, 6, and 48 hpi (left graph). The optimum virus titer was determined by decreasing dilutions from 16-fold to 1-fold (MOI of 2 TCID50/cell). PK15 cells treated with
heat-inacti-vated (HI) Sindbis virus did not induce interferon (right graph). (A to C) Sindbis virus replicates in CSFV-infected cells. Cells were fixed in methanol and stained for viral proteins. (A) Cells stained with rabbit anti-E2 glycoprotein from Sindbis virus and counterstained with goat anti-rabbit conjugated to Alexa 594 (red); (B) cell stained for E2 glycoprotein of CSFV and counterstained with goat anti-mouse Alexa 488 (green); (C) merge to show both viruses replicating in same cell. (b) CSFV inhibits interferon induction by the heterologous virus, Sind-bis virus and by dsRNA. Supernatants from noninfected PK15 cells (control) or PK15 cells induced to produce interferon by SV or syn-thetic dsRNA (pIpC) were tested on Mx-CAT reporter cells. PK15 cells were infected with CSFV for 48 h, and interferon was measured in supernatants from unstimulated cells (CSFV) or after induction with the heterologous virus SV (CSFV⫹SV) or with synthetic dsRNA (CSFV⫹pIC).
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.585.56.517.63.521.2]1a, left graph). Increasing incubation times or titer of Sindbis virus did not increase interferon production. For the interferon assay, supernatants from Sindbis virus-infected PK15 cells were heat treated before addition to MDBK cells to prevent the virus from inducing interferon from the reporter cell line. Heat treatment of Sindbis virus completely prevented its ability to induce interferon (Fig. 1a, right graph). To determine whether Sindbis virus could superinfect and replicate in PK15 cells infected with CSFV, cells were infected with CSFV for
24 h and then infected with Sindbis virus overnight. Infection of both PK15 cells and Max cells with CSFV at an MOI of 2 TCID50/cell resulted in 100% of cells infected with CSFV
[image:4.585.64.523.66.523.2](staining for CSFV E2 is shown in Fig. 2D and G). For double labeling cells infected with the two viruses, cells were stained for both the E2 antigen of Sindbis virus and the E2 glycopro-tein of CSFV (Fig. 1a, panels A to C). Both viral antigens were detected in the same cell, indicating that Sindbis virus could infect and replicate in the presence of CSFV. Sindbis virus E2
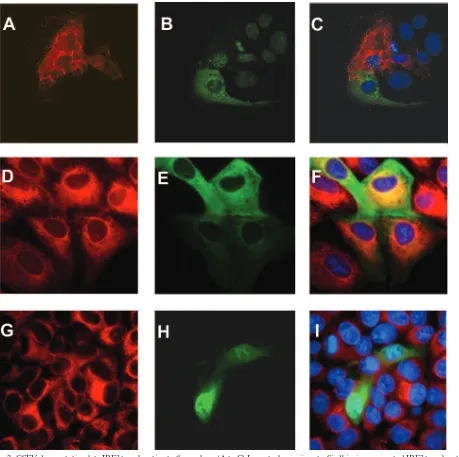
FIG. 2. CSFV does not stimulate IRF3 translocation to the nucleus. (A to C) In control experiments, Sindbis virus promoted IRF3 translocation to the nucleus. Max cells were transfected with GFP-IRF3 (B, green) and infected with Sindbis virus for 24 h (A, stained for Sindbis virus E2 in red). (C) IRF3 is located in the cytoplasm of uninfected cells but is seen as nuclear speckles in Sindbis virus-infected cells. (D to F) In CSFV-infected cells, IRF3 is located in the cytoplasm. CSFV-infected cells were detected with an anti-CSFV E2 antibody (D). Cells were transfected with a GFP-IRF3 plasmid (E), and IRF3 was located to the cytoplasm of CSFV-infected cells (F). (G to I) Constitutive shuttling of IRF3 to the nucleus is maintained in CSFV-infected cells. Max cells infected with CSFV for 48 h (G) and transfected with GFP-IRF3 (H) were treated with leptomycin B to block the nuclear export pathway. IRF3 accumulated in the nucleus, demonstrating no effect of CSFV on nuclear trafficking of IRF3 (I).
on November 8, 2019 by guest
http://jvi.asm.org/
levels and the titer of secreted virus were similar to those of cells infected with Sindbis virus alone. Interestingly, there was no change in the rate of cell death in cells infected with both viruses, with both CSFV-infected and uninfected PK15 cells dying between 24 and 48 hpi with Sindbis virus, showing that the inhibition of interferon by CSFV had no effect on Sindbis virus-induced cell death. These results are in agreement with work on BVDV and Semliki Forest virus, an alphavirus similar to Sindbis virus, where coinfection did not block Semliki Forest virus-induced apoptosis and also enhanced plaque size (1).
Sindbis virus or double-stranded RNA was then used to induce interferon from CSFV-infected cells. Both control PK15 cells and those infected with CSFV did not secrete in-terferon (Fig. 1b; 5). Infection with Sindbis virus (SV) for 2 h or transfection with pIpC for 2 h induced interferon, measured as pg/ml of CAT from the Mx-CAT reporter cells (Fig. 1b). Preinfection with CSFV inhibited interferon production in-duced by both the heterologous virus (SV) and dsRNA treat-ment (pIpC). We went on to characterize the mechanism of inhibition of interferon synthesis.
IRF3 does not translocate to the nucleus in CSFV-infected cells.It has been shown that in cells infected with the related pestivirus BVDV, IRF3 translocates to the nucleus but does not bind to DNA and induce interferon (1). We investigated the subcellular distribution of IRF3 in cells infected with CSFV. The localization of IRF3 was monitored using a plas-mid encoding an IRF3-GFP fusion protein (20). In control experiments, the IRF3 plasmid was transfected into cells over-night, and these cells were then infected for 24 h with Sindbis virus (Fig. 2A to C). In uninfected cells, IRF3 localization was predominantly in the cytoplasm. In each cell infected with Sindbis virus, IRF3 was translocated from the cytoplasm to the nucleus. In contrast, in CSFV-infected cells (Fig. 2D to F), IRF3 remained predominantly in the cytoplasm, although oc-casional cells show some nuclear localization. This suggests that there is no change in the localization of IRF3 in CSFV-infected cells compared to unstimulated cells, where IRF3 constitutively cycles between the cytoplasm and the nucleus, with cytoplasmic localization predominating. In the next ex-periment, we treated CSFV-infected cells with leptomycin B to prevent the nuclear export of IRF3. IRF3 accumulated in the nucleus of CSFV-infected cells (Fig. 2G to I). Uninfected PK15 cells treated with leptomycin B showed an accumulation of GFP-IRF3 in the nucleus identical to that of CSFV-infected cells (data not shown). These results show that IRF3 was able to shuttle into the nucleus in CSFV-infected cells, demonstrat-ing that traffickdemonstrat-ing of this factor is not blocked durdemonstrat-ing infection. Taken together, the results indicate that infection with CSFV does not provide an effective signal for IRF3 nuclear translo-cation and retention and that IRF3 distribution in CSFV-infected cells is similar to that seen in unCSFV-infected cells.
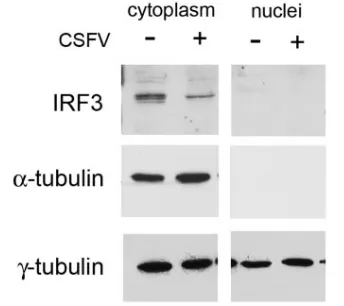
IRF3 is lost from PK15 cells infected with CSFV. We showed above that IRF3 is not translocated to the nucleus following CSFV infection. In the next experiments, IRF3 pro-tein was analyzed biochemically by Western blot analysis in the nuclear and cytoplasmic fractions. As with the human protein, porcine IRF3 was detected by Western analysis of PK15 cell lysates as a doublet of 53 to 55 kDa (Fig. 3). In uninfected cells, IRF3 was seen in the cytoplasm and not in the nuclear fraction (Fig. 3, top panel). The same samples were probed with an
anti-alpha tubulin antibody to confirm the purity of the cyto-plasmic fractions. In CSFV-infected cells, IRF3 was also seen in the cytoplasm and not the nucleus, and moreover, there was a decrease in IRF3 protein seen in the cytoplasm compared to uninfected cells. We did not detect multiple forms of IRF3 migrating faster than the two basal forms on denaturing or native gels in either the nuclear or cytoplasmic fraction follow-ing CSFV infection. The biochemical fractionation data con-firm the results with GFP-IRF3 (Fig. 2), showing that infection with CSFV does not stimulate translocation of IRF3 to the nucleus. In addition, it showed that IRF3 is lost directly from the cytoplasm of CSFV-infected cells without prior activation through phosphorylation and translocation to the nucleus
The expression of the two forms of IRF3 at 53 to 55 kDa was monitored over a time course of infection with CSFV (Fig. 4). The higher-molecular-weight protein was always more abun-dant than the smaller protein in these cells. In HeLa cells, two protein forms have been identified, the lower-molecular-weight form (form I) represented nonphosphorylated IRF3, while the higher-molecular-weight form (form II) may repre-sent a form phosphorylated on the N terminus, which does not appear to activate the factor (30). Interestingly, by 20 hpi, there was significantly less of the IRF3 protein doublet. By 22 hpi, the upper form had completely disappeared, with the lower form remaining but at a reduced level (Fig. 4, top). Expression of viral protein in these same samples was then checked using an antipeptide antibody to Npro. Npro is cleaved from the
CSFV polyprotein to give a final protein product of 19 kDa. A 19-kDa protein appeared at 8 hpi and increased up to 24 hpi. Hence, IRF3 protein levels decreased at a time when viral protein expression was increasing.
After activation by Sendai virus and double-stranded RNA, IRF3 has been shown to be degraded by the proteasome (20, 32). Therefore, we added the proteasome inhibitor MG132 and inhibitors of cellular proteases, including the cysteine
pro-FIG. 3. Subcellular fractionation of CSFV-infected cell lysates shows a loss of IRF3 from cytoplasm. Lysates from PK15 cells either uninfected (⫺) or 24 hpi with CSFV (⫹) were separated into nuclear and cytoplasmic fractions and analyzed by immunoblotting. (Upper panels) IRF3 was detected as a doublet at 53 to 55 kDa. There was a decrease in the cytoplasmic form of IRF3 following CSFV infection. (Middle panels) Subcellular fractionation was assessed using an anti-␣-tubulin antibody to show separation of the nuclear and cytoplasmic proteins. (Lower panels)␥-Tubulin shows equal lane loading of cyto-plasmic and nuclear extracts.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.330.502.70.223.2]tease inhibitor E64D and serine protease inhibitors, at 18 hpi with CSFV. None of these protease inhibitors was able to block loss of IRF3 (data not shown). We investigated whether the loss of IRF3 was due to transcriptional down-regulation of the
IRF3 gene. The promoter region of the IRF3 gene, either pIRF3-79 from KpnI-HindIII or pIRF3-80 from BamHI-Hin-dIII cloned upstream of luciferase, was transfected into PK15 cells (Fig. 4, bottom). The promoter was found to have basal expression in these cells similar to that seen in human cells (21). When transfected into cells infected with CSFV for 48 hpi, basal activity was inhibited to 20% of control values in infected cells. The loss of IRF3 protein from CSFV-infected cells was therefore due to down-regulation of tran-scription from theIRF3gene.
Expression of the N-terminal protease Npro
inhibits inter-feron production and promotes loss of IRF3.Previous work (27) using a recombinant virus with a deletion of the open reading frame of the N-terminal protein Nproshowed that this
virus no longer inhibited interferon production and dsRNA-induced apoptosis. These workers suggested that Nproplays an
important part in innate immune evasion. We investigated whether expression of Npro alone could inhibit interferon
in-duction by a heterologous virus (Fig. 5). Two types of cell lines were developed. In the first experiment, PK15 cells were co-transfected with a plasmid encoding Nproand with pBabe-puro
(experiment 1). In this experiment, PK15 cells expressing Npro
were selected in puromycin for 3 days [PK15(Npro)]. In
exper-iments 2 to 4, cells were transfected with Nproin pcDNA3 and
stable transfectants were selected for by treatment with G418 for several weeks. The experiments differed in the length of Sindbis virus infection (see the legend to Fig. 5). The cloned cell lines expressing Nprowere analyzed for constitutive
secre-tion of interferon by testing supernatants on the MDBKt2 Mx-CAT reporter cell line. Expression of Nprowithout
stimu-lation of PK15(Npro) cells did not induce interferon (Fig. 5).
Infection of PK15 cells with Sindbis virus stimulated interferon production, as shown in Fig. 1. Significantly, however, PK15(Npro) cells infected with Sindbis virus for 3.5 h
(experi-ments 1, 2, and 4) or 48 h (experiment 3) showed very little induction of interferon. Individual experiments are shown be-cause of the variation on Sindbis virus-induced interferon from control PK15 cells.
We checked that Nprowas being expressed in the cloned cell
lines by analyzing RNA and protein production (Fig. 6). RNA levels of Nprodetermined by reverse transcription-PCR were
[image:6.585.42.410.68.319.2]high in PK15(Npro) cells, expressing more NproRNA than an
FIG. 4. IRF3 loss from CSFV-infected cells is through inhibition of gene transcription. (Top) Time course of infection of PK15 cells with CSFV. (Upper panels) IRF3 protein, seen as a doublet at 53 to 55kDa, is lost from cells from 18 hpi with CSFV; (Middle panels) CSFV infection of cells was monitored using an antipeptide antibody to Npro
[image:6.585.306.533.485.621.2]and shows an increase in viral gene expression from 8 hpi; (Lower panels) equal lane loading is shown by probing the same samples for ␥-tubulin using mouse anti-␥-tubulin antibody. (Bottom) Transcrip-tional inhibition of the IRF3 gene. IRF3 promoter plasmids (pIRF3-79Luc and pIRF3-80Luc) were transfected into uninfected PK15 cells or PK15 cells 24 hpi with CSFV. CSFV infection inhibited IRF3 gene transcription to 20% of the basal level.
FIG. 5. Npro-expressing cells [PK(Npro)] show decrease in Sindbis
virus-induced interferon production. The graph shows secretion of interferon from control cells (PK15) or after induction with Sindbis virus (PK15⫹SV) and from cell stably expressing Nprowithout inducer
[PK15(Npro)] and following induction with Sindbis virus [PK(Npro)⫹SV]. In experiment 1, cells were selected for 3 days in 0.5 g/ml puromycin and challenged with Sindbis virus for 3.5 h. In ex-periments 2 to 4, cells were selected in G418 and challenged with Sindbis virus for 3.5 h (experiments 2 and 4) or 48 h (experiment 3). Interferon produced from supernatants of PK15 cells expressing Npro
were measured on Mx A-CAT reporter cells measured as pg/ml.
on November 8, 2019 by guest
http://jvi.asm.org/
equivalent amount of RNA from CSFV-infected cells (Fig. 6A). However Nproprotein levels were low in PK15(Npro) cells.
Protein levels were only detectable when cells were infected with vaccinia virus MVA T7, enabling the expression of T7 RNA polymerase to enhance the level of Nprobeing produced
by the Npro pcDNA3 plasmid (Fig. 6B). Lysates from
PK15(Npro) cells were probed for IRF3. Interestingly, very
little IRF3 was detected compared to control cells transfected with vector alone (Fig. 6C). In PK15(Npro) cells, the
lower-molecular-weight form has completely disappeared, leaving reduced amounts of the higher-molecular-weight form of IRF3. These results correspond to the effect seen in CSFV-infected cells. However, at 24 hpi with CSFV, the higher-molecular-weight form of IRF3 disappears, leaving reduced amounts of the lower-molecular-weight form of IRF3.
DISCUSSION
In this study, we have identified a novel mechanism for CSFV evasion of the host innate immune response to infec-tion. CSFV infection can potently block the synthesis of inter-feron induced by two pathways, a heterologous virus, Sindbis virus, and by double-stranded RNA. Sindbis virus stimulates
the translocation of IRF3 into the nucleus, where it plays a crucial role in the production of interferon by forming part of the enhanceosome complex that binds upstream of the beta interferon promoter. IRF3 is also a crucial regulator of other proinflammatory genes with ISRE sites, including RANTES, ISG15, ISG56, and interleukin-15. An IRF3-GFP fusion pro-tein was translocated into the nucleus following Sindbis virus infection and was found as discrete dots which may represent a distinct nuclear subcompartment. Although large amounts of interferon were induced by Sindbis virus, higher-molecular-weight phosphorylated forms of IRF3 were not detected even on native gels. Phosphorylation on C-terminal serine residues of IRF3 is a characteristic of activation by negative-stranded RNA viruses such as measles virus and Sendai virus (3). Sind-bis virus, a positive-stranded RNA virus, may activate different signaling pathways to IRF3 than paramyxoviruses. In contrast to Sindbis virus, CSFV did not change the cellular distribution of IRF3, which was seen predominantly in the cytoplasm of infected cells. However, IRF3 was able to translocate to the nucleus, since leptomycin B treatment led to nuclear accumu-lation of IRF3, thus indicating that CSFV infection did not impede nuclear trafficking. Significantly, not only did infection with CSFV fail to activate IRF3 but we also showed that this factor, essential for the transcription of interferon, was lost from the cytoplasm of infected cells by 24 hpi. Proteolytic inhibitors, including proteasome inhibitors, did not affect the rate of loss of IRF3, suggesting that IRF3 was not being de-graded. We show a novel transcriptional down-regulation of theIRF3gene following CSFV infection. The transcriptional down-regulation of IRF3 is a late event in infection, occurring after 18 hpi, at a time when viral proteins are synthesized. Importantly, this mechanism does not explain how the imme-diate induction of interferon on CSFV entry into cells is in-hibited. We have shown for Sindbis virus that interferon can be detected in supernatants as rapidly as 2 hpi. One would expect CSFV infection to have other mechanisms to inhibit this early increase in interferon secretion. Several viruses have been re-ported to have more than one mechanism to block host inter-ferons (22, 24). Since IRF3 is also important for mediating virus-induced apoptosis (15), loss of IRF3 may play an impor-tant part in the establishment of the persistence of CSFV in vitro. Persistently infected cells are resistant to dsRNA-in-duced apoptosis (5), and whether this is due to lack of IRF3 remains to be determined. Cells chronically infected with CSFV survive and divide normally for many weeks and have the same growth properties as uninfected cells (5). This is also the case for dominant-negative IRF3 cell lines, which survive as normal, except that they do not produce interferon in re-sponse to virus (17, 44). The mechanism of loss of transcription during CSFV infection is unknown. As well as effects on the IRF3 gene, there may be general effect on host transcription, as yet undefined. In the case of Rift Valley fever virus, the transcription factor TFIIH is targeted, leading to suppression of host cellular RNA synthesis (6, 19).
In contrast to other flaviviruses, members of thePestivirus
genus encode an extra protein at the N terminus, Npro, and we
show here for CSFV that this protein is crucial to suppressing interferon production. Npro is an important virulence factor,
since deletion leads to attenuation of the virus (23). Deletion of the Nprogene from the virus-induced interferon in
macro-FIG. 6. IRF3 is lost from cells stably expressing the CSFV Npro
gene. (A) Reverse transcription-PCR showing RNA levels of Nproin
PK15 cells (⫺), PK15 cells infected with CSFV (csfv), and PK15 stably transfected with Npro (Npro). (Upper panels) Npro-transfected cells
express high levels of NproRNA; (Lower panels) actin RNA shows
equal lane loading. (B) Western blot showing Nproprotein expression
in CSFV-infected cells (csfv), control cells transfected with vector alone (vector), and PK15(Npro) cells. Nproprotein is highly expressed
as a 19-kDa protein in CSFV-infected cells (csfv), not detectable in control cells transfected with vector alone (lower vector band is a nonspecific band seen with this antibody). Expression of Npro was
enhanced in PK15(Npro) cells from the T7 promoter using vaccinia
virus MVA T7 (MVA). (C) Western blot showing disappearance of IRF3 from PK15(Npro) cells. Control PK15 cells (⫺) or cells infected
with CSFV 24 hpi (csfv) or transfected with empty vector (vector) or Npro(Npro) were lysed, and equal amounts of protein were blotted
with anti-IRF3 antibody. IRF3 protein is detected as a doublet at 53 to 55 kDa. The higher-molecular-mass form of IRF3 disappears from CSFV-infected cells, while most of the higher form and all of the lower form disappear from PK15 cells expressing Npro but not from cells
expressing vector alone.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:7.585.44.284.70.254.2]phages and PK15 cells in the absence of dsRNA (27). Here we demonstrate that expression of Npro alone was sufficient to
cause inhibition of Sindbis virus and pIpC-induced interferon synthesis and loss of IRF3 protein. Nprois a cysteine-like
au-toprotease with subtilisin-like activity (28). There is no known inhibitor, and the only known site of cleavage is between itself and the core protein. Although it is possible that Npro
proteo-lytic activity degrades IRF3 directly in cells, the time course of expression suggests that Nprohas an indirect effect on the loss
of IRF3, possibly targeting a factor involved in its transcrip-tion.
The list of viral proteins which inhibit IRF3 is growing and includes the nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus, which block the activity of an IRF3-driven plasmid and phosphorylation of IRF3 (7) and the NS1 protein of influenza virus (37). The Ebola virus VP35 protein also inhibits activation of IRF3 through inhibition of IRF-3 phosphorylation and subsequent dimerization (4). In the
Bu-nyaviridaefamily, the NSs proteins of Bunyamwera virus and
Rift Valley hemorrhagic fever virus were shown to block IRF-3-dependent promoter activity (6, 17). This is the first demon-stration of transcriptional inhibition of IRF3 by a virus involv-ing a specific viral protein, Npro. It remains to be determined
which cellular factors are inactivated by this interesting pesti-virus protein.
ACKNOWLEDGMENTS
We thank the following people for reagents: Sondra Schlesinger (Washington University Medical School, St. Louis, Missouri) for Sind-bis virus reagents, John Hiscott (McGill University, Montreal, Canada) for GFP-IRF3, Paula Pitha Rowe (Johns Hopkins School of Medicine) for IRF3 reporter constructs, Bryan Charleston (IAH) for Mx-CAT T2 cells, Michael David (UCD, California) for the anti-IRF3 antibody, Silvia Soddu (Regina Elena Institute) for pBabe puro, and Armin Saalmuller (Tubingen, Germany) for the Max cell line.
This work was partly funded by the BBSRC and by DEFRA grant SE0773.
REFERENCES
1.Baigent, S. J., G. Zhang, M. D. Fray, H. Flick-Smith, S. Goodbourn, and J. W. McCauley.2002. Inhibition of beta interferon transcription by noncy-topathogenic bovine viral diarrhea virus is through an interferon regulatory factor 3-dependent mechanism. J. Virol.76:8979–8988.
2.Baigent, S. J., S. Goodbourn, and J. W. McCauley.2004. Differential acti-vation of interferon regulatory factors-3 and -7 by non-cytopathogenic and cytopathogenic bovine viral diarrhoea virus. Vet. Immunol. Immunopathol. 100:135–144.
3.Basler, C. F., and A. Garcia-Sastre.2002. Viruses and the type I interferon antiviral system: induction and evasion. Int. Rev. Immunol.21:305–337. 4.Basler, C. F., A. Mikulasova, L. Martinez-Sobrido, J. Paragas, E.
Muhl-berger, M. Bray, H.-D. Klenk, P. Palsae, and A. Garcia-Sastre.2003. The Ebola virus vp35 protein inhibits activation of interferon regulatory factor 3. J. Virol.77:7945–7956.
5.Bensaude, E., J. L. Turner, P. R. Wakeley, D. A. Sweetman, C. Pardieu, T. W. Drew, T. Wileman, and P. P. Powell.2004. Classical swine fever virus induces proinflammatory cytokines and tissue factor expression and inhibits apopto-sis and interferon syntheapopto-sis during the establishment of long-term infection of porcine vascular endothelial cells. J. Gen. Virol.85:1029–1037. 6.Billecocq, A., M. Spiegel, P. Vialat, A. Kohl, F. Weber, M. Bouloy, and O.
Haller.2004. NSs protein of Rift Valley fever virus blocks interferon pro-duction by inhibiting host gene transcription. J. Virol.78:9798–9806. 7.Bossert, B., S. Marozin, and K.-K. Conzelmann.2003. Nonstructural protein
NS1 and NS2 of bovine respiratory syncytial virus blocks activation of inter-feron regulatory factor 3. J. Virol.77:8661–8668.
8.Charleston, B., M. D. Fray, S. Baigent, B. V. Carr, and W. I. Morrison.2001. Establishment of persistent infection with non-cytopathic bovine viral diar-rhoea virus in cattle is associated with a failure to induce type I interferon. J. Gen. Virol.82:1893–1897.
9.Edwards, S., J. J. Sands, and J. W. Harkness.1988. The application of monoclonal antibody panels to characterize pestivirus isolates from rumi-nants in Great Britain. Arch. Virol.102:197–206.
10.Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz, D. T. Golenbock, A. J. Coyle, S.-M. Liao, and T. Maniatis.2003. IKKεand TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491–496.
11.Foy, E., K. Li, C. Wang, R. Sumpter, Jr., M. Ikeda, S. M. Lemon, and M. Gale, Jr.2003. Regulation of interferon regulatory factor 3 by the hepatitis C virus serine protease. Science300:1145–1148.
12.Fray, M. D., G. E. Mann, and B. Charleston.2001. Validation of an Mx/CAT reporter gene assay for the quantification of bovine type-I interferon. J. Im-munol. Methods249:235–244.
13.Goodbourn, S., L. Didcock, and R. E. Randall.2000. Interferons: cell sig-nalling, immune modulation, antiviral response and virus countermeasures. J. Gen. Virol.81:2341–2364.
14.Grandvaux, N., B. R. ten Oever, M. J. Servant, and J. Hiscott.2002. The interferon antiviral response: from viral invasion to evasion. Curr. Opin. Infect. Dis.15:259–267.
15.Heylbroeck, C., S. Balachandran, M. J. Servant, C. DeLuca, G. N. Barber, R. Lin, and J. Hiscott.2000. The IRF-3 transcription factor mediates Sendai Virus-induced apoptosis. J. Virol.74:3781–3792.
16.Karpova, A. Y., M. Trost, J. M. Murray, L. C. Cantley, and P. M. Howley. 2002. Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. USA99:2818–2823.
17.Kohl, A., R. F. Clayton, F. Weber, A. Bridgen, R. Randall, and R. Elliott. 2003. Bunymwera virus nonstructural protein NSs counteracts interferon regulatory factor 3-mediated induction of early cell death. J. Virol.77:7999– 8008.
18.Kumar, K. P., K. M. McBride, B. K. Weaver, C. Dingwall, and N. C. Reich. 2000. Regulated nuclear-cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol. Cell. Biol.20:4159–4168.
19.Le May, N., S. Dubaele, L. Proietti De Santis, A. Billecocq, M. Bouloy, and J.-M Egly.2004. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell116:541–550.
20.Lin, R., C. Heylbroeck, P. Pitha, and J. Hiscott.1998. Virus-dependent phosphorylation of the IRF3 transcription factor regulated nuclear translo-cation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol.18:2986–2996.
21.Lowther, W. J., P. A. Moore, K. C. Carter, and P. M. Pitha.1999. Cloning and functional analysis of the human IRF-3promoter. DNA Cell Biol. 18:685–692.
22.Macdonald, A., and M. Harris.2004. Hepatitis C virus NS5A: tales of a promiscuous protein. J. Gen. Virol.85:2485–2502.
23.Mayer, D., M. A. Hofmann, and J.-D. Tratschin.2004. Attenuation of clas-sical swine fever virus by deletion of the viral Npro
gene. Vaccine22:317–328. 24.Munoz-Jordan, J. L., G. G. Sanchez-Burgos, M. Laurent-Rolle, and A. Gar-cia-Sastre.2003. Inhibition of interferon signalling by Dengue virus. Proc. Natl. Acad. Sci. USA100:14333–14338.
25.Paton, D. J., and I. Greiser-Wilke.2003. Classical swine fever–an update. Res. Vet. Sci.75:169–178.
26.Paton, D. J., J. J. Sands, J. P. Lowings, J. E. Smith, G. Ibata, and S. Edwards.1995. A proposed division of the pestivirus genus using monoclo-nal antibodies, supported by cross-neutralisation assays and genetic sequenc-ing. Vet. Res.26:92–109.
27.Ruggli, N., J.-D. Tratschin, M. Schweizer, K. McCullough, M. A. Hofmann, and A. Summerfield.2003. Classical wine fever virus interferes with cellular antiviral defenses: evidence for a novel function of Npro. J. Virol.77:7645–
7654.
28.Rumenapf, T., R. Stark, M. Heiman, and H.-J. Theil.1998. N-terminal protease of pestiviruses: identification of putative catalytic residues by site-directed mutagenesis. J. Virol.72:2544–2547.
29.Schweizer, M., and E. Peterhans.2001. Noncytopathic bovine viral diarrhea virus inhibits double-stranded RNA-induced apoptosis and interferon syn-thesis. J. Virol.75:4692–4698.
30.Servant, M. J., B. ten Oever, C. LePage, L. Conti, S. Gessani, I. Julunen, R. Lin, and J. Hiscott.2001. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J. Biol. Chem. 276:355–363.
31.Servant, M. J., N. Grandvaux, and J. Hiscott. 2002. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem. Pharmacol.64:985–992.
32.Servant, M. J., N. Grandvaux, B. R. ten Oever, D. Duguay, R. Lin, and J. Hiscott.2003. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. J. Biol. Chem.278:9441–9447.
33.Sharma, S., B. R. ten Over, N. Grandvaux, G.-P. Zhou, R. Lin, and J. Hiscott.2003. Triggering the interferon antiviral response through an IKK-related pathway. Science300:1148–1151.
34.Smith, E. J., I. Marie, A. Prakash, A. Garcia-Sastre, and D. E. Levy.2001. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J. Biol. Chem.276:8951–8957. 35.Summerfield, A., S. M. Knoetig, and K. C. McCullough.1998. Lymphocyte
on November 8, 2019 by guest
http://jvi.asm.org/
apoptosis during classical swine fever: implication of activation-induced cell death. J. Virol.72:1853–1861.
36.Summerfield, A., S. M. Knoetig, R. Tschudin, and K. C. McCullough.2000. Pathogenesis of granulocytopenia and bone marrow atrophy during classical swine fever involves apoptosis and necrosis of uninfected cells. Virology 272:50–56.
37.Talon, J., C. M. Horvath, R. Polley, C. F. Basler, T. Muster, P. Palese, and A. Garcia-Sastre.2000. Activation of interferon regulatory factor 3 is inhib-ited by the influenza A virus NS1 protein. J. Virol.74:7989–7996. 38.Thiel, H. J., P. G. W. Plagemann, and V. Moennig.1996. Pestiviruses, p.
1059–1073.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
39.Van Gennip, H. G., A. C. Vlot, M. M. Hulst, A. J. De Smit, and R. J. Moormann.2004. Determinants of virulence of classical swine fever virus strain Brescia. J. Virol.78:8812–8823.
40.Van Oirschot, J. T.1988. Description of the virus infection, p. 1–25.InB.
Liess (ed.), Classical swine fever and related viral infections. Martinus Nij-hoff, Boston, Mass.
41.Wathelet, M. G., C. H. Lin, L. Parekh, L. V. Ronco, P. M. Howley, and T. Maniatis.1998. Virus infection induces the assembly of coordinately acti-vated transcription factors on the IFN-beta enhancer in vivo. Mol. Cell 1:507–518.
42.Weidman, M. K., R. Sharma, S. Raychaudhuri, P. Kunda, W. Tsai, and A. Dasgupta.2003. The interaction of cytoplasmic viruses with the nucleus. Virus Res.95:75–85.
43.Wensvoort, G., C. Terpstra, E. P. de Kluijver, C. Kragten, and J. C. War-naar.1989. Antigenic differentiation of pestivirus strains with monoclonal antibodies against hog cholera virus. Vet. Microbiol.21:9–20.
44.Yoneyama, M., W. Suhara, Y. Fukuhara, M. Fukada, K. Nishida, and T. Fujita.1998. Direct triggering of the type I interferon system by virus infec-tion: activation of a transcription factor complex containing IRF3 and CBP/ p300. EMBO J.17:1087–1095.