active site of the HCV RdRp (S. Bressanelli, L. Tomei, F. A. Rey, and R. De Francesco, J. Virol 76:3482–3492, 2002). To determine its physiological importance, we performed a systematic mutagenesis analysis of the rGTP-specific binding pocket by amino acid substitutions. Effects of mutations of the rGTP-specific binding site on enzymatic activity were determined by an in vitro RdRp assay, while effects of mutations on HCV RNA replication were examined by cell colony formation, as well as by transient replication of subgenomic HCV RNAs. Results derived from these studies demonstrate that amino acid substitutions of the rGTP-specific binding pocket did not significantly affect the in vitro RdRp activity of purified recombinant NS5B proteins, as measured by their abilities to synthesize RNA on an RNA template containing the 3ⴕuntranslated region of HCV negative-strand RNA. However, most mutations of the rGTP-specific binding site either impaired or completely ablated the ability of subgenomic HCV RNAs to induce cell colony formation. Likewise, these mutations caused either reduction in or lethality to transient replication of the human immunodeficiency virus Tat-expressing HCV replicon RNAs in the cell. Collectively, these findings demonstrate that the rGTP-specific binding site of the HCV NS5B is not required for in vitro RdRp activity but is important for HCV RNA replication in vivo.
Hepatitis C virus (HCV) is a medically important pathogen infecting approximately 4 million people in the United States and 170 million people worldwide (7, 37). The majority of HCV-infected individuals develop chronic hepatitis that can progress to liver cirrhosis and hepatocellular carcinoma (7). HCV is a small enveloped RNA virus that belongs to the
Hepacivirusgenus of theFlaviviridaefamily (33). It contains a single-stranded and positive-sense RNA genome, approxi-mately 9.6 kb in length, which is composed of the 5⬘ untrans-lated region (5⬘UTR), a single open reading frame, and the 3⬘UTR (9, 30). The viral proteins are translated as a single large polyprotein precursor of 3,010 to 3,040 amino acids, which is co- or posttranslationally processed by cellular and viral proteases into individual mature structural (C, E1, E2, and possibly p7) and nonstructural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) viral proteins (30). Additional viral proteins are also produced by a ribosomal frameshift from the core (C) coding region, whereas their biological significance has not yet been determined (35, 38).
Replication of HCV RNA occurs in a membrane-bound replication complex that consists of viral RNA and the non-structural (NS) proteins NS3, NS4A, NS4B, NS5A, and NS5B, as well as cellular proteins (10, 34). The key component of the
HCV replication complex is the virus-encoded RNA-depen-dent RNA polymerase (RdRp) that catalyzes the polymeriza-tion of ribonucleoside triphosphates (rNTPs) during RNA rep-lication. The HCV RdRp (nonstructural protein 5B [NS5B]) contains functional motifs characteristic to all known RNA polymerases (14, 23, 27). Biochemical studies demonstrated that all the conserved functional motifs of NS5B are important for the RdRp activity in vitro (17, 19). The N-terminal portion of NS5B is very critical to its RdRp activity, whereas the C-terminal hydrophobic region of 21 amino acid residues is dis-pensable for in vitro RdRp activity (11, 21). Purified recombi-nant NS5B protein is able to catalyze in vitro RNA synthesis on both HCV-specific and nonviral RNA templates, implying that NS5B itself lacks template specificity (2, 11, 17, 21, 25). Both primer-dependent and primer-independent (de novo) RNA syntheses were observed for purified recombinant HCV NS5B in vitro. In the absence of a primer, NS5B either extends the 3⬘ end of the RNA template itself (self priming or copy back) or initiates RNA synthesis de novo (2, 21, 42). Although purified recombinant NS5B is capable of catalyzing primer-dependent RNA synthesis in vitro, RNA synthesis de novo is the most likely mechanism used for HCV RNA replication in vivo (20). The atomic structure of the HCV NS5B has been deter-mined (1, 5, 16). It resembles the canonical structure of other polymerase with the characteristic finger, palm, and thumb subdomains. The polymerase active site is encircled in a 15-Å-wide and 18-Å-deep cavity at the center of the molecule with the palms as the base. The palm domain contains the con-served DXXXXD and GDD signature residues responsible for
* Corresponding author. Mailing address: Department of Microbi-ology, ImmunMicrobi-ology, and Molecular Genetics, University of Kentucky College of Medicine, 800 Rose St., MN477, Lexington, KY 40536-0298. Phone: (859) 257-5577. Fax: (859) 257-8994. E-mail: gluo0@uky .edu.
11607
on November 8, 2019 by guest
the nucleotidyl transfer reaction (16). The thumb subdomain is predominantly␣helical, suggesting a role in interaction with other viral and/or cellular proteins (5). The structures of NS5B in complex with rNTP have also been determined (4, 24). Interestingly, a low-affinity GTP-specific binding site was iden-tified on the surface of the enzyme about 30 Å away from the active site. The amino acid residues defining the low-affinity GTP-specific binding pocket include amino acid residues S29 and R32 from the fingertip and P495, P496, V499, and R503 from the thumb domain, which make direct or water-mediated contacts to the nucleotide (4). However, the physiological im-portance of the GTP-specific binding site in HCV RNA rep-lication has not been determined.
In an effort to determine the role of the rGTP-specific bind-ing pocket of the HCV NS5B in RNA replication, we per-formed a systematic mutagenesis analysis of the rGTP-specific binding site by amino acid substitutions. The effect of muta-tions of the rGTP-specific binding site on RdRp activity was determined by an in vitro RdRp assay, while their effects on HCV RNA replication were examined by cell-based HCV rep-licon replication systems. Findings derived from our studies demonstrate that mutations of the rGTP-specific binding site did not significantly affect in vitro RdRp activity but impaired or ablated HCV RNA replication in the cell. These findings indicate that the residues defining the rGTP-specific binding site of the HCV RdRp are important for HCV RNA replica-tion in vivo. Given the fact that none of the mutareplica-tions of the rGTP-specific binding site affect the RdRp activity of the pu-rified HCV NS5B, the role of the rGTP-specific binding pocket in HCV replication is probably to modulate the structure of NS5B for efficient interaction with other viral and/or cellular proteins of the HCV replication complex to allow efficient HCV RNA replication.
MATERIALS AND METHODS
Cell culture.A highly permissive cell line for HCV RNA replication, Huh7.5 (3), was kindly provided by Charles M. Rice (Rockefeller University). Huh7.5 cells were grown in Dulbecco’s modified essential medium (DMEM)
(Invitro-gen) supplemented with 100 units per ml of penicillin, 100g/ml of streptomycin,
nonessential amino acids, and 10% fetal bovine serum (FBS) (Invitrogen). Cells harboring a subgenomic HCV replicon were selected and maintained in DMEM containing 10% FBS and 0.5 mg/ml of G418 sulfate (6, 22). En5-3 cells were derived from Huh7 cells and contain a secreted alkaline phosphatase (SEAP) gene placed downstream of the long terminal repeat promoter of human immu-nodeficiency virus (HIV). En5-3 cells were maintained in culture as previously described (37).
DNA construction and site-specific mutagenesis.To express NS5B protein in
Escherichia coli, the NS5B cDNA was cloned into a pET21d vector (Novagen).
The NS5B cDNA was amplified by PCR using the vector pBR322/I377-NS3-3⬘/
S1179I as a template and synthetic oligonucleotides 5B-BspHI and 5B/His⌬C21
as primers (Table 1) (22). The PCR DNA was digested with BspHI and XhoI and inserted into the pET21d vector that was similarly digested with both NcoI (comparable cohesive end with BspHI) and XhoI restriction enzymes, resulting
in a construct designated pET21d/NS5B⌬21. The C-terminal hydrophobic
do-main of 21 amino acids (⌬21) of NS5B was replaced with a six-histidine tag to
facilitate protein purification (21). Site-specific mutations of the rGTP-specific binding site (Fig. 1) of the NS5B were introduced by a two-step PCR method. Nucleotides encoding mutant amino acids were introduced into synthetic oligo-nucleotides, which were used as primers for PCR (Table 1). For instance, a serine (Ser)-to-alanine (Ala) mutation at amino acid residue 29 of NS5B was created by mutating the Ser codon into an Ala codon by using two pairs of PCR primers (Table 1). The first PCR DNA fragment was amplified by using primer pairs 5B-BspHI and S29A/3, and the second one was amplified by primer pairs
S29A/5 and 5B/His⌬C21. Two PCR DNA fragments were then combined as
templates for another PCR using 5B-BspHI and 5B/His⌬C21 as primers. The
DNA fragment was subsequently digested with restriction enzymes BspHI and XhoI and ligated into the pET21d vector between NcoI and XhoI sites. The mutant plasmid was designated pET21d/5B-S29A, and the mutant NS5B protein was named after the mutation, in this case, S29A. Other mutations of the rGTP-specific binding site were introduced in the same way as the S29A tion except that different primer pairs were used for PCR (Table 1). All muta-tions were verified by DNA sequence analysis (Elim Biopharmaceuticals).
To facilitate the introduction of mutations of the rGTP-specific binding site
into a subgenomic HCV replicon vector, pBR322/I377-NS3-3⬘/S1179I (22), an
NsiI site at nucleotide (nt) 3670 of the Con1 replicon was destroyed by intro-ducing a silent nucleotide C-to-T mutation at position 3669, resulting in a
construct designated pBR322/I377-NS3-3⬘/S1179I/⌬NsiI3670(6). This
site-spe-cific mutation did not affect the replication of the subgenomic HCV RNA in the cell (6). Accordingly, the NsiI site at nt 7110 in the NS5B gene became a unique restriction enzyme site. All mutations of amino acids Ser29 and Arg32 were created by replacement of the DNA fragment between BclI (nt. 6011) and NsiI (nt 7110) sites with those derived from mutant pET21d vectors containing the corresponding mutations. DNA fragment inserts with specific mutations were amplified by PCR using HCV-BclI and 5B-NsiI/3 as primers and individual
mutant pET21d/NS5B⌬21 vector as a template. PCR DNAs were digested with
BclI and NsiI and inserted into the pBR322/I377/NS3-3⬘/S1179I/⌬NsiI3670vector
that was also cut by both BclI and NsiI enzymes. To mutate amino acid residues Pro495, Pro496, Val499, and Arg503, the above-described two-step PCR method was used. For example, an Arg503-to-Ala mutation was made by PCR using two pairs of primers. In the first PCR, oligonucleotides 5B-NsiI/5 and R503A/3
(Table 1) were used as primers, and the replicon pBR322/I377/NS3-3⬘/S1179I
vector DNA was used as a template. The second PCR was performed using oligonucleotides R503A/5 and PBR-Ase as primers. The two PCR DNA frag-ments were fused together by a third PCR with oligonucleotides 5B-NsiI/5 and PBR-Ase primers. The longer PCR product was digested with NsiI and PvuI and
ligated into the vector pBR322/I377/NS3-3⬘/S1179I/⌬NsiI3670, which was
[image:2.585.302.544.83.396.2]di-gested with the same enzymes. Other mutations at amino acid residues Pro495,
TABLE 1. Oligonucleotides used for PCR-directed mutagenesis and RT-PCR amplificationa
Oligonucleotide Sequence (5⬘to 3⬘)
5B-BspHI ...CGGGATCCTCATGAGCATGTCCTACACATGGAC AGGC
5B/His⌬C21 ...GCGCGACTAGTTCAGTGGTGGTGGTGGTGGTG
GCGGGGTCGGGCACGAGACAGGCTG S29A/3 ...ACGGAGCAAAGCGTTGCTCAGTGCATTGAT S29A/5 ...GCTTTGCTCCGTCACCACA R32K/3...TGTGGTGCTTGAGCAAAGAGTTGCTCAG R32K/5...AAGCACCACAACTTGGTCTATGC R32A/3 ...GTTGTGGTGAGCGAGCAAAGAGTTGCTCAG R32A/5 ...GCTCACCACAACTTGGTCTATG R32S/3 ...GTTGTGGTGACTGAGCAAAGAGTTGCTCAG R32S/5 ...AGTCACCACAACTTGGTCTA P495A/3 ...TCGCAAGGGCGCTACCCCAAGTTTCCTGAGG P495A/5 ...GCGCCCTTGCGAGTCTGGAG P496A/3 ...GACTCGCAAGGCCGGTACCCCAAGTTTCCTG P496A/5 ...GCCTTGCGAGTCTGGAGACA V499A/3 ...ATGTCTCCAGGCTCGCAAGGGCGGTACCCCA V499A/5 ...GCCTGGAGACATCGGGCCAGA V499G/3 ...ATGTCTCCAGCCTCGCAAGGGCGGTACCCCAAG V499G/5 ...GGCTGGAGACATCGGGCCAGAAG R503K/3...TTCTGGCCTTATGTCTCCAGACTCGCAA R503K/5...AAGGCCAGAAGTGTCCGCGCT R503A/3 ...ACTTCTGGCCGCATGTCTCCAGACTCGCA R503A/5 ...GCGGCCAGAAGTGTCCGCGCT HCV-Bcl-I...GACAGGCGCCCTGATCACG 5B-Nsi/3...GCCAGATGCATCGTGCGCGAC 5B-Nsi/5...CGCGCACGATGCATCTGGCA PBR-AseI ...TCCAGTCTATTAATTGTTGC HCV5935 ...AGCGACGGGTCTTGGTCTAC HCV7757 ...GAGTGTTTAGCTCCCCGTTC NsiI/3670/3 ...GCCGACATACATGCCATGATGTATTTG NsiI/3670/5 ...ATGTATGTCGGCTGACCTGGAG 3-Blunt...ACTTGATCTGCAGAGAGGCCA T7-minus-3 ...CGGAATTCTTAATACGACTCACTATAGGCGCGC CCTTTGGTTTTTCTTTGA Minus-3-EarI ...GCTCTAGACTCTTCGGCCAGCCCCCGATTG a
Nucleotide mutations are underlined. Oligonucleotides were synthesized by integrated DNA technology.
on November 8, 2019 by guest
http://jvi.asm.org/
Pro496, Val499, and Arg503 were created in the same way as the R503A muta-tion, but different primer pairs were used (Table 1). All mutations were verified by DNA sequence analysis (Elim Biopharmaceuticals).
A transient replication assay was carried out using modified replicons suitable for an enzyme reporter assay, as described by Yi et al. (39). To improve the low transient replication efficiency of the Con1-derived replicon, Btat2ANeo/SI, an
adaptive mutation (1202 E3G) in NS3 was introduced (15; M. Yi and S. M.
Lemon, unpublished data). In addition, a hepatitis delta virus ribozyme sequence was placed downstream of the HCV sequence, which enhances the replication
competence of the RNA by generating an authentic 3⬘end (40) (Yi and Lemon,
unpublished). The resulting replicon Btat2ANeo/EG/SI/delta replicated to a high level (see Fig. 7). To introduce mutations of the rGTP-specific binding site of NS5B, DNA fragments between XhoI (nucleotide 5570) and NheI (nucleotide
7919) sites of the pBR322/I377/NS3-3⬘/S1179I vector containing the
above-de-scribed mutations were excised and inserted into the same sites of the replicon Btat2ANeo/EG/SI/delta. Nucleotide mutations were confirmed by DNA se-quencing.
To produce a negative-stranded [(⫺)] HCV 3⬘UTR RNA template for the in
vitro RdRp assay, a cDNA of the (⫺)3⬘UTR RNA was amplified by PCR using
pBR322/I377/NS3-3⬘DNA as a template and oligonucleotides T7-minus-3 and
Minus-3-EarI as primers (Table 1). The PCR DNA fragment was digested with EcoRI and XbaI and cloned into a pUC19 vector between EcoRI and XbaI sites.
The resulting DNA construct was named pUC19/T7(⫺)3⬘UTR. A T7 promoter
sequence was placed immediately upstream of the cDNA encoding the
(⫺)3⬘UTR RNA so that a (⫺)3⬘UTR RNA could be generated by T7 RNA
transcription in vitro.
Expression and purification of NS5B proteins. E. coli strain BL21(DE3) (Novagen) was used to express recombinant HCV NS5B protein. BL21(DE3) cells were transformed with pET21d expression vector containing either wild-type or mutant NS5B cDNA, as described above. DNA-transformed BL21 was
grown at 37°C in LB medium containing 100g/ml of ampicillin to an optical
density at 600 nm of 1.0 to 1.2. NS5B protein expression was induced by the
addition of 1 mM isopropyl--D-thiogalactopyranoside (IPTG) at 25°C for 6 h.
The cell pellet was resuspended in a nickel buffer containing 50 mM sodium
phosphate, pH 8.0, 300 mM NaCl, 10 mM imidazole, 10 mM-mercaptoethanol,
10% glycerol, 0.5% Igepal CA630, 1 mM phenylmethylsulfonyl fluoride, 0.5
g/ml of leupeptin, 0.5g/ml of pepstatin A, and 2 mM benzamidine. After
being frozen at⫺80°C and thawed, cells were homogenized by sonication on ice.
The clarified supernatant was obtained by centrifugation at 13,000 rpm for 30
min. The supernatant was passed through a 0.22-m filter and loaded onto a
HiTrap chelating column charged with NiSO4(Amersham Pharmacia Biotech).
The column was washed sequentially with nickel buffer containing 0 to 100 mM imidazole. The bound NS5B protein was eluted out with an imidazole solution in a 100 to 500 mM linear gradient. The NS5B protein was confirmed by Western
blot analyses. The purity of the isolated protein was examined by electrophoresis on a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel, followed by Coo-massie blue staining. Purified recombinant NS5B proteins were aliquoted and
stored at⫺80°C.
Preparation of RNA transcripts.Subgenomic HCV replicon RNAs were tran-scribed in vitro by a T7 RNA polymerase from the above-detran-scribed DNA con-structs linearized by either the restriction enzyme ScaI (for
pBR322/I377-NS3-3⬘/S1179I-based replicons) or XbaI (for Btat2Aneo/EG/SI derived DNAs)
digestion. The (⫺)3⬘UTR RNA was transcribed from a pUC19/T7(⫺)3⬘UTR
DNA that was linearized by digestion with EarI. All RNA transcripts were prepared by using large-scale RNA production kits (Promega). After extensive treatment with RNase-free DNase I, the T7 RNA transcripts were purified by using an RNeasy RNA purification kit (QIAGEN). The RNA concentration was determined by spectrophotometer.
RNA synthesis by HCV NS5B in vitro.An RdRp assay was performed as previously described (21), with subtle modifications. Briefly, the assay was
per-formed in a 25-l reaction mixture containing 20 mM Tris-HCl, pH 7.4; 10 mM
MgCl2; 1 mM dithiothreitol; 10 mM KCl; 500M ATP, CTP, and GTP; 10M
UTP; 0.5Ci of [␣-32
P]UTP (ICN); 50 ng of (⫺)-strand 3⬘UTR RNA template;
20 U of RNasin; and 100 ng of purified NS5B protein. In the case of GTP stimulation experiments, different concentrations of GTP were added to the reaction mixture. The reaction mixtures were incubated at 30°C for either 30 min
or 2 h as indicated and then stopped by the addition of 25l stop buffer
containing 10g tRNA, 0.2% SDS, and 4g proteinase K. The reaction product
was extracted with phenol-chloroform, and RNA was precipitated with ethanol. RNA products were analyzed in a 6% polyacrylamide–7.7 M urea gel and quantitated with a PhosphorImager.
RNA transfection and selection of Huh7.5 cells resistant to G418 sulfate.
Wild-type and mutant subgenomic HCV RNAs were transfected into Huh7.5
cells by electroporation with a Gene Pulser system (Bio-Rad). Briefly, 1g of the
in vitro-transcribed and purified replicon RNAs was electroporated into 8⫻106
Huh7.5 cells in 0.4 ml (2⫻107cells/ml) of ice-cold phosphate-buffered saline.
After two pulses at 1.0 kV and 25F capacitance, the replicon-transfected
Huh7.5 cells were kept at room temperature for 10 min and then seeded in 100-mm dishes at different cell densities in DMEM containing 10% FBS. At 24 h posttransfection, cell culture medium was replaced with DMEM containing 10% FBS and 0.5 mg/ml G418 sulfate. The medium was changed every 3 to 4 days. After 4 weeks of selection with G418 sulfate, individual cell colonies were amplified for further characterization. Cell colonies in other dishes were fixed and stained by a solution containing 0.01% crystal violet and 19% methanol.
RNA extraction and quantitation by RPA.Total cellular RNA was extracted from HCV replicon-harboring cell lines using an RNeasy RNA isolation kit (QIAGEN). The levels of positive- and negative-strand HCV RNAs were
de-termined by an RNase protection assay (RPA) using [␣-32P]UTP-labeled
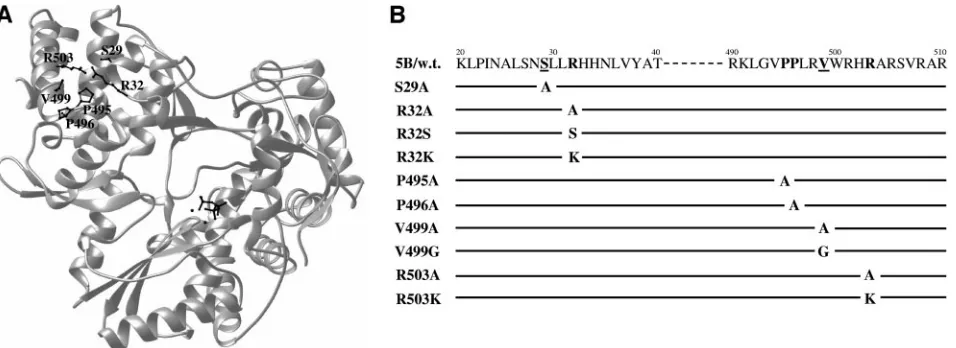
HCV-FIG. 1. (A) Ribbon structure of the HCV NS5B protein. The catalytic site of the enzyme is highlighted by a nucleotide triphosphate and two metals at the center of the molecule. The amino acid residues (S29, R32, P495, P496, V499, and R503) defining the low-affinity rGTP-specific binding pocket are highlighted by their side chains. (B) Diagram of mutations of the rGTP-specific binding site of NS5B. The name of the mutant RNA is indicated on the left, and the amino acid is indicated by single letters. The amino acid position is shown on the top. Residues involved in the GTP-specific binding are highlighted in boldface type. The underlined residues of the GTP binding site are less conserved among different HCV genotypes.
on November 8, 2019 by guest
[image:3.585.46.524.68.242.2]specific RNA probes, as described previously (6). A total of 10g of total RNA isolated from replicon-bearing Huh7.5 cells was used in RPA for hybridization
with 5⫻104cpm of [␣-32P]UTP-labeled-actin probe and 105cpm of either
(⫺)3⬘UTR or (⫹)5⬘UTR RNA probe. An RPA was performed by using an RPA
III kit following the manufacturer’s instructions (Ambion). RNA products were analyzed by electrophoresis in a 6% polyacrylamide–7.7 M urea gel. The levels of RNAs were determined by quantitation with a PhosphorImager (Molecular Dynamics).
Transient RNA replication assay.Transient replication assays were carried out with En5-3 cells transfected with Btat2ANeo/EG/SI/delta transcripts and tran-scripts derived from related mutants. RNA trantran-scripts were transfected into cells by electroporation, and RNA replication was monitored by assay of cell culture supernatant fluids for SEAP activity, as described previously (39).
RT-PCR and DNA sequence analysis.Nucleotide mutations introduced into subgenomic HCV replicon RNAs were determined by reverse transcription-PCR (RT-PCR) and sequence analysis. Recovered replicon RNAs extracted from G418-resistant Huh7.5 cells were reverse transcribed by SuperScript II reverse transcriptase (Invitrogen) with an HCV-specific primer, 3-Blunt (Table 1), which is complementary to nucleotides 7969 to 7989 of the replicon RNA
I377-NS3-3⬘/S1179I. The NS5B cDNA was amplified by PCR using primers HCV5935 and
HCV7757 (Table 1), and the PCR product was directly used for DNA sequence analysis (Elim Biopharmaceuticals).
RESULTS
Mutagenesis analysis of the rGTP-specific binding site of the HCV NS5B. X-ray crystallographic analysis of the HCV NS5B in complex with rNTP revealed a low-affinity rGTP-specific binding pocket lying on the surface of the enzyme between the thumb domain and the fingertip about 30 Å away from the enzyme active site (Fig. 1A) (4). A total of six amino acid residues were identified, which make direct or indirect contacts with the guanosine moiety of rGTP. These included a serine residue at amino acid 29 (S29), arginine at 32 (R32), prolines at 495 (P495) and 496 (P496), valine at 499 (V499), and arginine at 503 (R503) of the HCV NS5B (Fig. 1A) (4). To determine the importance of the rGTP-specific binding site in HCV RNA replication, we replaced each of these residues by either a structurally related (e.g., R-to-K mutation) or unre-lated (e.g., R-to-A mutation) amino acid (Fig. 1B). Each of these amino acid substitutions was individually introduced into both the HCV RdRp, which was expressed, purified, and mea-sured in vitro, and the NS5B that was encoded by a sub-genomic HCV replicon RNA. These mutations allow us to determine the functional role of the residues with respect to specific binding to rGTP in RdRp activity in vitro, as well as in HCV RNA replication in the cell.
Effects of mutations of the rGTP-specific binding residues on the RdRp activity.To determine the effects of mutations of the rGTP-specific binding pocket on the RdRp activity of HCV NS5B, wild-type and mutant NS5B proteins were expressed and purified to homogeneity (Fig. 2). As reported by others and found in our previous studies (11, 21), deletion of a C-terminal hydrophobic region of 21 amino acids of NS5B and addition of a six-histidine tag at the C terminus did not dras-tically compromise the RdRp activity in vitro but rather greatly increased the solubility of the protein. Therefore, we con-structed a vector that expresses NS5B protein with deletion of the C-terminal 21 amino acids and addition of a six-histidine tag at the C terminus to facilitate NS5B expression and puri-fication. NS5B proteins were expressed inE. coliand purified through a nickel column chromatograph. As shown in Fig. 2, wild-type and mutant NS5B proteins used for in vitro polymer-ase assays were of the highest purity.
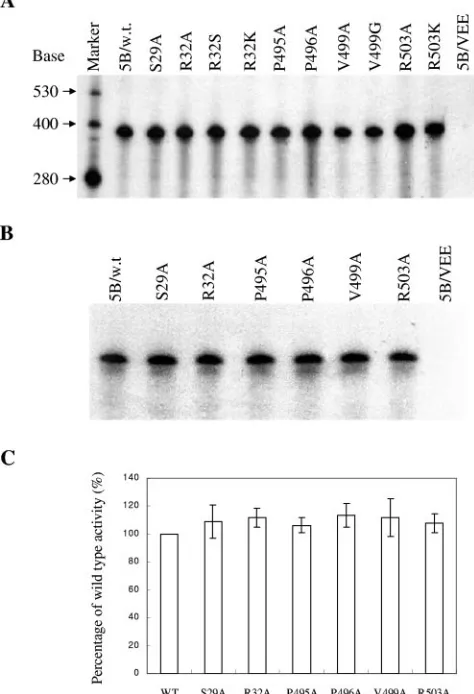
To determine the effects of mutations introduced into the low-affinity rGTP-specific binding site of the HCV NS5B on the RdRp activity, an in vitro assay for de novo RNA synthesis catalyzed by HCV RdRp was performed using the negative-sense HCV 3⬘UTR RNA as a template. As demonstrated in our previous studies (21), purified recombinant NS5B is able to initiate RNA synthesis de novo, which is the most likely mech-anism for HCV RNA replication in vivo (20). Therefore, the in vitro RNA synthesis experiments would reveal the effects of mutations on both the initiation and elongation activities of the HCV RdRp. We performed a time course experiment to first determine the correlation of RNA synthesis with incubation time (Fig. 3). Consistent with our previous results (21), RNA was faithfully synthesized by purified recombinant NS5B using HCV (⫺)3⬘UTR RNA as a template (Fig. 3). The RNA prod-ucts synthesized by NS5B were predominantly of the same size as the input RNA template (384 nt in length), indicating that the RNA was synthesized by de novo initiation (Fig. 3). Addi-tionally, the amount of RNA product increased proportionally with incubation time. There was a linear correlation between the amount of RNA product and the incubation time up to 2 h (Fig. 3). Using the same RdRp assay, we determined the effects of mutations of the rGTP-specific binding site on the RdRp activity of NS5B. Initially, the RdRp reactions were run for 2 h to determine the maximal levels of RNA synthesis (Fig. 4A). Surprisingly, the purified NS5B proteins with S29A, R32A, R32S, R32K, P495A, P496A, V499A, V499G, R503A, or R503K mutations all yielded RNA levels comparable to that of wild-type NS5B (Fig. 4A). The lower intensity of V499A and V499G bands was caused by the loss of RNA products, as determined by normalization with [32P]UTP-labeled -actin
RNA that was added into the tube after the RdRp reaction (data not shown). As a control, NS5B/VEE with amino acid GDD-to-VEE mutations in the active site of the enzyme did not catalyze any RNA synthesis (Fig. 4A and B, lanes 5B/ VEE), demonstrating that the RNA products were indeed the result of the authentic HCV RdRp activity. To confirm the finding that mutations of the low-affinity rGTP binding resi-dues do not affect the enzyme activity, the mutant NS5B pro-teins with each of the rGTP-binding residues mutated to
ala-FIG. 2. Electrophoretic analysis of purified recombinant NS5B proteins. Wild-type and mutant NS5B proteins with a deletion of the C-terminal 21 amino acids were expressed and purified as described in Materials and Methods. Briefly, 500 ng (each) of purified NS5B pro-teins was loaded in a 10% SDS-PAGE gel and visualized by Coomassie blue staining. The names of the purified NS5B proteins are indicated on the top, and the sizes of the protein molecule markers are shown on the left.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.585.302.544.71.188.2]nine were examined by an RdRp assay with a 30-min incubation time, at which the minimal level of RNA synthesis could be detected (Fig. 3). The short run of the RdRp reaction would allow the determination of the difference in the initial RNA synthesis between wild-type and mutant NS5B enzymes. Again, the levels of RNA products synthesized by wild-type and mutant NS5B proteins are very similar (Fig. 4B). Results derived from different independent experiments were com-pared (Fig. 4C). Although the RdRp activity of each mutant NS5B varied, none of the mutations of the rGTP-specific bind-ing residues had any significant effect on the in vitro RdRp activity when compared to the wild-type NS5B (Fig. 4C). These results suggest that amino acid residues of the low-affinity rGTP-specific binding pocket are not important for in vitro RdRp activity of purified NS5B.
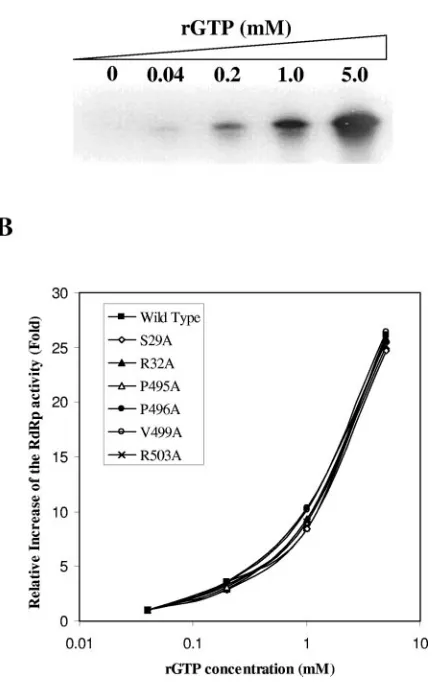
Effects of mutations of the rGTP-specific binding site on the stimulation of RNA synthesis by GTP in vitro.rGTP was found to specifically stimulate the in vitro RdRp activity of NS5B in a concentration-dependent manner (18). It was speculated that binding of rGTP to the rGTP-specific binding pocket might trigger a conformational rearrangement of the enzyme to allow alternative interactions between the thumb and finger domains of NS5B, which therein renders efficient initiation of RNA synthesis by HCV RdRp (4). To determine the role of the rGTP-specific binding site in GTP stimulation of in vitro RNA
synthesis, RNA synthesis experiments were carried out in the presence of increasing concentrations (0, 0.04, 0.2, 1.0, and 5 mM) of rGTP (Fig. 5A). The relative increase (n-fold) of RNA products was calculated considering the amount of RNA syn-thesized by wild-type NS5B as 1. Overall, the RdRp activities of purified NS5B proteins with an alanine mutation in each of the rGTP-specific binding pocket were all stimulated to a sim-ilar extent by increasing concentrations of rGTP (Fig. 5B). The stimulation curve of RNA synthesis by rGTP for each of these mutants was nearly superimposed to that of wild-type NS5B within the range of variation shown in Fig. 4C (Fig. 5B). This
[image:5.585.301.538.74.421.2]FIG. 3. (A) Time course of RNA synthesis by purified HCV NS5B. Wild-type NS5B (100 ng) was incubated with 50 ng of (⫺)3⬘UTR RNA template at 30°C for 10, 30, 45, 60, 90, and 120 min, respectively. The radiolabeled RNA products were analyzed in a 6% polyacrylamide–7.7 M urea gel, which was subsequently dried and subjected to autora-diography. (B) Correlation of RNA products with the incubation time of RdRp reaction. The RNA products shown in panel A were quan-tified by PhosphorImager (Molecular Dynamics). The amounts of RNA products (yaxis) are plotted against the incubation time (xaxis).
FIG. 4. Effects of mutations of the low-affinity rGTP-specific bind-ing site on the in vitro RdRp activity. A total of 100 ng of purified NS5B protein was incubated with 50 ng of HCV (⫺)3⬘UTR RNA template under the reaction conditions described in Materials and Methods. The RdRp reactions were run for either 120 min (A) or 30 min (B) RNA products were resolved in a 6% polyacrylamide–7.7 M urea gel and visualized by autoradiography. The levels of RNA prod-ucts were quantified by PhosphorImager analysis (Molecular Dynam-ics). Purified NS5B proteins are indicated on the top, and the sizes of the RNA markers are indicated on the left. (C) Comparison of relative in vitro RdRp activity between wild-type and mutant NS5B proteins. The amount of RNA synthesized by NS5B was quantified with a PhosphorImager. The RdRp activity of each mutant NS5B relative to wild-type NS5B was calculated as a percentage of wild-type activity, considering wild-type NS5B as 100%. The average percentages of three independent experiments are shown for each mutant NS5B, as indicated at the bottom.
on November 8, 2019 by guest
finding suggests that the rGTP-specific binding site has no significant role in rGTP stimulation of the in vitro RNA syn-thesis.
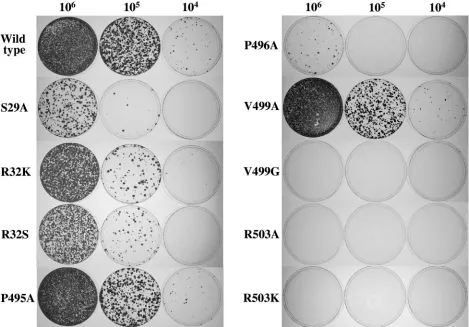
Effects of mutations of the rGTP-specific binding site on cell colony formation induced by HCV RNA replication.Purified recombinant NS5B lacks template specificity in vitro, as it is able to catalyze RNA polymerization on both HCV-specific RNA and nonviral RNA templates (2, 11, 13, 17, 21, 26). The lack of template specificity by purified NS5B in vitro suggests that viral replicase requires other nonstructural viral proteins and/or cellular factors to selectively replicate HCV RNA in vivo (20). Therefore, efforts were made to determine whether mutations of the rGTP-specific binding site affect HCV RNA replication in the cell. Subgenomic HCV RNAs with site-spe-cific mutations were transfected into Huh7.5 cells by electro-poration. Cells resistant to G418 sulfate as a result of HCV RNA replication were selected. Results are shown in Fig. 6 and Table 2. A serine-to-alanine mutation at residue 29 (S29A) reduced the efficiency of cell colony formation by nearly 50 fold. Likewise, mutations of amino acid 32 from arginine to lysine (R32K), serine (R32S), or alanine (R32A) resulted in about a four- to eightfold decrease in the efficiency of cell colony formation (Fig. 6 and Table 2). However, a proline-to-alanine mutation at residue 495 (P495A) caused only a subtle change (⬍2 fold) in the efficiency of cell colony formation. In contrast, a proline-to-alanine mutation at amino acid 496 re-duced the efficiency of cell colony formation by approximately 60 fold, suggesting that the conserved P496 residue is very important for HCV RNA replication. Interestingly, a valine to alanine mutation at residue 499 (V499A) had no significant (1.5-fold) effect on cell colony formation, whereas a valine-to-glycine mutation (V499G) completely ablated the ability of HCV RNA to replicate in the cell, as no cell colony formation was observed (Fig. 6). Surprisingly, mutation of the amino acid 503 from arginine to either lysine (R503K) or alanine (R503A) completely inactivated the replicon RNA to induce cell colony formation. Clearly, residue R503 cannot tolerate even a struc-turally conservative mutation, suggesting that it is critically important for the function of the HCV replication in the cell. Taken together, these findings demonstrate that all residues that make direct or indirect (water-mediated) contact with rGTP, as revealed by crystallographic study (4), are important for replication of HCV RNA in the cell, and mutation of each of these residues either impaired or ablated the ability of the viral replicase to catalyze RNA replication.
To determine whether the site-specific mutations were sta-ble or reverted back to wild-type sequence during G418 selec-tion, we carried out RT-PCR experiments to amplify portion of the NS5B gene of the replicon RNAs. RT-PCR DNA products were directly used for sequence analysis. Results from these experiments show that the recovered replicon RNAs all con-tain the expected mutations, suggesting that mutations intro-duced into the rGTP-specific binding site were stable once the RNA replicated (data not shown). However, we did not deter-mine whether supplemental mutations occurred in other re-gions of the replicons during G418 selection.
Effects of mutations of the rGTP-specific binding site on transient HCV RNA replication.The effects of mutations on HCV RNA replication, as determined by the above-described cell colony formation, could be complicated by compounding
[image:6.585.315.531.85.426.2]secondary mutations that potentially occurred during G418 selection or possible selection of Huh7.5 cells with clonally specific growth characteristics and/or permissiveness for viral RNA replication. To address this concern, the effects of mu-tations of the specific rGTP-binding site on RNA replication were further confirmed by using an HIV Tat-expressing sub-genomic HCV replicon, which allows monitoring of transient RNA replication without G418 selection. As demonstrated in a previous study (39), the HIV Tat-expressing replicon induces the expression of SEAP in proportion to the levels of HCV replicon RNA in the En5-3 cell, an Huh7 cell line with an integrated SEAP gene under the control of the HIV long terminal repeat. To enhance the efficiency of HCV RNA rep-lication, the original replicon Btat2ANeo/SI derived from the Con1 sequence was modified by introducing an additional
FIG. 5. Stimulation of RNA synthesis by rGTP. In vitro RdRp experiments were performed as described in Materials and Methods by the addition of increasing concentrations (0, 0.04, 0.2, 1.0, and 5.0 mM) of rGTP. Reaction mixtures were incubated at 30°C for 60 min. (A) Stimulation of wild-type RdRp by rGTP. The concentrations (in millimoles) of rGTP are shown on the top. (B) Effects of mutations of the rGTP binding site on the RdRp stimulation by rGTP. The amounts of RNA synthesized by wild-type and mutant NS5Bs (indicated in the graph) were quantified by PhosphorImager analysis (Molecular Dy-namics). The relative increase (n-fold) of the amount of RNA synthe-sized was calculated using the amount of RNA synthesynthe-sized by wild-type NS5B at 0.04 mM rGTP, set as 1.
on November 8, 2019 by guest
http://jvi.asm.org/
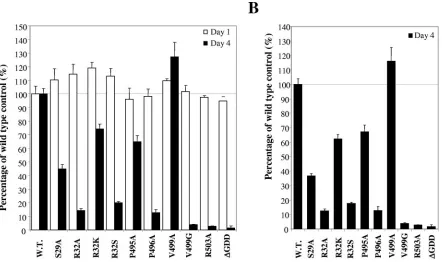
adaptive mutation (E1202 to G) in NS3 and the addition of a hepatitis delta virus ribozyme sequence at the 3⬘ end of the HCV replicon RNA, as described in Materials and Methods. The modified replicon Btat2ANeo/EG/SI/delta was replicated efficiently, as determined by a transient replication assay (Fig. 7). Mutations of the rGTP-specific binding site of NS5B were introduced into the replicon Btat2ANeo/EG/SI/delta. Upon transfection of replicon RNAs into En5-3 cells, the culture media were collected and changed every 24 h up to 4 days
posttransfection. The levels of SEAP in the culture media were determined to monitor the efficiency of HCV RNA replication in the cell. The results shown in Fig. 7 represent the levels of SEAP relative to that of wild-type control RNA, which is considered 100%. At 24 h posttransfection, replicon RNAs, including the one with a deletion of amino acids GDD in the active site of HCV RdRp that is defective in replication (⌬GDD), all produced high levels of SEAP (Fig. 7 and data not shown). The levels of SEAP at 24 h posttransfection rep-resent protein translation from input (transfected) RNAs in-dependently of RNA replication, which is used to normalize the amount of input RNA as well as the transfection efficiency. Without RNA replication, the levels of SEAP, as seen for the
[image:7.585.57.526.69.396.2]⌬GDD mutant replicon, gradually decreased over time and reached background levels (En5-3) (Fig. 7A and data not shown) at day 4 posttransfection. By contrast, replication of wild-type replicon RNA (Btat2ANeo/EG/SI/delta) resulted in increased levels of SEAP at day 4 (Fig. 7A and data not shown). The levels of SEAP expressed from RNAs with dif-ferent mutations in the rGTP-specific binding site varied de-pending on the specific mutation (Fig. 7). When ranked by the levels of SEAP, replicon RNAs fell in the following order (from high to low): V499A, wild-type, P495A, R32K, S29A, R32S, R32A, and P496A (Fig. 7B). Consistent with the results
FIG. 6. Effects of mutations of the rGTP-specific binding site on the efficiency of cell colony formation. Huh7.5 cells were transfected with in vitro T7 transcripts of subgenomic HCV replicons and seeded in 100-mm dishes at the cell density indicated on the top of the panel. Cell colonies were selected by culturing with 500g/ml of G418 sulfate for about 3 weeks, stained with a crystal violet solution, and photographed.
TABLE 2. Efficiency of cell colony formation by wild-type and mutant RNAs
Replicon RNA No. of cell colonies/
g of RNA
Wild type ... (5.8⫾0.8)⫻103
S29A ... (2.6⫾0.7)⫻102
R32K ... (1.6⫾0.2)⫻103
R32S ... (0.7⫾0.2)⫻103
R32A ...(0.85⫾0.15)⫻103
P495A... (3.7⫾0.4)⫻103
P496A... (1.1⫾0.25)⫻102
V499A ... (3.8⫾0.6)⫻103
V499G ... 0 R503K ... 0 R503A ... 0
on November 8, 2019 by guest
[image:7.585.44.284.596.724.2]derived from cell colony formation (Fig. 6; Table 2), no de-tectable level of transient RNA replication was observed for RNAs with V499G and R503A mutations, as SEAP expression from these RNAs was indistinguishable from that of the rep-lication-defective⌬GDD mutant (Fig. 7). These results further confirm that V499G and R503A are lethal mutations to HCV RNA replication. In contrast, the V499A mutation slightly enhanced the level of SEAP by about 15% of the wild-type activity (Fig. 7B), consistent with the fact that this amino acid was found to be an alanine in some HCV isolates (4). Similar to cell colony formation efficiency (Fig. 6 and Table 2), the P495A and R32K mutations resulted in about a 30 to 35% reduction in SEAP activity (Fig. 7B). Other mutations signifi-cantly decreased the levels of SEAP expression to a similar extent, as shown by cell colony formation assay (Fig. 6 and Table 2). The only discrepancy lay in the R32S and R32A mutations, which impaired the transient replication of the mu-tant RNA more than they were able to induce cell colony formation (Fig. 6 and 7). The exact reason for this is not clear. One possible interpretation might be the incompatibility of this mutation with the second adaptive E1202G mutation in the NS3 protein, which is present only in the RNA used for tran-sient RNA replication. Nevertheless, these findings demon-strate that amino acid residues defining the rGTP-specific
binding site of NS5B are important for efficient HCV RNA replication in the cell.
DISCUSSION
[image:8.585.81.523.68.330.2]In this study, we performed a systematic functional analysis of the amino acid residues involved in the low-affinity rGTP-specific binding site of the HCV NS5B, which was previously determined by a crystallographic study (4). Our findings dem-onstrate that the rGTP-specific binding site is not required for the in vitro RdRp activity of the HCV NS5B. This conclusion is strongly supported by the fact that none of the amino acid mutations of the rGTP-specific binding site had any significant effect on the in vitro RNA synthesis catalyzed by purified NS5B (Fig. 4). The RNA products synthesized by HCV RdRp from the HCV negative-sense 3⬘UTR RNA template (384 nt in length) are primarily monomer (Fig. 3 and 4), suggesting de novo RNA synthesis. This is consistent with previous findings that purified HCV RdRp is able to synthesize RNA in vitro by de novo (primer-independent) initiation (21, 25, 42). Thus, by using in vitro RdRp assay, we were able to determine the effect of mutations of the specific rGTP-binding site on either initi-ation or elonginiti-ation activity of the HCV RdRp. Given that purified NS5B proteins with each individual mutation of the
FIG. 7. Effects of mutations of the rGTP-specific binding site on transient HCV RNA replication in the cell. Wild-type and mutant RNAs were transfected into EN5-3 cells. The culture media were sampled at 24-h intervals and assayed for SEAP activity as a measure of the intracellular replicon RNA abundance (see Materials and Methods). The results represent the mean SEAP activities relative to that of the wild-type RNA. Error bars indicate the range of variation in duplicate transfection experiments. Open bars stand for the relative levels of SEAP as determined at 24 h (day 1) posttransfection, and solid bars indicate the relative levels of SEAP at 96 h (day 4) posttransfection. The names of amino acid mutations are indicated at the bottom. (A) Comparison of the relative SEAP activities at day 1 and day 4 between wild-type and mutant HCV RNAs. The percentage of wild-type control was calculated for each mutation considering the wild-type level as 100%. (B) Effects of mutations of the rGTP-specific binding site on transient HCV RNA replication. The levels of SEAP activities at day 1 (A) were used to normalize the input RNA as well as the transfection efficiency. After normalization, the levels of SEAP activities at day 4 are shown as a percentage of the wild-type control (100%).
on November 8, 2019 by guest
http://jvi.asm.org/
disrupting the rGTP binding would decrease the RdRp activity of mutant NS5B, provided that dimerization of NS5B was necessary for the in vitro RdRp activity. However, the findings derived from our studies do not support a role of the specific rGTP binding site in in vitro RdRp activity of purified recom-binant NS5B.
It was found that rGTP selectively stimulated in vitro RNA synthesis by purified recombinant NS5B (18). As to the ques-tion of whether the surface low-affinity rGTP binding site of NS5B is involved in the rGTP stimulation (18), our results demonstrate that mutations of the rGTP-specific binding site do not significantly influence the stimulation of RNA synthesis by rGTP (Fig. 5). Like that of wild-type NS5B, the RdRp activities of mutant enzymes were all stimulated by increasing concentrations of GTP (Fig. 5). Clearly, the low-affinity rGTP binding site on NS5B is not involved in the rGTP stimulation during in vitro RNA synthesis. We believe that binding of rGTP to the initiation site is probably responsible for rGTP stimulation and repression of primer-dependent RNA synthe-sis, which were observed for the in vitro RNA synthesis (18, 29). A recent study found that rGTP binds adjacent to the initiation NTP site of the RdRp of bovine viral diarrhea virus (BVDV), suggesting an important role of rGTP in the initia-tion of RNA synthesis by the BVDV RdRp (8, 12). Whether these in vitro observations bear physiological relevance in vivo remains unknown. In vitro, rGTP was found necessary for the initiation of de novo RNA synthesis catalyzed by purified re-combinant HCV and BVDV RdRps (12, 41, 42). However, replication of HCV negative-strand RNA is invariantly initi-ated with an rATP, and replication of the positive-strand RNA is initiated with either an rATP or rGTP in vivo (6).
Despite the fact that the rGTP-specific binding site of NS5B is not required for in vitro RdRp activity, mutations of the amino acid residues involved in the low-affinity rGTP binding either impaired or completely ablated the ability of sub-genomic HCV RNAs to replicate in the cell. Mutations of residues S29, R32, and P496 all resulted in a significantly lower efficiency of cell colony formation induced by HCV RNA rep-lication (Fig. 6); as well, lower levels of SEAP activities re-sulted from transient RNA replication (Fig. 7). In contrast to the P496A mutation, the P495A mutation only slightly lowered the efficiency of cell colony formation (⬍1.5 fold). Likewise, the P495A mutation only resulted in a slight decrease (30%) in transient RNA replication (Fig. 7B). These findings suggest
mation, suggesting that these mutant RNAs did not replicate in the cell. These findings were independently confirmed by ex-periments directly measuring transient HCV RNA replication. The R503A mutation was found to be lethal to transient HCV RNA replication (Fig. 7). Collectively, these findings demon-strate that the residues of the low-affinity rGTP-specific bind-ing site of NS5B are critical to HCV RNA replication in the cell. Mutations of the rGTP binding site impaired the ability of the HCV RNA to replicate in the cell to various extents, depending on the nature of mutation.
The question arose as to whether the mutations described in this study actually disrupted the GTP binding. Efforts were made to directly determine the rGTP binding to wild-type and mutant NS5B proteins by using photoactive rGTP and rATP analogues, 8-azidoguanosine 5⬘ [␥-32P]triphosphate and
8-azidoadenosine 5⬘-[␥-32P]triphosphate (ALT, Lexington,
KY). Results derived from the photoaffinity labeling experi-ment did not reveal any significant differences in cross-linking of rGTP and rATP to NS5B between wild-type and mutant proteins (data not shown). We have also tried to pull-down NS5B using rGTP-agarose beads, which were successfully used for column chromatography analysis of the poliovirus 3Dpol
(31). Nor did this assay detect any differences in rGTP binding between wild-type and mutant NS5B proteins. These could be due to either low efficiency of photoaffinity cross-linking, low concentrations of rGTP, or multiple NTP binding sites present in the HCV RdRp (4), which masked the subtle differences in rGTP binding. Considering the low-affinity nature of this rGTP binding pocket, high rGTP concentrations (in millimoles) are likely required for detection of significant rGTP binding (4). This makes it very difficult for any in vitro assay to determine changes in rGTP binding. Based on the X-ray structure of the rGTP-bound NS5B, mutations of residues S29, R32, and R503, which form hydrogen bonds with the guanine moiety of rGTP, are expected to disrupt the specific rGTP binding site. As shown by the structure of the NS5B-GTP complex (4), S29 was found to interact with the ribose of the guanosine through a water-mediated hydrogen bond. R32 forms hydrogen bonds between the quanidinium groups of its side chain and the 2⬘ hydroxyl group of the ribose and the N2 position of the gua-nine. R503 also makes a bidentate hydrogen bond to the N2 of the base and to the main chain carbonyl of S29. Considering the specific interaction with rGTP but not other nucleotides, R32 is therefore believed to be a specificity determinant, as it
on November 8, 2019 by guest
can only form a bidentate hydrogen bond with rGTP. Muta-tions of these residues, in particular residue R32, to alanine will result in the loss of hydrogen bonding and therefore dis-rupt rGTP binding. In this regard, the impairment of HCV RNA replication in the cell by these mutations could be due to disruption of the GTP binding.
However, the effects of mutations on HCV RNA replication cannot be satisfactorily explained by disruption of the rGTP-specific binding site alone, since the effects of different muta-tions on the replicon replication varied remarkably despite their predicted impacts on rGTP binding. For instance, muta-tions of residues S29 and R32 only caused a reduction in RNA replication, while V499G and R503A mutations completely ablated RNA replication in the cell. Failure of replication of the subgenomic RNAs with V499G and R503A mutations sug-gests that the residues involved in the specific rGTP binding might have additional roles besides rGTP binding in HCV RNA replication. In this scenario, interesting analogy could be found in poliovirus polymerase (3Dpol). Poliovirus polymerase
has two NTP binding sites, one site of which (Lys 61) was shown to be essential for RNA chain elongation activity by purified enzyme, while the second site (Lys 276), when mu-tated to leucine, was shown to have no impairment of poly-merase activity while showing severely reduced levels of NTP binding (31, 32). When this K276L mutation was created in a poliovirus genomic RNA background, mutated poliovirus RNA showed a minute plaque phenotype that rapidly reverted to a wild-type phenotype with a mutation of Leu 276 residue to Arg. Although it is premature to correlate the exact mecha-nism of action of a second NTP binding site of poliovirus 3Dpol
to the low-affinity rGTP binding site of HCV NS5B, this strik-ing phenotypic similarity points to the evolutionary linkage of utilizing NTP binding to viral polymerase in virus replication beyond viral polymerase activity.
Then, what could the roles of the low-affinity rGTP-specific binding site of NS5B in HCV RNA replication be? The exact reason for the reduction of HCV RNA replication in the cell by mutations of the low-affinity rGTP-binging site of the HCV RdRp is not clear. In the case of poliovirus revertant L276R mentioned above, an Arg 276 mutation restored rNTP binding at this second site, indicating that rNTP binding is necessary to preserve the characteristics of poliovirus 3Dpolto render
effi-cient viral replication. This, combined with the fact that both the 3Dpolsecond rNTP binding site and the low-affinity rGTP
binding site of HCV NS5B are located at the surface of the protein, indicates that they might be involved in protein-pro-tein interactions. Supporting evidence for this explanation came from our recent observation that a His-to-Leu mutation at amino acid 34, which is juxtaposed to the rGTP-specific binding site, of HCV NS5B exhibits cell tropism in HCV RNA replication (G. Luo et al., unpublished results). This observa-tion suggests that the␣-helical-rich thumb subdomain of the HCV RdRp is involved in interactions with a cellular pro-tein(s), which was speculated based on the crystal structure of NS5B (5). In this context, rGTP binding to this particular site of NS5B may alter or stabilize its conformation to allow it to interact with other viral and/or cellular proteins that are im-portant for efficient HCV RNA replication in the cell. It will be interesting to further determine whether these mutations affect
protein-protein interactions with other viral or cellular pro-teins.
ACKNOWLEDGMENTS
We thank Charlie Rice (Rockefeller University) for providing us with the Huh7.5 cell line, Byung Chul Ahn for assistance in DNA construction, and Fenghua Yuan for help in protein purification. We are grateful to David Rodgers (Department of Molecular and Cellular Biochemistry, University of Kentucky) for preparing the structure model of NS5B as shown in Fig. 1A and Jochin Jarjer (The Wadsworth Center) for critical reading of the manuscript.
This work was supported by an NIH/NCI grant (CA93712) and in part by a grant from the Kentucky Science and Engineering Founda-tion (KSEF-148-502-02-18). M.Y. is supported by NIH grant AI063451.
REFERENCES
1.Ago, H., T. Adachi, A. Yoshida, M. Yamamoto, N. Habuka, K. Yatsunami, and M. Miyano.1999. Crystal structure of the RNA-dependent RNA
poly-merase of hepatitis C virus. Structure Fold. Des.7:1417–1426.
2.Behrens, S. E., L. Tomei, and R. De Francesco.1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus.
EMBO J.15:12–22.
3.Blight, K. J., J. A. McKeating, and C. M. Rice.2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J.
Vi-rol.76:13001–13014.
4.Bressanelli, S., L. Tomei, F. A. Rey, and R. De Francesco.2002. Structural analysis of the hepatitis C virus RNA polymerase in complex with
ribonucle-otides. J. Virol.76:3482–3492.
5.Bressanelli, S., L. Tomei, A. Roussel, I. Incitti, R. L. Vitale, M. Mathieu, R. De Francesco, and F. A. Rey.1999. Crystal structure of the RNA-dependent
RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. USA96:13034–
13039.
6.Cai, Z.-H., J. Liang, and G. Luo.2004. Effects of mutations of the initiation
nucleotides on hepatitis C virus RNA replication in the cell. J. Virol.78:
3633–3643.
7.Centers for Disease Control and Prevention.1998. Recommendations for prevention and control of hepatitis C virus (HCV) infection and
HCV-related chronic disease. Morb. Mortal. Wkly. Rep.47:1–39.
8.Choi, K. H., J. M. Groarke, D. C. Young, R. J. Kuhn, J. L. Smith, D. C. Pevear, and M. G. Rossmann.2004. The structure of the RNA-dependent RNA polymerase from bovine viral diarrhea virus establishes the role of
GTP in de novo initiation. Proc. Natl. Acad. Sci. USA101:4425–4430.
9.Choo, Q. L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton.1989. Isolation of a cDNA clone derived from a blood-borne
non-A, non-B viral hepatitis genome. Science244:359–362.
10.El-Hage, N., and G. Luo.2003. Replication of hepatitis C virus RNA occurs in a membrane-bound replication complex containing nonstructural viral
proteins and RNA. J. Gen. Virol.84:2761–2769.
11.Ferrari, E., J. Wright-Minogue, J. W. Fang, B. M. Baroudy, J. Y. Lau, and Z. Hong.1999. Characterization of soluble hepatitis C virus RNA-dependent
RNA polymerase expressed inEscherichia coli. J. Virol.73:1649–1654.
12.Kao, C. C., A. M. Del Vecchio, and W. Zhong.1999. De novo initiation of RNA synthesis by a recombinant flaviviridae RNA-dependent RNA
poly-merase. Virology253:1–7.
13.Kao, C. C., X. Yang, A. Kline, Q. M. Wang, D. Barket, and B. A. Heinz.2000. Template requirements for RNA synthesis by a recombinant hepatitis C
virus RNA-dependent RNA polymerase. J. Virol.74:11121–11128.
14.Koonin, E. V.1991. The phylogeny of RNA-dependent RNA polymerases of
positive-strand RNA viruses. J. Gen. Virol.72:2197–2206.
15.Krieger, N., V. Lohmann, and R. Bartenschlager.2001. Enhancement of hepatitis c virus RNA replication by cell culture-adaptive mutations. J. Virol.
75:4614–4624.
16.Lesburg, C. A., M. B. Cable, E. Ferrari, Z. Hong, A. F. Mannarino, and P. C. Weber.1999. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol
6:937–943.
17.Lohmann, V., F. Korner, U. Herian, and R. Bartenschlager.1997. Biochem-ical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic
activity. J. Virol.71:8416–8428.
18.Lohmann, V., H. Overton, and R. Bartenschlager.1999. Selective stimula-tion of hepatitis C virus and pestivirus NS5B RNA polymerase activity by
GTP. J. Biol. Chem.274:10807–10815.
19.Lohmann, V., A. Roos, F. Korner, J. O. Koch, and R. Bartenschlager.2000. Biochemical and structural analysis of the NS5B RNA-dependent RNA
polymerase of the hepatitis C virus. J. Viral Hepat.7:167–174.
20.Luo, G.2004. Molecular virology of hepatitis C virus, p. 67–85.InJ. M. Colacino and B. A. Heinz (ed.), Hepatitis prevention and treatment. Birkhauser, Basel, Switzerland.
on November 8, 2019 by guest
http://jvi.asm.org/
and computer predictions of secondary structure. Virology252:287–303. 28.Qin, W., H. Luo, T. Nomura, N. Hayashi, T. Yamashita, and S. Murakami.
2002. Oligomeric interaction of hepatitis C virus NS5B is critical for catalytic
activity of RNA-dependent RNA polymerase. J. Biol. Chem.277:2132–2137.
29.Ranjith-Kumar, C. T., L. Gutshall, R. T. Sarisky, and C. C. Kao.2003. Multiple interactions within the hepatitis C virus RNA polymerase repress
primer-dependent RNA synthesis. J. Mol. Biol.330:675–685.
30.Reed, K. E., and C. M. Rice.2000. Overview of hepatitis C virus genome structure, polyprotein processing, and protein properties. Curr. Top.
Micro-biol. Immunol.242:55–84.
31.Richards, O. C., S. Baker, and E. Ehrenfeld.1996. Mutation of lysine resi-dues in the nucleotide binding segments of the poliovirus RNA-dependent
RNA polymerase. J. Virol.70:8564–8570.
32.Richards, O. C., and E. Ehrenfeld.1997. One of two NTP binding sites in
ribosomal frameshift. EMBO J.20:3840–3848.
39.Yi, M., F. Bodola, and S. M. Lemon.2002. Subgenomic hepatitis C virus replicons inducing expression of a secreted enzymatic reporter protein.
Vi-rology304:197–210.
40.Yi, M., and S. M. Lemon.2003. Structure-function analysis of the 3⬘ stem-loop of hepatitis C virus genomic RNA and its role in viral RNA replication.
RNA9:331–345.
41.Zhong, W., E. Ferrari, C. A. Lesburg, D. Maag, S. K. Ghosh, C. E. Cameron, J. Y. Lau, and Z. Hong.2000. Template/primer requirements and single nucleotide incorporation by hepatitis C virus nonstructural protein 5B
poly-merase. J. Virol.74:9134–9143.
42.Zhong, W., A. S. Uss, E. Ferrari, J. Y. Lau, and Z. Hong.2000. De novo initiation of RNA synthesis by hepatitis C virus nonstructural protein 5B
polymerase. J. Virol.74:2017–2022.