Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Coding Sequences Upstream of the Human Immunodeficiency Virus
Type 1 Reverse Transcriptase Domain in Gag-Pol Are Not Essential
for Incorporation of the Pr160
gag-pol
into Virus Particles
Hsu-Chen Chiu, Szu-Yung Yao, and Chin-Tien Wang*
Institute of Clinical Medicine, National Yang-Ming University School of Medicine, and Department of Medical Research and Education, Taipei Veterans General Hospital, Taiwan
Received 9 October 2001/Accepted 27 December 2001
Incorporation of the human immunodeficiency virus type 1 (HIV-1) Gag-Pol into virions is thought to be mediated by the N-terminal Gag domain via interaction with the Gag precursor. However, one recent study has demonstrated that the murine leukemia virus Pol can be incorporated into virions independently of Gag-Pol expression, implying a possible interaction between the Pol and Gag precursor. To test whether the HIV-1 Pol can be incorporated into virions on removal of the N-terminal Gag domain and to define sequences required for the incorporation of Gag-Pol into virions in more detail, a series of HIV Gag-Pol expression plasmids with various extensive deletions in the region upstream of the reverse transcriptase (RT) domain was constructed, and viral incorporation of the Gag-Pol deletion mutants was examined by cotransfecting 293T cells with a plasmid expressing Pr55gag. Analysis indicated that deletion of the N-terminal two-thirds of thegagcoding
region did not significantly affect the incorporation of Gag-Pol into virions. In contrast, Gag-Pol proteins with deletions covering the capsid (CA) major homology regions and the adjacent C-terminal CA regions were impaired with respect to assembly into virions. However, Gag-Pol with sequences deleted upstream of the protease, or of the RT domain but retaining 15 N-terminalgagcodons, could still be rescued into virions at a level about 20% of the wild-type level. When assayed in a nonmyristylated Gag-Pol context, all of the Gag-Pol deletion mutants were incorporated into virions at a level comparable to their myristylated counterparts, suggesting that the incorporation of the Gag-Pol deletion mutants into virions is independent of the N-terminal myristylation signal.
During the late stage of the human immunodeficiency virus (HIV) life cycle, viral capsid protein precursor Pr55gagis
trans-ported to the plasma membranes, where the Pr55gagmolecules
self-assemble into virus particles and bud out from the cell membrane (6, 10, 34). After virus budding, the Pr55gag is
cleaved by the virus-encoded protease (PR) into matrix (MA; p17), capsid (CA; p24), p2, nucleocapsid (NC; p7), p1, and the C-terminal p6 domain (8, 12, 15, 18). PR-mediated Gag pro-cessing is not required for particle assembly and release but is essential for both virus maturation and infectivity (7, 13, 25, 26). Apart from the PR, the reverse transcriptase (RT) and integrase (IN) required for virus replication are also encoded by pol. HIV Pol is translated initially as a Pr160gag-pol by a
ribosomal⫺1 frameshift event which occurs at a frequency of about 5% duringgagencoding (11). Within the Gag-Pol, the p6 domain is truncated and replaced by a transframe domain referred to as p6* (23).
The Pr160gag-pol is thought to be incorporated into virus
particles by interaction with assembling Pr55gag through its
N-terminalgagdomain (9, 22, 27, 28). To identify which re-gions in Pr160gag-polare responsible for its incorporation into
virions, a system involving transient coexpression of Pr55gag
and Pr160gag-polfrom separate plasmids has been used widely
(9, 17, 22, 27, 28). In this system, PR in the encoded Pr160gag-pol
would cleave the coexpressed Pr55gagintrans, and the
incor-poration of Gag-Pol into virus particles would then be moni-tored by the appearance of the particle-associated p24gag. The
level of incorporated Gag-Pol was quantified by in vitro RT assays. One of these studies showed that substitution mutations in the CA major homology region (MHR) of Pr160gag-polhave
no detectable effects on Gag-Pol incorporation into the Pr55gag
particles (17). Different results were reported by another group, demonstrating that deletion of the whole MHR signif-icantly impaired the incorporation of Gag-Pol into virus par-ticles (28). Another study using a chimeric HIV/MLV (murine leukemia virus) Gag-Pol expression system showed that both the HIV type 1 (HIV-1) CA MHR and adjacent CA terminal sequences are required for efficient incorporation of Gag-Pol into virus particles (9).
Interestingly, HIV RT and IN proteins fused to the Vpr, an HIV accessory protein, could be rescued into virus particles, presumably via interaction with Pr55gagthrough the fusion Vpr
(5, 24, 35, 36). Although these results strongly suggest that the incorporation of HIVpol-encoded products into virus particles can be independent of Pr160gag-polexpression, a certain protein
sequence like Vpr, which can interact with Pr55gag, is required
to be present in order to mediate the incorporation ofpolgene products into virions. However, one more recent study has shown that the MLV Pol could be incorporated into virus particles, implying that the Gag domain in MLV Gag-Pol is not absolutely required for its assembly into particles and raising the hypothesis that putative interactions between the Pol and
* Corresponding author. Mailing address: Department of Medical Research and Education, Taipei Veterans General Hospital, No. 201, Sec. 2, Shih-pai Rd., Shih-pai, Taipei 112, Taiwan. Phone: 886-2-2871-2121, ext. 2655. Fax: 886-2-2874-2279. E-mail: [email protected].
3221
on November 8, 2019 by guest
http://jvi.asm.org/
Gag precursors may exist to facilitate the incorporation of Gag-Pol into virus particles (3).
It is not known whether HIV Gag-Pol can be incorporated into particles without any need for the presence of the N-terminal Gag domain. It remains controversial whether the CA MHR in Pr160gag-pol is essential for viral incorporation of
Pr160gag-pol. To address this issue and to map the sequences
responsible for the Pr160gag-polincorporation in more detail,
we constructed a series of HIV Gag-Pol constructs with various deletion mutations in thegag-polcoding region. The incorpo-ration of the mutant Gag-Pol product into virus particles was assayed by coexpressing the mutants with the Pr55gag. We
found that HIV Gag-Pol, with coding sequences deleted up-stream of the PR or RT regions but retaining only 15 N-terminal MA residues, could still be incorporated into virus independently of the N-terminal myristylation signal.
MATERIALS AND METHODS
Plasmid construction.The parental HIV-1 proviral plasmid DNA in this study was HXB2. All the plasmids were expressed in the HIVgpt backbone, which carries a simian virus 40oriandgptgene in theenvregion (19). To construct the Gag-Pol frameshift mutant, five T nucleotides were deleted in thegagandpol
overlap region by PCR-mediated mutagenesis. This resulted in placingpoland
gagin the same reading frame, leading to expression of the Pr160gag-polonly. The
sequence in the region of the juncture was nucleotide (nt) 2070-5⬘GAGAGAC AGGCTAATAGGGAA 3⬘. This Pr160gag-pol-expressing plasmid designated
GPfs was used as a wild-type (wt) control, and all the other Gag-Pol fusion constructs were derived from the GPfs (Fig. 1). To make a series of deletion mutations in the GPfs, we initially created aBamHI site at the following nucle-otide positions: 835, 1222, 1423, 1630, 1705, 1753, 1876, 2071, and 2538. Re-placement of the fragment nt 831-ClaI to nt 1705-BamHI with the fragment nt 831-ClaI to nt 1630-BamHI resulted in a complete deletion of the MHR (nt 1641 to 1694), yielding the construct⌬MHR. Similarly, deletion of theBamHI frag-ment nt 1876 to 2071 generated⌬NC. Recombination of theBamHI linker constructs 1423 and 1753, 1222 and 1753, 1423 and 1876, and 1222 and 1876 yielded the capsid deletion mutants⌬CA-1,⌬CA-2,⌬CA-3, and⌬CA-4, respec-tively. The more extensive deletion mutants⌬(CA⫹NC), ⌬(MA⫹2/3 CA),
⌬(MA⫹CA), and⌬Gag were made by deletion of the fragmentsBamHI-1222 to
BamHI-2071,BamHI-835 to BamHI-1876, and BamHI-835 to BamHI-2071, respectively. The⌬MA mutation, as described previously (33), was generated by deletion of the fragment nt 831-ClaI to nt 1147-PvuII and insertion of aSalI linker in the deleted region. The Pr55gag-expressing construct pGAG was created
by deletion of thepolgene fragment fromBclI-2429 toSalI-5786. To block the N-terminal myristylation of Pr160gag-pol, a myristylation-deficient mutation
(Myr⫺) (29), in which the second amino acid, glycine, of Gag was changed to
alanine, was introduced into each of the Gag-Pol fusion constructs. The juncture sequences for the Gag-Pol deletion mutants are shown in Fig. 1B. Each mutant construct was confirmed by DNA sequencing.
Cell culture and transfection.293T cells were maintained in Dulbecco’s mod-ified Eagle’s medium supplemented with 10% fetal calf serum. Confluent 293T cells were trypsinized, split 1:10, and seeded onto 10-cm plates 24 h before transfections. For each construct, 293T cells were transfected with 15g of plasmid DNA by the calcium phosphate precipitation method, with the addition of 50M chloroquine to enhance transfection efficiency. When the wt or mutant GPfs was cotransfected with the pGAG, 2g of each construct and 10g of pGAG were used, with addition of 8g of pBlueScript SK to a final amount of 20g of plasmid DNA. For cotransfection of PR-defective (PR⫺) GPfs
con-structs with the pGAG, 1g of each construct and 10g of pGAG were used, with addition of 9g of pBlueScript SK to a total amount of 20g of plasmid DNA. Culture media and cells were harvested for protein analysis or RT activity assays at 48 h posttransfection.
Western immunoblot analysis. Culture media from transfected 293T cells were filtered through 0.45-m-pore-size filters, followed by centrifugation through 2 ml of 20% sucrose in TSE (10 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1 mM EDTA) plus 0.1 mM phenylmethylsulfonyl fluoride (PMSF) at 4°C for 40 min at 274,000⫻g(SW41 rotor at 40,000 rpm). Viral pellets then were sus-pended in IPB (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 0.1% sodium dodecyl sulfate [SDS], 0.5% sodium deoxycholate, 1% Triton X-100,
0.02% sodium azide) plus 0.1 mM PMSF. Cells were rinsed with ice-cold PBS (phosphate-buffered saline), scraped from the plates, collected in 1 ml of PBS, and pelleted at 1,000⫻gfor 5 min. The cell pellets were resuspended in 250l of IPB plus 0.1 mM PMSF and then subjected to microcentrifugation at 4°C for 15 min at 13,700⫻g(14,000 rpm) to remove cell debris. Supernatant and cell samples were mixed with equal volumes of 2⫻sample buffer (12.5 mM Tris-HCl [pH 6.8], 2% SDS, 20% glycerol, 0.25% bromophenol blue) and -mercapto-ethanol to 5% and boiled for 5 min. Samples were subjected to SDS-polyacryl-amide gel electrophoresis (PAGE) and electroblotted onto nitrocellulose mem-branes. The membranes were blocked with 3% gelatin in Tris-buffered saline containing 0.05% Tween 20 (TBST), followed by incubation with the primary antibody in 1% gelatin–TBST for 1 h on a rocking platform at room temperature. The membranes were then washed three times for 10 min each with TBST and rocked for 30 min with the secondary antibody in 1% gelatin–TBST. The blots were again washed three times in TBST for 10 min each, and the membrane-bound antibody-conjugated enzyme activity was detected by an enhanced chemi-luminescence detection system or by a colorimetric method. For detection of HIV Gag proteins, we used an anti-p24gag(mouse hybridoma clone 183-H12-5C)
or anti-p17gag(catalog no. HB-8975; American Type Culture Collection,
Rock-ville, Md.) monoclonal antibody at a 1:5,000 dilution from ascites. The secondary antibody was a horse anti-mouse immunoglobulin G (IgG)-alkaline phosphatase conjugate at a 1:5,000 dilution (Vector Laboratories), and the Gag proteins were visualized by a color reaction solution of nitroblue tetrazolium–5-bromo-4-chloro-3-indolylphosphate (Promega). For detection of the HIV Gag-Pol, an HIV-positive human serum was used at 1:10,000 dilution as the primary anti-body. The secondary antibody was a goat anti-human IgG horseradish peroxidase (HRP)-conjugated antibody at 1:10,000 dilution, and the procedures used for HRP activity detection followed the manufacturer’s protocol (Amersham Phar-macia).
In vitro RT assay.Culture supernatants of transfected 293T cells were har-vested, filtered, and pelleted as described above. Viral pellets were resuspended in 30l of TSE buffer. A 10-l aliquot of each sample was used for Western immunoblot analysis. The remaining samples were further diluted with TSE, and a 10-l aliquot of each of the diluted samples was mixed with 40l of reaction cocktail containing 0.1% Triton X-100, 5 mM dithiothreitol, 10 mM MgCl2, 50
mM Tris-HCl (pH 8.0), 1.2 mM poly(rA)-(dT)15(Amersham Pharmacia), and 25
Ci of [3H]TTP/ml (30). Reactions were allowed to proceed at 37°C for 2 h,
followed by the addition of 5l of tRNA (10 mg/ml). The reaction mixtures then were precipitated with ice-cold 10% trichloroacetic acid and filtered with GF/C filters. After washing and drying, the filters were counted in a Beckman scintil-lation counter to determine RT activity.
Sucrose density gradient fractionation.Culture supernatants of transfected 293T cells were collected, filtered, and centrifuged through a 2-ml 20% sucrose cushion as described above. Viral pellets were suspended in TSE buffer and layered on top of premade 20-to-60% sucrose gradients consisting of 1-ml layers of 20, 30, 40, 50, and 60% sucrose in TSE, which had been allowed to mix by sitting for 2 h. Gradients were centrifuged at 274,000⫻g(SW50.1 rotor; 40,000 rpm) for 16 to 18 h at 4°C, and 500-l fractions were collected from top to bottom. Each fraction was measured for density and analyzed for Gag proteins and RT activity.
RESULTS
Pr160gag-polmutants with extensive deletions in thegag
cod-ing sequence, or in combination with additional removal of the N-terminal myristic acid moiety, could still be incorporated into virus particles.To define more completely the boundaries of HIVgag-pol sequences required for its incorporation into virus particles, a series of HIV Gag-Pol fusion constructs con-taining various deletion mutations in thegagcoding sequences was engineered and introduced into an HIV Pr160gag-pol
-ex-pressing plasmid, GPfs. GPfs, derived from an HIV replica-tion-defective vector HIVgpt (19), hadgagandpolpositioned in the same reading frame due to a frameshift mutation at the gag-poljunction. Figure 1A shows a schematic representation of the GPfs construct and its derivatives withgag-polsequences deleted to different extents. Except for ⌬(NC⫹PR) and
⌬(GAG⫹PR), all constructs retained an intact PR and should have been able to cleave the coexpressed Pr55gag in trans,
on November 8, 2019 by guest
http://jvi.asm.org/
yielding the mature Gag protein p24CA. Thus, incorporation of the Pr160gag-polinto virus particles could be monitored by
Western immunoblot analysis of the culture medium. How-ever, it should be noted that a very low level of incorporated Gag-Pol might provide enough PR activity to process the Gag. It is also not known whether PR action on Gag requires normal incorporation of Pol proteins. Therefore, this is only a measure of the incorporation of some minimal amount of PR.
We transfected 293T cells with each of the wt or mutant GPfs constructs alone or together with a Pr55gag expression
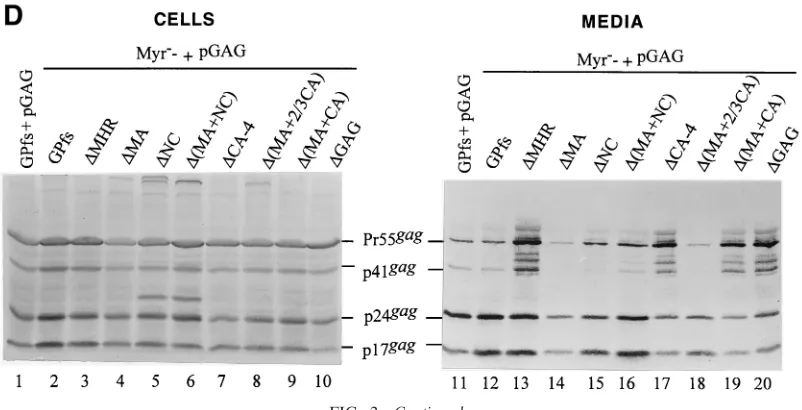
plasmid, pGAG. At 48 h posttransfection, cells and culture supernatants were collected and subjected to Western immu-noblot analysis as described in Materials and Methods. Ex-pressed and released Gag proteins were probed with an anti-p24gagand/or an anti-p17gagmonoclonal antibody. As shown in
Fig. 2A, bands corresponding to the intermediate p41gagand
mature p24gagand p17gagwere detected in the GPfs
transfec-tants (lane 1) while the Pr55gag was, as expected, assembled
and released from cells transfected with pGAG (lanes 9 and 19). Bands corresponding to the p17gagwere not seen in cells
transfected with the⌬MA or⌬(MA⫹NC), consistent with a deletion of the MA domain (lanes 2 and 4). Both⌬NC and
⌬(MA⫹NC) transfectants produced a p24-associated Gag product migrating slower than the p24gag(Fig. 2A). This
incom-pletely processed Gag product might correspond to the
CA-⌬NC-p6*, but further experiments were required to test this hypothesis. In agreement with the previous observation (27, 28), the Pr160gag-polor its deletion mutation derivatives could
not form virus particles by themselves, and Gag proteins were not detected in the medium of cells transfected with the GPfs,
[image:3.587.48.279.74.686.2]⌬MA,⌬NC, or⌬(MA⫹NC) (Fig. 2A, lanes 11 to 14). Mature processed Gag proteins were only observed when the wt and mutant GPfs were cotransfected with pGAG (lanes 15 to 18). This indicates that the Gag-Pol of the wt and mutant GPfs was
FIG. 1. HIV-1 Pr160gag-polmutations. (A) Mature Gag protein
do-mains andpol-encoded p6*, PR, and RT domains. The name of each construct is shown at the left. pGAG was constructed by deletion of the polgene. GPfs was engineered by placing thegagandpolin the same
open reading frame via a five-T nucleotide deletion in the overlap region of gag-pol. The other mutants were derived from GPfs by deletion ofgag-polsequences to different extents (see Materials and Methods for specific details). The pGAG and the wt and mutant GPfs were all expressed on the HIVgpt backbone.⌬MA contains a deletion of 105 codons and a replacement of 4 codons in the MA protein. For ⌬NC, sequences encoding the p2 and most of NC were deleted and replaced by two amino acid residues. Combination of the⌬MA and ⌬NC mutations generated the construct⌬(MA⫹NC). In the construct ⌬MHR, 28 codons of the CA domain (CA codon 148 to 177), covering the major homology region (codon 153 to 172) were deleted and replaced by three amino acid residues.⌬CA-1 contains a deletion of 111 codons (codon 81 to 191) and insertion of one amino acid residue into the deleted region.⌬CA-2 is identical to ⌬CA-1 except that it contains a further 3⬘deletion downstream of the N terminus of p2. A further 5⬘deletion in⌬CA-1, deleted from CA codons 81 to 12, yielded ⌬CA-3. Combination of the⌬CA-2 and⌬CA-3 mutations generated the ⌬CA-4, and introduction of the ⌬NC mutation to the ⌬CA-4 yielded⌬(CA⫹NC).⌬(MA⫹2/3 CA) contains a deletion ofgagcoding sequences including the N-terminal two-thirds of CA and most of the MA (deletion from MA codon 17 to CA codon 150). Constructs ⌬(MA⫹CA) and ⌬GAG were derived from combinations of ⌬(MA⫹2/3 CA) with ⌬CA-4 or with ⌬(CA⫹NC), respectively. ⌬(NC⫹PR) has a deletion of sequences encoding the NC, p2, and the PR, and⌬(GAG⫹PR) was derived from a combination of⌬(NC⫹PR) and⌬GAG. (B) Viral DNA sequences and encoded protein sequences of mutated HIV regions. HIV nucleotide positions are indicated, and inserted or altered amino acid residues are shown in boldface.
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 2. Western immunoblotting analysis of HIV proteins expressed and released from cells coexpressing Pr55gagand Pr160gag-pol. 293T cells
were transfected or cotransfected with the designated plasmids. Fifteen micrograms of each plasmid was used for individual transfections, and 10 g of pGAG and 2g for each of the GPfs constructs were used for cotransfection. At 48 h posttransfection, cells and supernatants were collected and prepared for protein analysis as described in Materials and Methods. Cell samples corresponding to 4% of the total samples and supernatant samples corresponding to 50% of the total samples were fractionated by SDS–10% PAGE and electroblotted onto a nitrocellulose filter. HIV Gag proteins were detected with an anti-p24gagmonoclonal antibody (B) or together with an additional anti-p17gagmonoclonal antibody (A, C, and D)
at 1:5,000 dilution, followed by a secondary alkaline phosphatase-conjugated horse anti-mouse antibody at 1:5,000 dilution, and alkaline phosphatase activity was determined. The positions of molecular size markers (Std.) and those of HIV Gag proteins Pr55, p41, p24, and p17 are indicated. The arrow in panel A indicates the p24-associated Gag product produced by both⌬NC and⌬(MA⫹NC) that migrated slower than the p24gag.
on November 8, 2019 by guest
http://jvi.asm.org/
incorporated into virus particles and provided a functional PR to cleave the Pr55gag intransinto mature Gag products. The
levels of Pr55gag and mature p24gag produced by the mutant
GPfs cotransfectants were roughly comparable to those of wt GPfs cotransfectants (lanes 15 to 18). Removal of the MHR alone or together with the adjacent CA sequence from the Pr160gag-poldid not block its incorporation into virus particles,
as the p24gag and p17gag were clearly detected in the media
from cells cotransfected with pGAG plus the⌬MHR or the
⌬CA-1 (Fig. 2B). The absence of p24-associated Gag products in both⌬MHR and⌬CA-1 transfectants (Fig. 2B, lanes 3 and 4) was not due to expression problems but was most likely due to a deletion of the antigen epitopes, since the mature p17gag
could be detected readily when probed with an anti-p17gag
monoclonal antibody (data not shown).
When the GPfs constructs with more extensive deletions in the Gag region were cotransfected with pGAG, p24gag and
p17gagwere still readily detected in the cotransfectant medium
(Fig. 2C). Moreover, one mutant (⌬GAG), in which the whole gagcoding sequence upstream of thegag/poljunction was de-leted except for the N-terminal 15 codons, could still be res-cued into Gag particles at least at some minimal level, since some PR function was present to form to process the Pr55gag
(lane 18). That the band of p24gagwas derived from cleavage of
Pr55gagby the GPfs deletion mutants was evident because it
was not seen either intracellularly or extracellularly when each mutant was expressed alone (data not shown).
Although the N-terminal myristylation signal has been shown to be essential for Pr55gagplasma membrane targeting
and assembly (2, 7, 20), it is dispensable for Pr160gag-pol
incor-poration into particles (22, 27), and it is unknown whether the Myr⫺mutation has any adverse effects on Gag-Pol
incorpora-tion in the presence of large sequence deleincorpora-tions downstream. To address this issue, we introduced the Myr⫺mutation into
the wt and mutant GPfs constructs and transfected 293T cells with each of the resulting plasmids alone or together with pGAG. We found that mature p24gagcould be readily detected
in the medium of cells cotransfected with each of the
nonmyri-stylated mutants plus pGAG (Fig. 2D, lanes 12 to 20). Consis-tent with previous reports (22, 27), cotransfection of the Myr⫺
GPfs and pGAG mainly produced p24gag(lane 12), suggesting
an efficient incorporation of the Myr⫺Pr160gag-pol. Myr⫺ ⌬CA-4, Myr⫺⌬(MA⫹CA), and Myr⫺⌬GAG, with more
ex-tensive deletion mutations, were still capable of being incor-porated into virus particles when cotransfected with pGAG (lanes 17, 19, and 20), since at least some PR was available to form to process the Pr55gag. Cotransfections of pGAG plus the
Myr⫺⌬CA-1, -2, or -3, or the Myr⫺⌬(CA⫹NC) also produced
an extracellular Pr55gag processing pattern similar to that of
their myristylated counterparts (data not shown). These results suggest that removal of the N-terminal myristylation signal has no markedly adverse effects on incorporation of GPfs deletion mutants into virus particles.
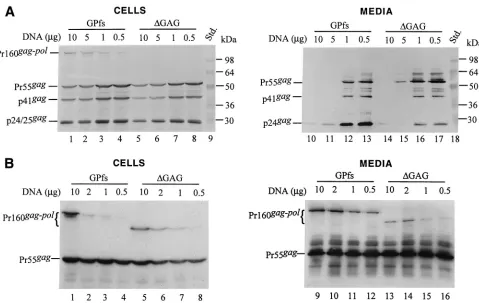
The amount of GPfs plasmid DNA used for cotransfection could significantly affect the level of particle production and Gag-Pol incorporation.It is interesting that Pr160gag-polwith
the gag coding sequence almost deleted (⌬GAG) was still capable of being incorporated into virus particles (Fig. 2C, lane 18, and 2D, lane 20). The results shown in Fig. 2 suggest that Gag-Pol products of the GPfs derivatives could be recruited into virions when cotransfected with pGAG. However, the level of particle-associated p24gagproduced by the
cotransfec-tants cannot reflect the level of incorporated Pr160gag-pol
be-cause the expression level of the Gag-Pol carrying an active PR could affect virus assembly and budding (1, 14, 21, 31). To test this possibility, various quantities (10, 5, 1, and 0.5g) of GPfs or⌬GAG were cotransfected with 10g of pGAG. Figure 3A shows that the level of Pr55gag and p24gag recovered in the
medium was inversely proportional to the quantity of GPfs DNA (lanes 10 to 13) or⌬GAG DNA (lanes 14 to 17) present in the cotransfection mixture. A relatively higher level of p24gag
was detected in the cotransfection medium samples when the DNA ratio of Pr55gag-to-Pr160gag-pol-expressing plasmids was
kept at 10:1 or 20:1 (lanes 12 to 13 and lanes 16 to 17). Both GPfs and ⌬GAG also displayed a relatively higher level of particle-associated RT activity under these cotransfection
con-FIG. 2—Continued.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.587.93.494.74.279.2]ditions. The particle-associated RT activity of ⌬GAG was about 10%⫾ 2% (mean⫾standard deviation) of wt (GPfs) activity in parallel experiments. These data suggest that the Pr160gag-pol would be more efficiently incorporated into virus
particles when the ratio of Pr55gag-to-Pr160gag-pol-expressing
plasmids resembles the physiological expression ratios of Pr55gagand Pr160gag-polin host cells.
Quantification of Gag-Pol incorporation by in vitro RT ac-tivity assay.The results shown above indicated that the⌬GAG with a native PR could be incorporated into virions at about 1/10 of the wt level. However, the extent to which the PR affects particle budding and Gag-Pol incorporation is not known. To avoid the effects of PR activity on particle produc-tion, GPfs and⌬GAG constructs were introduced into a PR-inactivated mutant in which the Asp residue for proteolytic activity had been replaced by an Arg residue (31). When co-transfected with pGAG plasmid DNA at various DNA ratios, the Gag-Pol of GPfs and⌬GAG could be clearly detected in the medium (Fig. 3B). Based on the results of RT assays, the level of incorporated Gag-Pol for⌬GAG was about 20%⫾5% of the wt GPfs level in parallel experiments. We observed repeatedly that the PR-inactivated⌬GAG exhibited a slightly higher level of efficiency of entry into virus particles than its PR-native counterpart when cotransfected with pGAG at a DNA ratio of 1:10 or 1:20. This discrepancy could be due in
part to a differential PR activity between GPfs and⌬GAG, which might result in virus assembly and budding being af-fected to different extents.
The results shown in Fig. 3B indicate that the level of virus-associated Pr160gag-polfor GPfs and⌬GAG increased when the
amounts of DNA used for cotransfection were increased; how-ever, no significant differences in the efficiency of⌬GAG in-corporation were detected among the different DNA ratio-cotransfection experiments when the particle-associated RT activity for ⌬GAG had been normalized to that of GPfs in parallel experiments. Nevertheless, to minimize the effect of protein expression level on the specific incorporation of the Pr160gag-polinto virus particles, each of the GPfs constructs was
cotransfected with pGAG at a DNA ratio of 1:10. Aliquots of cotransfection supernatant samples were analyzed by both Western blotting and in vitro RT assays.
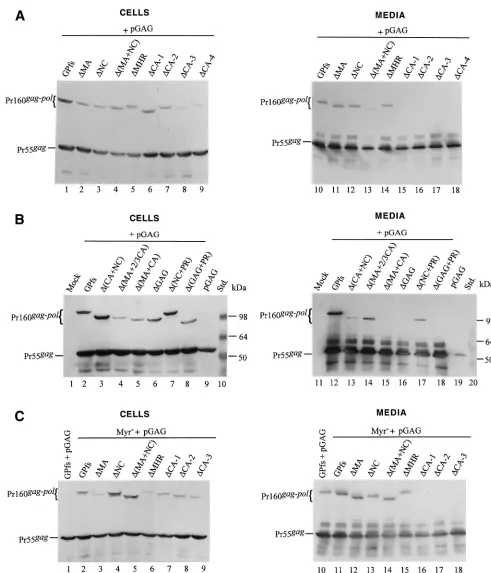
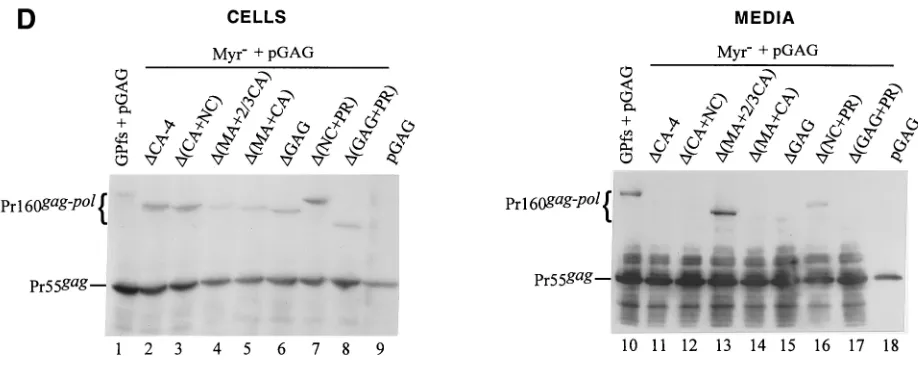
[image:6.587.53.532.70.373.2]As shown in Fig. 4A, Gag-Pol of the mutants⌬MA and⌬NC was released into the medium at a level comparable to that of wt GPfs (lanes 10 to 12) when cotransfected with pGAG, suggesting efficient incorporation of the mutants into virus particles. Similarly, Gag-Pol of⌬(MA⫹NC) was also readily detected in the cotransfection medium sample (lane 13). Sur-prisingly, a significant amount of⌬MHR Gag-Pol was released into the medium on cotransfection with pGAG (lane 14). In contrast, Gag-Pol in the medium was barely detectable when
FIG. 3. Pr160gag-pollacking most of thegagcoding sequence could be incorporated into virus particles. (A) 293T cells were cotransfected with
10g of pGAG plus the indicated amount of GPfs or⌬GAG plasmid DNA. The total DNA in each transfection mixture was kept constant by addition of pBlueScript SK. At 48 h posttransfection, culture supernatants and cells were collected, prepared, and resolved on SDS–10% (A) or SDS–8% (B) PAGE gels. Viral proteins were probed with an anti-p24gagmonoclonal antibody (A) or an HIV-positive human serum (B). The
positions of molecular size markers (Std.) and those of HIV Gag-Pol precursor and Gag proteins Pr55, p41, and p24 are indicated.
on November 8, 2019 by guest
http://jvi.asm.org/
cells were cotransfected with pGAG and mutants lacking the central CA portion including the MHR [⌬CA-1, ⌬CA-2,
⌬CA-3,⌬CA-4,⌬(MA⫹CA), and⌬GAG] (Fig. 4A, lanes 15 to 18, and B, lanes 15 to 16). Nevertheless, these mutant Gag-Pol products in the cotransfection medium could still be detected occasionally, as in the case of⌬(CA⫹NC) (Fig. 4B, lane 13), or they became visible after a longer exposure of the blots (data not shown). Interestingly, cells cotransfected with
⌬(MA⫹2/3 CA) plus pGAG released substantial amounts of Gag-Pol (Fig. 4B, lane 14). It is unclear why the level of intracellular Gag-Pol of⌬(MA⫹2/3 CA) was relatively lower than those of the other mutants (Fig. 4B). One possibility is that the expressed mutants may, under certain circumstances, be rapidly transported and incorporated into virus particles, resulting in a relatively lower intracellular level. Similar phe-nomena were occasionally observed in the case of GPfs (Fig. 3B and 4C and D). Another possibility is that the mutant was simply degraded very rapidly within the cells.
The PR domain is apparently not essential for Gag-Pol in-corporation, since Pr160gag-pollacking the PR and NC domains
was efficiently released from the cotransfectants (Fig. 4B, lane 17). When converted to a nonmyristylated form and cotrans-fected with pGAG, some of the mutants could be detected in the medium by Western blotting, with a profile similar to that of their myristylated counterparts (Fig. 4C and D).
To quantify the particle-associated Pr160gag-pol accurately,
aliquots of identical samples for Western blotting were sub-jected to in vitro RT activity analysis. To optimize RT assay conditions and to ensure that the RT level could reflect the level of incorporated Gag-Pol exactly, resuspended superna-tant pellets derived from GPfs (PR⫺)-plus-pGAG
cotransfec-tion were serially diluted and analyzed for RT activity as de-scribed in Materials and Methods. In one set of experiments, RT reaction mixtures were aliquoted and measured for RT activity at 0, 0.5, 1, 1.5, and 2 h of incubation. We found that RT activities (in counts per minute) increased linearly with time and sample concentrations when the reading was not over 2.4⫻ 105 cpm. Higher counts-per-minute readings could be
obtained by increasing sample concentrations; however, RT activities were no longer increased in a geometric fashion, possibly due either to limitation of substrate or to inhibition of reverse transcription by accumulated RT products, or to both. Thus, to hold the detected RT activity within a linear range, we used only 10% of the recovered supernatant pellets for RT assay.
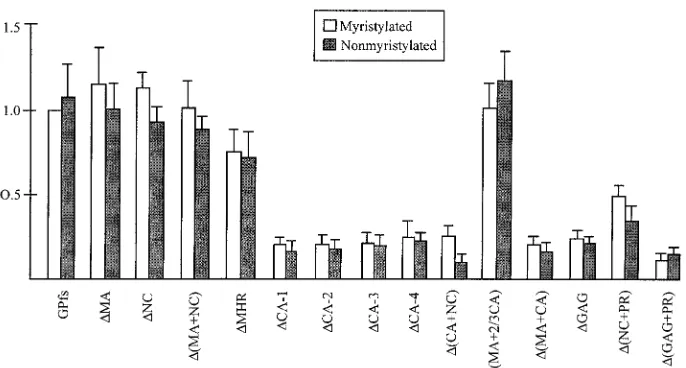
As shown in Fig. 5, mutants⌬MA,⌬NC,⌬(MA⫹NC), and
⌬MHR released remarkable amounts of RT activity, at a level comparable to or over 50% of the wt GPfs level. The relatively lower level of particle-associated Gag-Pol of ⌬(MA⫹NC) shown in Western blotting (Fig. 4A, lane 13) was due to partial sample loss, since a stronger signal comparable to that of wt was observed in repeat experiments. For mutants with a dele-tion involving the MHR and the adjacent C-terminal CA region [⌬CA-1, -2, -3, or -4,⌬(CA⫹NC),⌬(CA⫹NC),⌬(MA⫹CA),
⌬GAG, and ⌬(GAG⫹PR)], the released RT activity levels were all below 30% of that of wt GPfs. This result suggests that removal of the CA central portion including the MHR from the Gag-Pol product may impair its interaction with Pr55gag,
leading to subsequent deficient incorporation of Gag-Pol into virus particles. Conversely, the RT level produced by
⌬(MA⫹2/3 CA) was comparable to that of the wt. This result suggests that the N-terminal two-thirds of the Gag in Pr160 gag-polcould be deleted without significantly affecting the
incorpo-ration of Gag-Pol into virus particles. ⌬(NC⫹PR) released significant amounts of RT activity, indicating that the NC and PR regions are not essential for Gag-Pol incorporation. In accordance with previous studies (22, 27), the level of incor-porated nonmyristylated Pr160gag-pol(Fig. 5) was comparable
to that of the myristylated one. Each of the nonmyristylated constructs, when cotransfected with pGAG, could also release RT activity at a level comparable to or slightly lower than that of its myristylated counterpart, suggesting that the incorpora-tion of the Gag-Pol mutants into virus particles is independent of the N-terminal myristylation signal.
Although some mutants, when expressed alone, occasionally secreted RT activity two- to threefold higher than the back-ground level, significant amounts of RT activity were only detected on cotransfection with pGAG, with an average level at least 20- to 100-fold higher than the background level. This suggests that the RT activity recovered from the cotransfectant medium pellets was particle associated. To confirm that the RT activity released from the cotransfectants was associated with the virus particles, medium pellets from the cotransfection were centrifuged through a 20-to-60% sucrose density gradient as described in Materials and Methods. The results showed that both the RT activity and the Pr55gagpeaked at the same
fraction with a density of 1.16 to 1.18 g/ml (data not shown), suggesting that the mutant was indeed associated with virus particles.
DISCUSSION
We have shown that at least low levels of all the GPfs constructs could be incorporated into virus particles when co-transfected with a Pr55gagexpression plasmid at a DNA ratio
of 1:5, as judged by the presence of PR function that processes Gag and releases p24gagin the medium (Fig. 2). We have also
demonstrated that the levels of released Pr55gag and p24gag
were markedly reduced when the DNA ratio of cotransfected Pr160gag-pol-to-Pr55gagexpression plasmids was increased to 2:1
or 1:1 (Fig. 3A). This is not surprising, since a number of studies have shown that overexpression of the PR or Gag-Pol can suppress virus budding, presumably due to premature cleavage of the Gag precursors (1, 4, 14, 21, 31). This result also reflects the fact that it would be inadequate to assess the incorporation of the Gag-Pol carrying an active PR by using the two-plasmid coexpression system, since the amount of DNA used or the expression ratio of PR could affect the level of released Pr55gagand incorporated Pr160gag-pol. Additionally,
removal of the upstreamgagcoding sequence may affect PR activity (37), resulting in an alteration in the level of virus production.
The results shown in Fig. 4 and 5 suggest that neither MA nor NC is required for efficient incorporation of the Pr160gag-pol
into virus particles. These data agree with a previous study show-ing that replacement of HIV MA or NC with the MLV counter-parts had no detectable effect on the incorporation of chimeric Gag-Pol into HIV particles (9). Efficient incorporation of HIV Gag-Pol into virus particles has been suggested to require the presence of both the HIV CA MHR and the adjacent
on November 8, 2019 by guest
http://jvi.asm.org/
FIG. 4. Expression and incorporation of protease-defective (PR⫺) Pr160gag-poldeletion mutants into Pr55gagparticles. (A and B) 293T cells were
transfected with the plasmid indicated. The HIV PR in all GPfs constructs was either inactivated or truncated. For cotransfection with pGAG, 1 g of each plasmid DNA and 10g of pGAG were used, with addition of 9g of pBlueScript SK to a total amount of 20g of plasmid DNA. Two days after transfection, cells and supernatants were collected for protein analysis as described in Materials and Methods. Samples were fractionated by SDS–8% PAGE and then subjected to Western immunoblot analysis. Membrane-bound HIV proteins were probed with an HIV-positive human serum at a dilution of 1:10,000, followed by a secondary goat antihuman HRP-conjugated antibody at 1:10,000 dilution. (C and D) Incorporation of nonmyristylated Gag-Pol into virus particles. Methodology and plasmids used for analysis were identical to those in panels A and B, except that the GPfs constructs were expressed on a nonmyristylated backbone. The positions of the molecular size markers (Std.) and those of the HIV Pr55gagand Pr160gag-polare indicated.
on November 8, 2019 by guest
http://jvi.asm.org/
terminal CA sequences (9). This proposal has been corrobo-rated by our data showing that both deletion mutants⌬(MA⫹2/3 CA) and ⌬(NC⫹PR), which retain an intact MHR and C-terminal CA domain, were more efficiently incorporated into virus particles than most of the CA deletion mutants. Mutants lacking the MHR could still be rescued into particles to about 50% or over 50% of the wt level, which contrasts with one previous study showing that deletion of the MHR can signifi-cantly impair the incorporation of Pr160gag-polinto virus
parti-cles (28). This discrepancy may be due to different systems employed by different groups. Alternatively, the three foreign residues Arg-Ile-Arg replacing the deleted sequence in our
⌬MHR mutant may somehow contribute in a positive sense to the incorporation of Gag-Pol into virus particles.
Our present results indicate that the MHR and the adjacent C-terminal one-third of CA in the Pr160gag-pol is the most
important region for incorporation of Gag-Pol into virus par-ticles. However, all of the capsid deletion mutants could re-lease RT activity at least 20-fold higher than the background level when cotransfected with pGAG, at a minimum level of about 10% of wt level (Fig. 5). Moreover, a deletion that removed all but the 15 N-terminalgag codons did not com-pletely prevent the incorporation of the encoded Gag-Pol into virus particles (Fig. 3). This suggests that thegag coding se-quence in Pr160gag-polis not absolutely required for its
incor-poration into virus particles. It is possible that the expression levels of Gag and Gag-Pol may affect the specific interaction between the two molecules. There is also a possibility that some of the Gag-Pol deletion mutants may interfere with the assembly of the coexpressed Pr55gag, resulting in a reduction in
virus budding and Gag-Pol incorporation. To avoid these ef-fects induced by the overexpressed Gag-Pol, each of the GPfs constructs was cotransfected with pGAG at a DNA ratio of 1:10. However, efficient uptake of the two plasmids by 293T cells may occur under some circumstances, which could also reach a high expression level. Thus, minor but deleterious effects of the mutations may be masked by using a high-level expression system.
HIV RT and IN have been shown to be capable of being incorporated into virus particles independent of Pr160gag-pol
expression (5, 35, 36). In these studies, the RT or IN was fused to Vpr, and the incorporation of fusions Vpr-RT and Vpr-IN into virus particles presumably depends on interaction of the N-terminal Vpr with Pr55gag (24). It is possible that the
N-terminal 15 residues may be responsible for rescue of the mutant [⌬GAG or ⌬(GAG⫹PR)] into particles via putative interactions with the Pr55gag. This is unlikely, however, because
HIV Gag–-galactosidase (HIV GBG) fusion proteins, con-structed by fusion of the-Gal gene to HIV gag at the N-terminal 15th codon or at the C terminus of MA, were ex-cluded from HIV particles (32), suggesting that the N-terminal 15 residues are not involved in the incorporation of Gag-Pol into virus particles. In contrast, HIV GBG (analogous to the HIV Gag-Pol) fused at the end of the NC domain was effi-ciently incorporated into virus particles (32). Perhaps the HIV Pol possesses some uncharacterized property distinct from that of the-Gal. In support of this hypothesis, it has been dem-onstrated clearly that the HIV GBG (32), not HIV Pr160gag-pol,
is severely impaired in its incorporation into virions upon re-moval of the N-terminal myristylation signal. Thus, it is con-ceivable that HIV Pol may have as-yet-undefined functions associated with it which are involved in the process of Gag-Pol transport and incorporation.
There is no direct evidence suggesting that an interaction occurs between the HIV Gag and Pol. However, the human foamy viruspolgene is encoded as Pro-Pol, and the Pol prod-ucts can be incorporated into virus particles without the for-mation of a Gag-Pol (16). One more recent study also dem-onstrated that the free MLV Pol could be incorporated into virus particles when MLV Pol and MLV Gag were coexpressed from separate plasmids (3). These data lead to the speculation that there may be some unidentified conserved regions residing in the retroviral Pol, which may contribute to facilitating the incorporation of Pol into virions. The HIV PR does not appear to play a crucial role in the incorporation of Pol into virus particles, since significant amounts of particle-associated
[image:9.587.59.518.73.257.2]⌬(NC⫹PR) could be released into the medium on cotransfec-tion with pGAG. It would be of interest to test whether the GPfs deletion mutants can complement RT- or IN-deficient HIV virions intransto produce infectious virus particles.
FIG. 4—Continued.
on November 8, 2019 by guest
http://jvi.asm.org/
ACKNOWLEDGMENTS
We thank C.-C. Chang for technical assistance and Steve S.-L. Chen for helpful discussions and suggestions. The hybridoma clone 183 H12-5C and HIV-positive human serum were provided by the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID. This work was supported by grant NSC89-2320-B-010-106 from the National Science Council and by grant DOH89-DC-1014 from the Department of Health, Taiwan, Republic of China.
REFERENCES
1.Arrigo, S. J., and K. Huffman.1995. Potent inhibition of human immuno-deficiency virus type 1 (HIV-1) replication by inducible expression of HIV-1 PR multimers. J. Virol.69:5988–5994.
2.Bryant, M., and L. Ratner.1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA
87:523–527.
3.Buchschacher, G. L., Jr., L. Yu, F. Murai, T. Friedmann, and A. Miyano-hara.1999. Association of murine leukemia virus Pol with virions, indepen-dent of Gag-Pol expression. J. Virol.73:9632–9637.
4.Burstein, H., D. Bizub, and A. M. Skalka.1991. Assembly and processing of avian retroviral gag polyproteins containing linked protease dimers. J. Virol.
64:6156–6172.
5.Fletcher, T. M., III, M. A. Soares, S. McPhearson, H. Hui, M. Wiskerchen, M. A. Muesing, G. M. Sham, A. D. Leavitt, J. D. Boeke, and B. H. Hahn.
1997. Complementation of integrase function in HIV-1 virions. EMBO J.
16:5123–5138.
6.Freed, E. O.1998. HIV Gag proteins: diverse functions in the virus life cycle. Virology251:1–15.
7.Gottlinger, H. G., J. G. Sodroski, and W. A. Haseltine.1989. Role of capsid precursor processing and myristylation in morphogenesis and infectivity of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA86:5781–5785. 8.Henderson, L. E., M. A. Bowers, R. C. Sowder II, S. A. Serabyn, D. G. Johnson, J. W. Bess, Jr., L. O. Arthur, D. K. Bryant, and C. Fenselau.1992. Gag proteins of the highly replicative MN strain of human immunodeficiency virus type 1: posttranslational modifications, proteolytic processing, and complete amino acid sequences. J. Virol.66:1856–1865.
9.Huang, M., and M. A. Martin.1997. Incorporation of Pr160gag-polinto virus
particles requires the presence of both the major homology region and adjacent C-terminal capsid sequences within the Gag-Pol polyprotein. J. Vi-rol.71:4472–4478.
10.Hunter, E.1994. Macromolecular interactions in the assembly of HIV and other retroviruses. Semin. Virol.5:71–83.
11.Jacks, T., M. D. Power, F. R. Masiarz, P. A. Luciw, P. J. Barr, and H. E.
Varmus.1988. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature (London)331:280–283.
12.Kaplan, A. H., M. Manchester, and R. Swanstorm.1994. The activity of the protease of human immunodeficiency virus type 1 is initiated at the mem-brane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J. Virol.68:6782–6786. 13.Kohl, N. E., E. A. Emini, W. E. Schleif, L. J. Davis, J. C. Heimbach, R. A. F.
Dixon, E. M. Scolnick, and I. S. Sigal.1988. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA
85:4686–4690.
14.Krausslich, H.-G.1991. Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc. Natl. Acad. Sci. USA88:3213–3217.
15.Leis, J., D. Baltimore, J. B. Bishop, J. Coffin, E. Fleissner, S. P. Goff, S. Oroszlan, H. Robinson, A. M. Skalka, H. M. Temin, and V. Vogt.1988. Standardized and simplified nomenclature for proteins common to all ret-roviruses. J. Virol.62:1808–1809.
16.Lochelt, M., and R. M. Flugel.1996. The human foamy virus pol gene is expressed as a Pro-Pol polyprotein and not as a Gag-Pol protein. J. Virol.
70:1033–1040.
17.Mammano, F., A. Ohagen, S. Hoglund, and H. G. Gottlinger.1994. Role of major homology region of human immunodeficiency virus type 1 in virion morphogenesis. J. Virol.68:4927–4936.
18.Mervis, R. J., N. Ahmad, E. P. Lillehoj, M. G. Raum, F. H. R. Salazar, H. W. Chan, and S. Venkatesan.1988. Thegaggene products of human immuno-deficiency virus type 1: alignment within thegagopen reading frame, iden-tification of posttranslation modifications, and evidence for alternativegag
precursors. J. Virol.62:3993–4002.
19.Page, K. A., N. R. Landau, and D. R. Littman.1990. Construction and use of a human immunodeficiency virus: vector for analysis of virus infectivity. J. Virol.64:5270–5276.
20.Pal, R., M. S. Reitz, Jr., E. Tschanchler, R. C. Gallo, M. G. Sarngadharan, and F. D. M. Veronese.1990. Myristylation ofgagproteins of HIV-1 plays an important role in virus assembly. AIDS Res. Hum. Retrovir.6:721–730. 21.Park, J., and C. D. Morrow.1991. Overexpression of the gag-pol precursor
from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J. Virol.
65:5111–5117.
22.Park, J., and C. D. Morrow.1992. The nonmyristylated Pr160gag-pol
polypro-tein of human immunodeficiency virus type 1 interacts with Pr55gagand is
incorporated into virus-like particles. J. Virol.66:6304–6313.
23.Partin, K., H. G. Krausslich, L. Ehrlich, E. Wimmer, and C. Carter.1990. Mutational analysis of a native substrate of the human immunodeficiency virus type 1 proteinase. J. Virol.64:3938–3947.
[image:10.587.122.465.73.257.2]24.Paxton, W., R. I. Connor, and N. R. Landau.1993. Incorporation of Vpr into
FIG. 5. Release of HIV RT activity from cells cotransfected with the Pr160gag-poldeletion mutants and pGAG. 293T cells were cotransfected
with 1g of each PR-defective GPfs construct and 10g of pGAG. At 48 h posttransfection, supernatants were collected, filtered, and pelleted through a 20% sucrose cushion as described in Materials and Methods. Viral pellets were suspended in TSE, and about 40% of the suspensions were subjected to Western immunoblot analysis. The remaining suspensions were further diluted by addition of TSE and were then aliquoted for in vitro RT assays. RT activities (open bars) in each experiment were normalized to that obtained with GPfs plus pGAG, which was set at 100. Relative levels of released RT activity for each nonmyristylated GPfs deletion mutant were determined (shaded bars) by dividing the mutant GPfs RT activity by that of wt GPfs in parallel experiments. Values for each construct are derived from at least three independent experiments. Error bars indicate standard deviations.
on November 8, 2019 by guest
http://jvi.asm.org/
human immunodeficiency virus type 1 virions: requirement for the p6 region of gag and mutational analysis. J. Virol.67:7229–7237.
25.Peng, C., B. K. Ho, T. W. Chang, and N. T. Chang.1989. Role of human immunodeficiency virus type 1-specific protease in core particle maturation and viral infectivity. J. Virol.63:2550–2556.
26.Rose, J. R., L. M. Base, and C. S. Craik.1995. Defining the level of human immunodeficiency virus type 1 (HIV-1) protease activity required for HIV-1 particle maturation and infectivity. J. Virol.69:2751–2758.
27.Smith, A. J., N. Srinivasakumar, M.-L. Hammarskjold, and D. Rekosh.
1993. Requirements for the incorporation of Pr160gag-polfrom human
im-munodeficiency virus type 1 into virus-like particles. J. Virol.67:2266–2275. 28.Srinivasakumar, N., M. L. Hammarskjold, and D. Rekosh.1995. Charac-terization of deletion mutations in the capsid region of human immunode-ficiency virus type 1 that affect particle formation and Gag-Pol precursor incorporation. J. Virol.69:6106–6114.
29.Wang, C.-T., and E. Barklis.1993. Assembly, processing, and infectivity of human immunodeficiency virus type 1gagmutants. J. Virol.67:4264–4273. 30.Wang, C.-T., H.-Y. Lai, and J.-J. Li. 1998. Analysis of minimal human immunodeficiency virus type 1gagcoding sequences capable of virus-like particle assembly and release. J. Virol.72:7950–7959.
31.Wang, C.-T., Y.-C. Chou, and C.-C. Chiang.2000. Assembly and processing
of HIV gag mutants containing a partial replacement of the matrix domain by the viral protease domain. J. Virol.74:3418–3422.
32.Wang, C.-T., J. Stegeman-Olsen, Y. Zhang, and E. Barklis.1994. Assembly of HIV gag--galactosidase fusion proteins into virus particles. Virology
200:524–534.
33.Wang, C.-T., Y. Zhang, J. McDermott, and E. Barklis.1993. Conditional infectivity of a human immunodeficiency virus matrix domain deletion mu-tant. J. Virol.67:7067–7076.
34.Wills, J. W., and R. C. Craven.1991. Form, function, and use of retroviralgag
proteins. AIDS5:639–654.
35.Wu, X., H. Liu, H. Xiao, J. A. Conway, E. Hunter, and J. C. Kappes.1997. Functional RT and IN incorporated into HIV-1 particles independently of the Gag/Pol precursor protein. EMBO J.16:5113–5122.
36.Wu, X., H. Liu, H. Xiao, J. Kim, P. Seshaiah, G. Natsoulis, J. D. Boeke, B. H. Hahn, and J. C. Kappes.1995. Targeting foreign proteins to human immu-nodeficiency virus particles via fusion with Vpr and Vpx. J. Virol.69:3389– 3398.
37.Zybarth, G., and C. Carter.1995. Domains upstream of the protease (PR) in human immunodeficiency virus type 1 Gag-Pol influence PR autoprocessing. J. Virol.69:3878–3884.