JOURNAL OFVIROLOGY, 0022-538X/97/$04.0010
Mar. 1997, p. 2233–2240 Vol. 71, No. 3
Copyrightq1997, American Society for Microbiology
Accumulation of Defective Viral Genomes in Peripheral Blood
Mononuclear Cells of Human Immunodeficiency
Virus Type 1-Infected Individuals
GISELLE SANCHEZ, XIAOYUAN XU,† JEAN-CLAUDE CHERMANN,ANDIVAN HIRSCH*
INSERM U322, Unite´ de Recherches sur les Re´trovirus et Maladies Associe´es, Parc Scientifique et Technologique de Luminy, BP33, 13273 Marseille ce´dex 09, France
Received 8 July 1996/Accepted 9 December 1996
Human immunodeficiency virus type 1 (HIV-1) genomes present in peripheral blood mononuclear cells (PBMCs) of infected persons or in lymphocytes infected in vitro were studied by long-distance PCR (LD-PCR) using primers localized in the HIV-1 long terminal repeats. The full-length 9-kb DNA was the only LD-PCR product obtained in peripheral and cord blood lymphocytes from seronegative donors infected in vitro. However, a high proportion (27% to 66%) of distinct populations of extensively deleted HIV-1 genomes of variable size was detected in PBMCs of 15 of 16 HIV-1-infected persons. Physical mapping of defective genomes showed that the frequency of deletions is proportional to their proximity to the central part of HIV-1 genome, which is consistent with a deletion mechanism involving a single polymerase jump during reverse transcrip-tion. Sequencing of deletion junctions revealed the presence of short direct repeats of three or four nucleotides. The number of defective HIV-1 genomes decreased after in vitro activation of PBMCs. Persistence of full-length and deleted genomes in in vitro activated PBMCs correlated with isolation of an infectious virus. Our results represent the first quantitative assessment of intragenomic rearrangements in HIV-1 genomes in PBMCs of infected persons and demonstrate that, in contrast to in vitro infection, defective genomes accumulate in PBMCs of infected persons.
The accumulation of cell-associated defective viruses is gen-erally the consequence of frequent sequential passages of virus at high multiplicities of infection (30) and of the substantially longer lifetime of nonproductively compared to productively infected cells. Production of large amounts of human immu-nodeficiency virus type 1 (HIV-1) particles and rapid turnover of HIV-1-infected cells and virions (17, 27, 31) form a natural base for appearance of defective HIV-1 genomes. Besides a high local multiplicity of infection in lymphoreticular tissue, where the majority of virus population is believed to replicate (13, 24), the immune system rapidly depletes productively in-fected cells (17, 31) and selects cells inin-fected with defective viruses that do not express the HIV-1 genome. The occurrence of defective retroviruses is related to the frequency of point
mutations, estimated to range from 1025to 1024per
nucleo-tide per cycle (reviewed in references 6 and 7), and in-tragenomic rearrangements, including deletions, duplications, and inversions. The frequency of intragenomic rearrangements is supposed to be high but remains unknown (6, 7) since all the data have been derived from cloning and sequencing limited numbers of proviruses or their fragments. In lentiviruses, re-peated attempts to isolate full-length genomes yielded exten-sively deleted DNA fragments of HIV-1 (18), human adult T-cell leukemia virus type 1 (16), caprine arthritis encephalitis virus (14), and visna virus (21).
The number of peripheral blood mononuclear cells (PBMCs) containing HIV-1 DNA detected by PCR greatly exceeds the number of cells producing infectious HIV-1 in end point dilu-tion cultures (4, 8, 9). This quantitative difference between the total virus load and the amount of infectious virus could at
least partially be explained by the existence of defective virus particles. To determine the proportion of defective viruses in the HIV-1 population is actually very difficult because of un-known sensitivity of the indicator cell systems used for infec-tivity assays in vitro and because of obscuring epigenetic fac-tors such as the presence of neutralizing antibodies in the host plasma and of cytotoxic cells in PBMCs. In this study, we have used long-distance (LD) PCR to amplify HIV-1 sequences located between both long terminal repeats (LTR) present in PBMCs infected in vitro and in PBMCs from infected individ-uals. This experimental approach permitted us to observe the entire population of HIV-1 genomes. A high percentage of extensively deleted defective viral genomes was detected in PBMCs from infected persons but not after in vitro infection. The population complexity of defective HIV-1 genomes tended to decrease after in vitro activation of PBMCs.
MATERIALS AND METHODS
Subjects.A total of 16 HIV-1-seropositive informed consenting individuals, aged from 21 to 66 years, treated at Hoˆpital de la Conception and at Hoˆpital St. Marguerite, Marseilles, France, were studied. The general and virological char-acteristics of patients are shown in Table 1. Ten of them had no detectable infectious virus in their PBMCs and two, SUFDE and QEVNE, were persons with a long-term (.10 years) nonprogressive HIV-1 infection. PBMCs of two persons, NUSEP and HETNE, were obtained at two occasions separated by about 1 year.
Cell culture and virus isolation.PBMCs of HIV-seropositive donors were separated on Ficoll-Hypaque gradients. Monocyte-depleted peripheral blood lymphocytes (PBL) were activated with phytohemagglutinin (Difco; diluted 500 times) for a period of 3 days, and virus production was monitored as described by Barre´-Sinoussi et al. (1).
Virus infection of tissue cultures in vitro.HIV-1 PAR, a strain isolated from the cerebrospinal fluid of an HIV-1-seropositive man suffering from acute en-cephalopathy (15), was prepared on cord blood lymphocytes (CBL) from a healthy donor. HIV-1 LAV, a subtype B prototype virus (1), and HIV-1 NDK, a highly cytopathic subtype D Zairian virus (12), were propagated in PBL from a healthy HIV-1-negative donor, in the permissive T-cell line MT4 or in the intestinal line HT29 (11).
* Corresponding author. Phone: (33)4 91 82 75 40. Fax: (33)4 91 41 92 50.
† Permanent address: Division of Infectious Diseases, Lanchou Medical College, Lanchou, 730000 China.
2233
on November 9, 2019 by guest
http://jvi.asm.org/
PCR amplification of viral DNA.For PCR analysis, 53106
cells were lysed in 2 ml of lysis buffer containing 100 mM NaCl, 10 mM Tris-HCl (pH 7.6), 10 mM EDTA, and 1% Sarcosyl and incubated overnight at 568C in the presence of 100 mg of proteinase K per ml. After phenol extraction and ethanol precipitation, DNA was dissolved in water and its concentration was determined by optical density at 260 nm. To amplify HIV-1 provirus, DNA samples (1mg) were subjected to LD-PCR with 2.6 U of enzyme (Expand Long Template PCR System, Boehringer Mannheim) in a 50-ml reaction mixture containing 50 mM
Tris-HCl (pH 9.2), 14 mM (NH4)2SO4, 1.75 mM MgCl2, 350mM each
[image:2.612.153.462.73.293.2]de-oxynucleoside triphosphate, and 15 pmol each of LTR(U5) upstream primer, 59-GTCTGTTGTGTGACTCTGGT-39 (nucleotides [nt] 112 to 131) and LTR(R) downstream primer, 59-GAGGCTTAAGCAGTGGGTTC-39(nt 9185 to 9204), described previously by Bukrinsky et al. (3), (Fig. 1); all annotated positions of oligonucleotide primers and probes in this work correspond to the sequence of HIV-1 LAI (22). Reaction mixtures were then submitted to 25 amplification cycles comprising a denaturing step of 10 s at 948C, a primer-FIG. 1. HIV-1 specific oligonucleotide primers and probes for PCR analysis. The primers and their orientation are indicated by arrows. Numbers in parentheses correspond to nucleotide positions in the HIV-1 LAI sequence (22). LTR-derived oligonucleotides are present in both 59and 39LTRs. In consequence, LTR(R) (nt 9185 to 9204) corresponds also to nucleotide positions 53 to 72. Primers LTR(U5)-LTR(R) and 59NCS-nefwere used in the LD-PCR, and primersgagA1-gagA2 were used in the classical PCR for quantification of HIV-1 copies. Oligonucleotides 59NCS andnefcontaining restriction sequencesBssHII andXhoI, respectively, were used as primers or probes. The figure is not drawn to scale. (A) Molecular organization of HIV-1 provirus. (B) LTR-LTR junction of the two-LTR circle.
TABLE 1. Characteristics of HIV-1 infected individuals
DNA
no.a Code Age Sexb
Detection of HIV positivity (y)
Antiretroviral therapyc
No. of CD4 cells perml
CD4 cell declined
gagcopies/mg of DNA/105PBMCe
Virus isolation (PBL)
1 NUSEP 31 M 1993 1 41 1 NDf 1
2 IVDNE 39 F 1989 2 558 2 60 2
3 NIDIW 30 F 1990 1 199 2 60 1
4 WOEKI 54 M 1985 2 450 2 3 1
5 FOCEP 35 F 1993 2 328 2 ND 2
6 SUFDE 32 F 1985 2 610 2 25 2g
7 TEMAE 21 M 1992 2 1,075 2 ND 2
8 QEVNE 34 M 1985 2 813 2 60 2
9 HETNE 45 F 1991 2 433 2 30 2
10 FYNWO 26 M 1984 1 23 1 3 1
11 CESMO 34 M ND 1 87 1 ND 2
12 DJOUM 31 M 1985 1 2 1 50 1
13 TEMNO 49 M 1990 1 121 1 20 2
14 HETNE 45 F 1991 2 433 2 2 2
15 FESFU 41 F 1989 2 706 2 300 2g
16 NIPDJ 38 F 1991 2 234 2 300 2
17 NUSEP 31 M 1993 1 26 1 150 1
18 DECNO 56 M ND 1 48 1 100 1
aDNA samples were used in three types of experiments: no. 1 to 10 were used in physical mapping of defective proviruses; no. 7 and 12 to 18 in the study of defective
proviruses in in vitro activated PBMCs; and no. 5, 11, and 16 in the cloning of defective proviruses.
bM, male; F, female.
c1, mono- or alternating chemotherapy;2, no chemotherapy.
d1,.50% regular decline of CD4 cells during the last year;2, no significant change during the last year.
eNumber of copies of HIV-1gagper 1mg of DNA extracted from PBMCs of infected individuals was determined by semiquantitative PCR as described in Materials
and Methods. Onemg of DNA is contained in about 105PBMCs.
fND, not determined.
gPositive after cocultivation with CBL.
on November 9, 2019 by guest
http://jvi.asm.org/
annealing step of 30 s at 648C, and a primer extension step of 8 min at 688C. The primer extension step of the last 15 amplification cycles was gradually extended by 20 s at each cycle. A final step of 7 min at 728C was performed. To increase sensitivity, 10 additional amplification cycles (without time extension) were per-formed with 1.75 U of enzyme. At least three aliquots of each DNA sample were amplified in parallel. In addition to the whole internal sequence of HIV-1 provirus, LTR(U5) and LTR(R) primers span the LTR-LTR junction of the two-LTR circle (Fig. 1B) and may generate a shorter amplification product (595 nt in the case of HIV-1 LAI) (3).
The number of input target molecules for LD-PCR in DNA extracted from PBMCs of infected individuals was estimated by semiquantitative PCR of the HIV-1gaggene (Table 1). Decadic dilutions of DNA were assayed in a classical PCR reaction withTaqpolymerase andgagA1 primer, 59-GATTTAAACACC ATGCTAAACACAGTGG-39(nt 882 to 909, sense), andgagA2 primer, 59-TT TGGTCCCTGTCTTATGTCCAAAATGC-39(nt 1177 to 1204, antisense), for the presence of a 322-nt amplification product (29). Decadic dilutions of the plasmid pNL4-3 standard DNA were amplified in the parallel.
To avoid potential PCR product contamination, all stages of the PCR were carried out in a PCR room, distinct from the laboratory where the amplification products were analyzed. HIV DNA-bearing plasmids, including those used as positive controls, were manipulated strictly outside the PCR room.
Analysis of PCR products.Southern blots were hybridized with the32
P-labeled oligonucleotide probes 59NCS, 59-TTGCTGAAGCGCGCACGGCAA-39 (nt 249 to 269, sense,NDK1) (10);gag, 59-TTTGTTCCTGAAGGGTACTAGTAG TTCC-39(nt 1047 to 1074, antisense,gagDTRA) (29);pol, 59-TGCCCACACTA ATGATGTAAAACAATTA-39, (nt 3208 to 3235, sense,polA1) (29);tat, 59-TT GGGTGTCGACATAGCAGAATAGGCGT-39(nt 5361 to 5388, sense,tatA2) (29); env, 59-ATCCTCAGGAGGGGACCCAGAAATT-39 (nt 6906 to 6930, sense); nef, 59-TTTCCAGGTCTCGAGATACTGCTCC-39 (nt 8480 to 8504, antisense); and LTR(U3), 59-CTACAAGGGACTTTCCGCTGG-39(nt 9022 to 9042, sense).
Cloning of HIV-1 DNA fragments.LD-PCR products were separated in aga-rose gel, electroeluted, and reamplified by LD-PCR using the nested primers 59NCS andnefto obtain a sufficient quantity of DNA for molecular cloning. The primers 59NCS andnefcontainBssHII andXhoI restriction sites, respectively, conserved in HIV-1 nucleic acid sequence database (22). The obtained LD-PCR products were gel purified, digested withBssHII andXhoI to generate cohesive ends, and inserted by homologous recombination into plasmid pNL4-3 (10, 11).
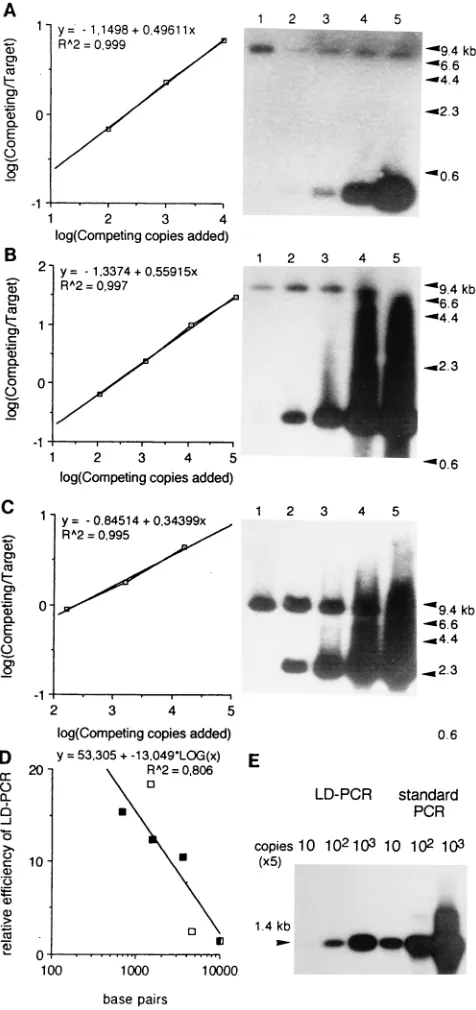
Quantitation of deleted genomes.The relative efficiency of LD-PCR for DNA fragments of different size was determined by a modification of quantitative competitive PCR (27). Different amounts of cloned HIV-1 DNA fragments were amplified with a constant amount of plasmid pNL4-3 by using the set of primers LTR(U5) and LTR(R) as well as the primers 59NCS andnef. LD-PCR products were separated in an agarose gel, blotted, and hybridized with the32
P-labeledgag probe. The intensity of radioactive signal in obtained bands was determined by densitometric tracing of autoradiograms by using a computer-based video image system (The Imager; Oncor Appligene, Inc.) and matched custom software. The log10of intensity ratios of the bands corresponding to the deleted templates over
the bands corresponding to the full-length templates were plotted against the log10of the number of deleted molecules introduced in the LD-PCR reaction.
An intensity ratio of 1 (log10of intensity ratio50) corresponds to the original
amount of deleted molecules that is necessary to obtain the same number of copies as with the complete molecules.
RESULTS
Full-length HIV-1 DNA is the major LD-PCR product in primary lymphocytes infected in vitro. DNA extracted from PBL and CBL infected in vitro with HIV-1 LAV and PAR, respectively, was amplified with oligonucleotide primers (Fig. 1) located in the LTR(U5) region (upstream primer) and in the LTR(R) region (downstream primer) in order to span the whole internal sequence of the provirus, enclosed by truncated LTRs. In both cases, the expected 9-kb band was the major LD-PCR product (Fig. 2). Similar results were obtained with DNA extracted from the T-cell line MT4 infected with HIV-1 LAV and NDK and from the intestinal cell line HT29 persis-tently infected with HIV-1 NDK or directly with the plasmid
pNL4-3 prepared in Escherichia coli. The minor additional
bands observed in the amplified DNA extracted from HIV-1-infected cells (Fig. 2, lines 1, 4, and 5) were not found in the amplification product of plasmid pNL4-3. They may represent LD-PCR product of aberrant HIV-1 DNA formed during HIV-1 replication in vitro. Similar results demonstrating the recovery of full-length HIV-1 provirus have recently been de-scribed by Salminen et al. (28). The presence of a 9-kb band as
the only or major LD-PCR product suggests that, under the reaction conditions used, the polymerase system amplified faithfully its template consisting predominantly of full-size HIV-1 DNA.
Both full-length and extensively deleted HIV-1 genomes are present in PBMCs of the majority of infected persons. LD-PCR was then used to analyze HIV-1 DNA present in PBMCs of infected persons at different levels of development of HIV-1 infection (Table 1). Southern blot hybridization of LD-PCR
products withgag,pol,tat, and nefprobes revealed the
pres-ence of the full-length HIV-1 DNA in 8 of 10 individuals studied (Fig. 3). LD-PCR failed to reveal the full-length HIV-1 DNA in one person with progressive infection (FYNWO) and detected only a shorter 7-kb product in one nonprogressor (QEVNE) (Fig. 3, lanes 8 and 10). LD-PCR products of all individuals but one (SUFDE) (Fig. 3, line 6) yielded 2 to 7 discrete bands in the range from 600 nt to full-length HIV-1 DNA. A control LD-PCR of HIV-1 PAR DNA extracted at the peak of virus production from CBL infected in vitro and analyzed in parallel to tested samples produced a single band of 9 kb. The full-length HIV-1 DNA was also a major ampli-fication product of DNA extracted from phytohemagglutinin-activated PBMCs 4 days postinfection with HIV-1 LAV (Fig. 2). This indicates that shorter HIV-1 DNA fragments present in PBMCs of HIV-1 infected persons are not LD-PCR artifacts and suggests that a high proportion of extensively deleted proviruses appears to be present in vivo.
The frequency of deletions increases with their proximity to the center of the HIV-1 genome.The use of oligonucleotide probes derived from seven distant regions spread over all the HIV-1 genome permitted us to determine HIV-1 DNA se-quences present in defective proviruses and to construct their physical map (Fig. 4). Each probe was checked for the absence of significant sequence homology with the rest of the HIV-1
genome. The probe LTR(U3) can detect both the 39end of the
HIV-1 genome (Fig. 1A) and the LTR-LTR junction of the two-LTR circle (0.6-kb fragment, Fig. 1B). Thus, LD-PCR products of 0.6 kb detected only by the probe LTR(U3) could correspond to the two-LTR circles rather than to deleted pro-viruses. However, DNA bands with this characteristic
repre-FIG. 2. HIV-1 DNA present in primary cells and cell lines infected in vitro with HIV-1. One microgram of DNA, equivalent to 105
human cells, was am-plified by LD-PCR. The resulting product was separated by agarose gel electro-phoresis, transferred to a nylon membrane, and probed by32
P-labeledgag oli-gonucleotide. Lanes 1 to 3 and 4 to 6: LD-PCR of decatic dilutions of DNA extracted from PBL and CBL 4 days postinfection with HIV-1 LAV and PAR, respectively. Lanes 10 to 12: LD-PCR of decadic dilutions of purified pNL4-3. The total amount of DNA was adjusted to 1mg by DNA extracted from PBL cells from a healthy donor.
VOL. 71, 1997 DEFECTIVE HIV-1 GENOMES IN PBMCs 2235
on November 9, 2019 by guest
http://jvi.asm.org/
sented only 10% of the total number of bands detected by the LTR(U3) probe (LTR(U3) column, Fig. 4B).
The majority of bands hybridized with probes specific for both extremities of the HIV-1 provirus. In most cases the deleted sequences were adjacent one to another suggesting that most defective genomes result from a single deletion event. However, signals from the most centrally located probes progressively disappeared with decreasing molecular weight of defective DNA fragments. The implications of these findings appear more clearly in a histogram (Fig. 4C) in which the total number of bands that hybridized with a given HIV-1 probe was plotted as a function of the probe location in the genome. This revealed a nonrandom pattern of deletions: it appears that the frequency of deletions is proportional to their proximity to the
center of the HIV-1 genome, around thetatgene.
Short direct repeats are present at the deletion junctions.
To analyze deletion junctions in further detail, LD-PCR prod-ucts from DNAs of individuals FOCEP, CESMO, and NIPDJ
were cloned into BssHII and XhoI sites of plasmid pNL4-3.
Sequencing of the resulting plasmids pDCESMO, pDFOCEP, and
pDNIPDJwas performed from 59NCS to thenefgene, across the
deleted regions (Fig. 5). Comparison with the HIV-1 LAI sequence identified in all three plasmids simple deletions with characteristic short direct repeats at the deletion junction. The
extent of deletions corresponded to 7759 (pDCESMO), 7014
(pDFOCEP), and 5394 (pDNIPDJ) nucleotides, and the sizes of
direct repeats were of three (pDFOCEP and pDNIPDJ) or four
(pDCESMO) nucleotides. The remainder of the nucleotide
se-quences did not differ significantly from the HIV-1 LAI ge-nome.
Deleted viruses represent a high proportion of the HIV-1 DNA in PBMCs of infected patients.The efficiency of the PCR
technique varies with the size of the DNA template. To quan-titate the proportion of deleted and complete HIV-1 genomes in PBMCs of HIV-1-infected persons, it was necessary to de-termine the efficiency of LD-PCR for DNA fragments of dif-ferent sizes. A reconstruction experiment was carried out for
this purpose, in which plasmids pDFOCEP, pDCESMO, and
pDNIPDJwere mixed in different proportions with a constant
amount of plasmid pNL4-3 (Fig. 6A, B, and C). All plasmids
were linearized by digestion with AatII in the vectorial part
before LD-PCR. Linearization precluded amplification of the vectorial part of the plasmid by primers localized in LTR sequences (Fig. 1) and a possible bias in amplification of the mixture of circular and linear forms present in plasmid prep-arations. The relative efficiency of LD-PCR for templates of different sizes was determined as described previously (27) and plotted against size of deleted genomes (Fig. 6D). Our results show that LD-PCR of different deleted fragments is competi-tive (Fig. 6A, B, and C), that its outcome is proportional to the original number of deleted molecules and that its efficiency is a logarithmic function of the fragment size (Fig. 6D). This function (Fig. 6D) provides the correction factor required for quantitation of HIV-1-deleted genomes of different sizes.
[image:4.612.127.485.64.350.2]PBMCs of 3 of 10 infected persons presented only deleted or rearranged genomes (Fig. 4B, individuals 8, 9, and 10) and in one case (Fig. 4B, no. 6) only the full-length genome was detected. Quantification of deleted genomes in six remaining individuals was performed by densitometric tracing of autora-diograms of Southern blots hybridized with the probe LTR(U3) by using correction factors shown in Fig. 6D. LTR(U3) probe was used because it hybridized with more than 90% of all defective genomes (Fig. 4B). The proportion of extensively deleted molecules among six analyzed individuals
FIG. 3. HIV-1 DNA present in PBMCs of HIV-1-infected persons. One microgram of DNA, equivalent to 105PBMCs, was amplified by LD-PCR. The resulting
product was separated by agarose gel electrophoresis, transferred to a nylon membrane, and reprobed by32P-labeled oligonucleotidesgag,pol,tat, andnef. Lanes 1
to 10: individuals 1 to 10 (Table 1).
on November 9, 2019 by guest
http://jvi.asm.org/
varied between 27% and 66% with an average of 46% (Fig. 4B). These values highly exceed the background frequency of recombination during PCR which is no greater than 5% (20).
From 2 to 300 copies of thegaggene were detected in 1mg
of DNA from PBMCs of infected persons by classical PCR (Table 1). The efficiency of amplification of the defective
frag-ment cloned in the plasmid pDFOCEP by using the LD
prim-ers was about 15 times higher in standard PCR than in LD-PCR (Fig. 6E). This shows that with respect to the sensitivity of LD-PCR, the HIV-1 DNA from PBMCs of some infected persons is present in end point concentrations. As expected based on Poisson distribution, the pattern of deleted genomes amplified by LD-PCR was not identical in all parallel experi-ments. The population of defective genomes is therefore more complex than represented by the LD-PCR pattern of a single aliquot.
The quantity of defective HIV-1 genomes decreases after in vitro activation of PBMCs. The quantity of deleted HIV-1 genomes was gradually reduced after activation of PBMCs with phytohemagglutinin (Fig. 7). In activated PBMCs nega-tive for the presence of infectious virus, the LD-PCR signal was completely lost (HETNE, NIPDJ, TAMAE) or persisted but decreased (TEMNO). Also, full-length genomes disap-peared from activated PBMCs negative for virus isolation (HETNE, NIPDJ, TAMAE). Isolation of an infectious virus correlated with persistence of the full-length LD-PCR product in activated PBMCs (Fig. 7; DJOUM, NUSEP, DECNO, and FESFU, negative for HIV-1 isolation from PBL but positive in coculture with CBL). Only in these cases did significant quan-tities of deleted HIV-1 genomes persist 28 days after cell stim-ulation.
DISCUSSION
Our results demonstrate that, in contrast to in vitro infec-tion, defective genomes accumulate in PBMCs of infected per-sons. It is likely that a major cause for the different outcomes of infection in both systems is the elimination of productively infected cells by the immune system. The possibility that de-fective viruses accumulate in vivo due to their inability to perpetrate direct cell killing is unsatisfactory because this phe-nomenon would equally occur in vitro. Replication competent viruses were positively selected and the complexity of the pop-ulation of defective HIV-1 genomes decreased after in vitro activation of PBMCs (Fig. 7). In a similar way at the level of point mutations, a more complex population of quasispecies has been detected after evolution of HIV-1 infection in vivo than in vitro (19). Taken together, our results suggest that in vitro activated PBMCs represent a transient state between the presence of defective viruses in vivo and their subsequent elim-ination by adaptation of replication competent virus to tissue cultures.
[image:5.612.66.277.73.710.2]The persistence of the LD-PCR signal after in vitro activa-tion of PBMCs correlates with the isolaactiva-tion of an infectious HIV-1. The concomitant disappearance of defective and full-length genomes from activated PBMCs that do not contain
FIG. 4. Physical mapping of defective viruses from 10 HIV-1-infected per-sons (no. 1 to 10, Table 1). (A) Organization of HIV-1 genome. Position and orientation of LD-PCR primers are shown by arrows. For the exact positions of the oligonucleotide probes, see Fig. 1. (B) Hybridization results from Fig. 3 are shown for each patient:1, positive signal;2, no signal. *The size of deleted genomes was estimated from their electrophoretic mobility (Fig. 3). †Estimated molar proportion of extensively rearranged genomes to the full-length 9-kb LD-PCR product. (C) Relative frequency of positive hybridizations with differ-ent probes per total quantity of defective viral genomes.
VOL. 71, 1997 DEFECTIVE HIV-1 GENOMES IN PBMCs 2237
on November 9, 2019 by guest
http://jvi.asm.org/
infectious virus suggests that the recovery of defective genomes depends on their complementation or recombination with rep-lication-competent viruses. Both full-length and defective HIV-1 genomes have been detected in persons with no detect-able infectious virus in PBMCs. It is highly probdetect-able that rep-lication incompetent or highly attenuated viruses are present in PBMCs of these individuals (4, 25).
The quantity of defective HIV-1 genomes decreased after in vitro activation of PBMCs and full-length genomes disap-peared from activated PBMCs without detectable infectious virus (HETNE, NIPDJ, TAMAE). Thus, in contrast to repli-cation-competent viruses, defective genomes are aborted from PBMC cultures. This abortion could be rationalized in differ-ent ways. Defective viruses could be toxic for activated PBMCs or could be present predominantly in the monocyte fraction of PBMCs depleted by adherence from analyzed PBL. The ma-jority of HIV-1 DNA in quiescent T cells is present in a non-integrated form and integrates into the host genome after cell activation (2, 5, 32). Therefore, the abortion of deleted ge-nomes could also result from a lower integration efficiency. However, this hypothesis is unlikely since LTRs of defective proviruses appear essentially intact.
The frequency of deletions was found to be proportional to their proximity to the central part of the genome which sug-gests that the majority of defective virus is formed by a “copy choice” mechanism in vivo. According to this model, transcrip-tion is prematurely terminated, the polymerase moves with the nascent strand to another template site and resumes transcrip-tion. As the deleted DNA region increases in size, the proba-bility that it contains central genomic parts increases more rapidly than the probability that it contains peripheral parts. In addition, the distribution of deletions along the HIV-1 genome is slightly asymmetric (Fig. 4C). The frequency of sequences
present at the 39end of defective genomes [LTR(U3)] is higher
than that at the 59 end (59NCS). Acknowledging that a
[image:6.612.62.299.71.182.2]tran-scription stop has equal probability to occur at any position, then the first stop is more likely of occurring at the beginning of transcription than at the end. Therefore, the asymmetry in Fig. 4C is consistent with a model involving a single polymerase jump during reverse transcription of the minus strand,
pro-ceeding from 39 to 59 end of HIV-1 RNA. It is worth noting
[image:6.612.318.556.76.581.2]that polymerase detachment can also occur without the restart
FIG. 5. DNA sequence of cloned defective proviruses at the deletion junc-tion. The DNA sequences determined by dideoxy sequencing from plasmids pDCESMO, pDFOCEP, and pDNIPDJby using primers 59NCS andnefwere
com-pared with the corresponding sequences of HIV-1 LAI (22). Numbers in paren-theses correspond to the length of DNA band amplified by primary LD-PCR using primers LTR(U5) and LTR(R). The directly repeated sequences from HIV-1 LAI and defective proviruses are underlined. Percentages bellow the sequences of defective proviruses, upstream and downstream from the deletion junctions, correspond to the homology between sequenced region and HIV-1 LAI sequence. For complete distribution of deleted fragments of FOCEP see Fig. 3 and 4 (individual no. 5); for NIPDJ, see Fig. 7.
FIG. 6. Quantitative aspects of LD-PCR. (A, B, C) Plasmid pNL4-3 (3,000 copies), was amplified with 10, 102
, 103
, 104
, and 105
copies of competing tem-plates pDCESMO(A), pDFOCEP(B), and pDNIPDJ(C) using the primers 59NCS
andnef. All plasmids were linearized byAatII prior to LD-PCR. The resulting product was separated in an agarose gel, transferred to a nylon membrane, and probed by32
P-labeledgagoligonucleotide. Plots represent log10of the ratio of
intensity of the bands corresponding to the competing deleted templates over the bands corresponding to the complete target pNL-4-3 template as a function of the log10of the number of copies of competing templates added. (D) Relative
efficiency of LD-PCR for HIV-1 DNA fragments of different size was calculated as the ratio of complete to deleted input molecules resulting after amplification in an equimolar output. Relative efficiency was plotted against the size of LD-PCR products. The efficiency of LD-LD-PCR for the full size 9-kb product was set arbitrarily at 1. Open squares, LD-PCR primers LTR(U5) and LTR(R), closed squares, LD-PCR primers 59NCS andnef. (E) Comparison of the efficiency of LD-PCR and standard PCR for theAatII linearized plasmid pDCESMOby using
primers LTR(U5) and LTR(R) at three decadic dilutions.
on November 9, 2019 by guest
http://jvi.asm.org/
of synthesis or without termination in the LTR. Such events cannot be detected in our assay.
In a similar way as was described for other retrovirus ge-nomes (23, 26; see also reference 7 for review), we have found that deletions of HIV-1 involve short stretches of identical sequence at the deletion junctions. Only little sequence iden-tity is needed for the recombination event. No additional in-sertions of short sequences at the deletion junctions, docu-mented by Pathak and Temin (26) for spleen necrosis virus, were detected in deleted HIV-1 genomes analysed in the present study.
Whereas a considerable body of data has been accumulated on HIV-1 sequence variability (22), the presence of large de-letions and other extensive alterations in HIV-1 genomes is much less well documented. Defective viruses involving large deletions have been found after direct cloning of HIV-1 DNA from human brain tissue without selection in vitro (18). How-ever, in this case only a few molecules could be analyzed. In comparison with previous approaches, LD-PCR reveals a rep-resentative distribution of the entire HIV-1 population con-taining flanking LTR sequences and it permits a quantitative assessment of intragenomic rearrangements in the viral popu-lation.
Our findings have direct relevance with respect to the usual approaches used to detect and quantify HIV-1 genomes and to follow the different steps of retrotranscription of the HIV-1 genome, including its start and completion, which rely on dif-ferent modifications of standard PCR. Our work shows that these approaches may be in some cases inaccurate or mislead-ing, as they rely on the selected amplification primers and distribution of the defective viruses in the analyzed sample.
The amplification of whole internal HIV sequences by PCR could be useful in different areas of HIV research. LD-PCR allows for the amplification and direct cloning of HIV DNA for functional studies without biological adaptation and selection of HIV in tissue cultures. An acceptably low LD-PCR-introduced error rate of 0.14% has recently been deter-mined (28). LD-PCR could also help establish correlations between the quantity of defective viruses and HIV transmis-sion from mother to child or variations in rates of HIV-1 replication in different tissues (e.g., secondary lymphatic or-gans and brain). Although we did not find any clear correlation between the quantity of defective genomes and the clinical
status of patients, the value of LD-PCR for diagnostic pur-poses is certainly worthy of a closer examination. It could be noted in this context that three out of four individuals in whose PBMCs we detected only highly rearranged HIV-1 genomes (QEVNE, HETNE, TEMNO) were also negative for the iso-lation of infectious virus.
ACKNOWLEDGMENTS
We thank R. H. Bassin and J. Svoboda for stimulating discussions and F. Silvy, S. Allione, and F. Nevie`re for excellent technical assis-tance in virus isolation. We are indebted to C. Santini and A. Chenine for providing us with cloned defective HIV-1, technical help, and discussion. This study would not have been possible without the col-laboration of clinicians from Hoˆpital de la Conception (H. G. Gallais) and Hoˆpital St. Marguerite (J.-A. Gastaut).
This work was financially supported by ANRS and INSERM.
REFERENCES
1.Barre´-Sinoussi, F., J. C. Chermann, F. Rey, M. T. Nugeyre, S. Chamaret, J. Gruest, C. Dauguet, C. Axler-Blin, F. Brun-Vezinet, C. Rouzioux, W. Rozen-baum, and L. Montagnier.1983. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science220:868–871.
2.Bukrinsky, M. I., T. L. Stanwick, M. P. Dempsey, and M. Stevenson.1991. Quiescent T lymphocytes as an inducible virus reservoir in HIV-1 infection. Science254:423–427.
3.Bukrinsky, M. I., N. Sharova, T. L. McDonald, T. Pushkarskaya, W. G. Tarpley, and M. Stevenson.1993. Association of integrase, matrix, and reverse transcriptase antigens of human immunodeficiency virus type 1 with viral nucleic acids following acute infection. Proc. Natl. Acad. Sci. USA
90:6125–6129.
4.Cao, Y., L. Qin, L. Zhang, J. Safrit, and D. D. Ho.1995. Virologic and immunologic characterization of long-term survivors of human immunode-ficiency virus type 1 infection. N. Engl. J. Med.332:201–208.
5.Chun, T. W., D. Finzi, J. Margolick, K. Chadwick, D. Schwartz, and R. F. Siliciano.1995.In vivofate of HIV-1 infected T cells: quantitative analysis of the transition to stable latency. Nat. Med.12:1284–1290.
6.Coffin, J. M.1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science267:483–489.
7.Coffin, J. M.1996. Retroviridae: the viruses and their replication, p. 1767– 1847.InB. N. Fields, D. M. Knipe, P. M. Howley, et al. (ed.), Fields Virology, 3rd ed. Lippincott—Raven Publishers, Philadelphia, Pa. 8.Connor, R. I., H. Mohri, Y. Cao, and D. D. Ho.1993. Increased viral burden
and cytopathicity correlate temporally with CD41T-lymphocyte decline and clinical progression in human immunodeficiency virus type 1-infected indi-viduals. J. Virol.67:1772–1777.
9.Daar, E. S., T. Chernyavskiy, J. Q. Zhao, P. Krogstad, I. S. Y. Chen, and J. A. Zack.1995. Sequential determination of viral load and phenotype in human immunodeficiency virus type 1 infection. AIDS Res. Hum. Retroviruses
[image:7.612.100.512.69.244.2]11:3–9.
FIG. 7. HIV-1 DNA present 0, 14, and 28 days after phytohemagglutinin activation of PBMCs of infected persons. One microgram of DNA, equivalent to 105
PBMCs, was amplified by LD-PCR. The resulting product was separated by agarose gel electrophoresis, transferred to a nylon membrane, and probed by32P-labeled gagoligonucleotide. *HIV-1 of FESFU could be isolated by coculture of patient PBMCs with CBL.
VOL. 71, 1997 DEFECTIVE HIV-1 GENOMES IN PBMCs 2239
on November 9, 2019 by guest
http://jvi.asm.org/
10. de Mareuil, J., B. Brichacek, D. Salaun, J. C. Chermann, and I. Hirsch.
1992. The human immunodeficiency virus (HIV)gaggene product p18 is responsible for enhanced fusogenicity and host range tropism of the highly cytopathic HIV-1-NDK strain. J. Virol.66:6797–6801.
11. de Mareuil, J., N. Guettari, C. Bolmont, D. Salaun, J. G. Baillon, Z. Hos-tomsky, and I. Hirsch.1995. Restriction of HIV-1 replication in intestinal cells is genetically controlled by the gag-pol region of the HIV-1 genome. Virology207:160–167.
12. Ellrodt, A., F. Barre´-Sinoussi, P. Le Bras, M. T. Nugeyre, L. Palazzo, F. Rey, F. Brun-Vezinet, C. Rouzioux, P. Segond, R. Caquet, L. Montagnier, and J. C. Chermann.1984. Isolation of a new human T-lymphotropic retrovirus (LAV) from a married couple of Zairians, one with AIDS, the other with prodromes. Lanceti:1383–1385.
13. Embretson, J., M. Zupanic, J. L. Ribas, A. Burke, P. Racz, K. Tenner-Racz, and A. T. Haase.1993. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature362:
359–362.
14. Gazit, A., R. Sarid, P. Mashiah, D. Archambault, J. E. Dahlberg, S. R. Tronick, and A. Yaniv.1992. Defective viral particles in caprine arthritis encephalitis virus infection. Virology189:344–349.
15. Gout, O., B. Rouquette, E. Tournier-Lasserve, F. Barre´-Sinoussi, O. Lyon-Caen, and J. C. Chermann.1988. Acute and regressive encephalopathy coincident with transient isolation of human immunodeficiency virus from cerebrospinal fluid of a seropositve man. Biomed. Pharmacother.42:15–20. 16. Hiramatsu, K., and H. Yoshijura.1986. Frequent partial deletion of human adult T-cell leukemia virus type 1 proviruses in experimental transmission: pattern and possible implication. J. Virol.58:508–512.
17. Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz.1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature373:123–126.
18. Li, Y., J. C. Kappes, J. A. Conaway, R. W. Price, G. M. Shaw, and B. H. Hahn.1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and -defective viral genomes. J. Virol.65:3973–3985. 19. Meyerhans, A., R. Cheynier, J. Albert, M. Seth, S. Kwok, J. J. Sninsky, L. Morfeldt-Manson, B. Asjo¨, and S. Wain-Hobson.1989. Temporal fluctua-tions in HIV quasispecies in vivo are not reflected by sequential HIV isola-tion. Cell58:901–910.
20. Meyerhans, A., J.-P. Vartanian, and S. Wain-Hobson.1990. DNA recombi-nation during PCR. Nucleic Acids Res.18:1687–1691.
21. Molineaux, S., and J. E. Clements.1983. Molecular cloning of unintegrated visna viral DNA and characterization of frequent deletions in the 39 termi-nus. Gene23:137–148.
22. Myers, G., B. H. Hahn, J. W. Mellors, L. E. Henderson, B. Korber, K.-T. Jeang, F. E. McCutchan, and G. N. Pavlakis.1995. Human retroviruses and AIDS: a compilation and analysis of nucleic acid and amino acid sequences. Report T-10. Los Alamos National Laboratory, Los Alamos, N.Mex. 23. Omer, C. A., K. Pogue-Geile, R. Guntaka, K. A. Staskis, and A. J. Faras.
1983. Involvement of directly repeated sequences in the generation of dele-tions of the avian sarcoma virussrcgene. J. Virol.47:380–382.
24. Pantaleo, G., C. Graziosi, J. F. Demarest, L. Butini, M. Montroni, C. H. Fox, J. M. Orenstein, D. P. Kotler, and A. S. Fauci.1993. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of dis-ease. Nature362:355–358.
25. Pantaleo, G., S. Menzo, M. Vaccarezza, C. Graziosi, O. J. Cohen, J. F. Demarest, D. Montefiori, J. M. Orenstein, C. Fox, L. K. Schrager, J. B. Margolick, S. Buchbinder, J. V. Giorgi, and A. S. Fauci.1995. Studies in subjects with long-term nonprogressive human immunodeficiency virus in-fection. N. Engl. J. Med.332:209–216.
26. Pathak, V. K., and H. M. Temin.1990. Broad spectrum ofin vivoforward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc. Natl. Acad. Sci. USA87:6024–6028.
27. Piatak, M., Jr., M. S. Saag, L. C. Yang, S. J. Clark, J. C. Kappes, K. C. Luk, B. H. Hahn, G. M. Shaw, and J. D. Lifson.1993. High levels of HIV-1 plasma during all stages of infection determined by competitive PCR. Science259:
1749–1754.
28. Salminen, M. O., C. Koch, E. Sanders-Buell, P. K. Ehrenberg, N. L. Michael, J. K. Carr, D. S. Burke, and F. E. McCutchan.1995. Recovery of virtually full-length HIV-1 provirus of diverse subtypes from primary virus cultures using the polymerase chain reaction. Virology213:80–86.
29. Sauvaigo, S., V. Barlet, N. Guettari, P. Innocenti, F. Parmentier, C. Bastard, J. M. Seigneurin, J. C. Chermann, R. Teoule, and J. Marchand.1993. Standardized nested polymerase chain reaction-based assay for detection of human immunodeficiency virus type 1 DNA in whole blood lysates. J. Clin. Microbiol.31:1066–1074.
30. von Magnus, P.1954. Incomplete forms of influenza virus. Adv. Virus. Res.
2:59–86.
31. Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. A. Hahn, M. S. Saag, and G. M. Shaw.1995. Viral dynamics in human immunodeficiency virus type 1 infec-tion. Nature373:117–122.
32. Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Y. Chen.1990. HIV-1 entry into quiescent primary lymphocytes: molecular
analysis revels a labile, latent viral structure. Cell61:213–222.