Copyright ©1993, AmericanSocietyforMicrobiology
Mutation of
Large
T-Antigen-Binding
Site
A,
but Not Site
B
or
C, Eliminates Stalling by RNA Polymerase
II in
the
Intergenic
Region of Polyomavirus DNA
JOHN BERTIN,tNOELLE-ANN SUNSTROM,i ANDNICHOLAS H. ACHESON*
Departmentof Microbiology andImmunology, McGillUniversity, Montreal, Quebec, Canada H3A 2B4 Received 29 October1992/Accepted 25 June 1993
Duringtranscription of the late strand of
polyomavirus
DNA, RNApolymerase II stalls and accumulates nearby the binding sites on viral DNA recognized by polyomavirus large T antigen. Stalling by RNApolymerasesiseliminated when thermolabile large T antigenis inactivated by usingatemperature-sensitive
virus mutant (J. Bertin, N.-A. Sunstrom, P. Jain, and N. H. Acheson, Virology 189:715-724, 1992). To determine whether stalling by RNApolymerases is mediated through theinteraction of large T antigen with one or more of itsbinding sites, viable polyomavirus mutantsthat contain altered large-T-antigen-binding sites were constructed. Point mutations were introduced by site-directed mutagenesis into the multiple, clustered
G(A/G)GGC pentanucleotides known to be the target sequence forlargeT-antigen binding. Mutation of the
G(A/G)GGCpentanucleotides in the first two binding sites encountered by RNA polymerases in the intergenic region (sites C and B) had no detectable effect on stalling as measured by transcriptional run-on
analysis.
However,mutationof the twoGAGGC pentanucleotides in binding site A, which lies adjacent to the origin of viral DNAreplication, eliminated stalling by RNA polymerases. We conclude that binding of large T antigen tosite A blockselongation by RNA polymerase II. Furthercharacterizationof virus containing mutated site A didnotreveal any effectsonearly transcription levelsor onvirus DNAreplication. However, themutantvirus gaverise to small plaques,suggesting impairment insomestageof virusgrowth. Stalling of RNA polymerases by largeTantigen boundto theintergenic region of viral DNA may functiontoprevent transcription from displacingproteins whose binding is required for the normal growth of polyomavirus.Termination of transcription by RNA polymerase II is a
complex, multistep process that involves the cessation of
elongation and the releaseof theRNAtranscript and RNA
polymerase fromthe DNAtemplate. Inhighereukaryotes, the 3' ends of all mRNAsaregenerated byRNAprocessing events rather than by transcription termination (70). The results of transcriptional run-on experiments have shown
that RNA polymerase II continues to transcribe for dis-tances ranging from several hundred to several thousand nucleotides beyondthe site whichdeterminesthe 3' end of the mRNAtranscript(57). However, terminationatthe end ofsomegenestranscribed byRNApolymeraseII has been showntobedependentonthe presence of upstream 3'-end processing signals(13, 16, 44, 45, 72). Sequence analysis of the various regions of termination has not revealed the
existenceofacommonterminator element, and in onlyafew cases have specific elements involved in terminating
tran-scriptionby RNApolymerase II been identified (3, 12, 13,
30, 60,
66).
RNApolymerase II has been observed to stall or termi-natetranscription within several viral and cellular transcrip-tion units in regions on the template DNA which contain
bindingsites forproteins (3,9, 13, 17, 18, 47, 58,62). It has been proposedthat DNA-binding proteins may function as
stallingorterminationsignals which impede the progression of RNApolymerasesthroughspecific regions ofa transcrip-tionunit. Interestingly, not all proteins have the ability to
* Correspondingauthor.
tPresentaddress:Institute for MolecularVirology,Universityof Wisconsin-Madison, Madison,WI53706-1596.
tPresent address: John Curtin School of Medical Research, Australian National University, CanberraACT2601,Australia.
impede elongationby RNApolymerase II, suggesting that
blockage may be a specific property of only some
DNA-binding proteins (17, 18, 41). The mechanism used by DNA-binding proteins to block elongation by RNA poly-merase II is currently not understood. However, several DNA-binding proteinshave been showntoblockelongation
in an orientation-dependent manner, suggesting that the
blockingproteinmustbespecificallyoriented with respectto the transcribing RNA polymerase (3, 18). DNA-binding proteinshavebeen showntostallorterminatetranscription byother RNApolymerases, suchasEscherichia coli RNA
polymerase(24, 52, 59, 61), eukaryotic RNApolymerase I
(5, 49),andmitochondrialRNApolymerase(41). Therefore, bindingofspecific proteinstotemplateDNA appearstobea signal used by both prokaryotes and eukaryotes to block elongation byRNApolymerases during transcription.
Theearly and late genes ofpolyomavirus aretranscribed byRNApolymeraseIIindivergent directionsfromwithina 300-nucleotide (nt) intergenic regionthat contains
transcrip-tional enhancer elements and the viraloriginofDNA
repli-cation (29). Because polyomavirus DNA is circular, RNA
polymerasesareabletotranscribearound the DNAtemplate
and through the intergenic region. Although most RNA
polymerases transcribing the late DNA strand terminate
upstream of the intergenic region (9, 62), some RNA poly-merases do transcribe through thisregion and traverse the entirecircular genome,producingmultigenome-length tran-scripts (1). However, transcription through the intergenic regionon someviral DNAtemplatesisblocked,resultingin theaccumulation of stalled RNApolymerases(62). Stalling occurswithin a 164-nt region which lies between the early transcription startsite and theorigin of viral DNA
replica-tion
(9)
(Fig. 1A).Thisregion containsfouradjacent sites (C,5766
on November 9, 2019 by guest
http://jvi.asm.org/
A
LATE TRANSCRIPTION
402
LARGE TANTIGEN
BINDINGSITES
C B A 1/2
EARLY LATE 150 1/6315 5128
l
STALLINGREGION
175 1,1
1L 2L
FRAGMENT
169 C
I -
-TCTAGAGGCTGAAATGAGGCGGGAAGAGGCGGTTGGGGCTTAATTATAT
B
CGATTTGGGTGGCCCCACAGCGCCTCCAAGGCGCCAGTCCTGTGTCGA A
I * *
CAAGTTGCCTCTGGAATGCCTCTCTTCTTTTTCTCCAGAGCATG
1/2
CGGAGGCCAGGGGCCCCCGGCCTCTGCTTAATACTAAAAAAAACAGCTG
[image:2.612.59.299.92.334.2]1/5315 5295
FIG. 1. (A) Locations of stalling-region and large T-antigen-binding sites on polyomavirus DNA. The early and major late transcriptionstartsites,andfragments1Land2Lusedto assayfor stalling levels by transcriptional run-on analysis, are shown. (B) Nucleotidesequenceoftheregion containing large T-antigen-bind-ing sites C, B,A,and1/2invirusstrain AT3-Modori.Differencesin nucleotide sequence compared with virus strain AT3 (62) are indicated withasterisks. The regions protected bylarge Tantigen binding in DNase I protection assays (20) and the G(A/G)GGC pentanucleotidesequences(arrows)recognizedby large Tantigen in eachbindingsite areindicated.
B, A, and1/2)towhichlarge T antigen binds(20, 28, 33, 54), leading to the initiation of viral DNA replication (55) and probablytotherepressionofearly gene expression (15, 23,
31,32, 40). Inactivation ofpolyomavirus largeTantigenby useofatemperature-sensitivemutantvirus eliminates
stall-ingby RNApolymerases in this region, suggesting that the interaction of large T antigen with these DNA-binding sites is responsible for the block to transcription elongation (9). However,it remainedpossiblethatlarge T antigen interferes with transcription elongation by a mechanism that is not dependent on the binding of large T antigen to the DNA
template.
Togaininsight into the mechanism used by polyomavirus
large Tantigentoblockelongation by RNA polymerase II, we have introduced mutations into the pentanucleotide
G(A/G)GGC
sequences thatare knownto be the targetfor DNAbinding(21, 53). Inthis study, we show that mutation of both GAGGCpentanucleotidesinlargeT-antigen-bindingsite A, which is locatedadjacenttotheorigin of viralDNA replication, eliminates stalling by RNA polymerases. In contrast, mutation of thefirsttwobinding sites (sites C and
B) encountered by RNA polymerases transcribing the late DNAstrand hadno effecton the level of RNApolymerase
stalling.Viruscontaining mutated site A replicated its DNA at ratescomparable to those ofwild-type virus and did not show any change in early transcription levels. However, mutation of site A causedasignificantreduction in the size ofplaques formed by themutantvirus. Therefore, stalling of
RNApolymerasesin the intergenic regionmaybe necessary
for the normal growthof polyomavirus.
MATERIALSANDMETHODS
Cells and viruses.Growth of mouse 3T6 and primary baby mouse kidney cells and production of virus stocks were carriedout asdescribedpreviously(9).Thewild-type poly-omavirus strain (AT3-Modori) used in this st'ddy contains
four additional restriction sites which were introduced, by oligonucleotide-directed mutagenesis, into strain AT3 adja-cent to large T-antigen-binding sites C, B, A, and 1/2 (63).
The locations of the additional restriction sites are as fol-lows:SphI, nt 32;Sall,nt72;ClaI, nt 118; andXbaI,nt164
(Fig.
1B).
Construction ofpolyomavirusmutants.Mutant polyomavi-rus strains were constructed by site-directed mutagenesis (43), using a procedure provided by Bio-Rad, Inc. All
mutations were introduced into polyomavirus strain AT3-Modori clonedviaanEcoRIsite intoplasmidpGEM-3Zf(-) (PromegaBiotec, Inc.). The sequences of the
oligonucleo-tides used formutagenesisare asfollows:oligonucleotideC, 5'-GAT ATA ATT AAT TCC CAA CCT TCT CTT CCC TTC TCA TTl CATTCT CTA GACC-3'; oligonucleotide
B, 5'-CAC AGG ACT GAA GCC TTG GAG AAG CTG TGG GAA CAC CCA AATC-3';oligonucleotideA,5'-AGA AGA GAT GTA TTC CAG AGT TAACTTGTC-3';
oligo-nucleotide
AO,
5'-AAA AAG AAG AGA TGAATTCCAGAG GC-3'; oligonucleotide Al, 5'-CAT TCC AGA GTT
AAC TTG TCG A-3'; oligonucleotide A2, 5'-AAA AAG AAG AAA GGC ATT CCA GAG GC-3'; and
oligonucleo-tideA1A2,5'-AAA AAG AAG AAA GGCATTCCA GAG
1TA ACT TGT CGA-3'. Plasmids containing the desired
mutations were screened as indicated: C, additional AseI
site; B, missing BglI site; A, additional HpaI site;
AO,
additional EcoRI site;Al,
additional HpaI site; A2, no restriction site altered, mutant identified by sequencing; A1A2, additional HpaI site. The polyomavirus genomecontainingthe desiredmutationwasreleased frompGEM by digestion with EcoRI and transfected into baby mouse kidney cells(100ngof DNA per 107cells) to producemutant polyomavirus stocks. Presence of desired mutations and absence of defective DNA were confirmed by restriction endonuclease digestion and sequencing of DNA extracted from cells infectedathighmultiplicity (1to50 PFU percell)
with each virus stock.
Transcriptional run-on assay. Viral transcription com-plexeswere isolated frominfected cellsas described previ-ously(62). Transcriptionalrun-onassayswereperformed by extendingnascentRNAchains for 2 minat26°Cina250-,ul reaction mixcontaining 0.35 ,M
[a-32P]CrP
(3,000 Ci/mmol; Amersham).32P-RNA
was hybridized in solution to an excess ofsingle-stranded M13 DNA containingthe late or earlystrand of variousfragments ofpolyomavirusDNA(9,62).
Hybridizations tofragment 2, which contains thelarge T-antigen-binding sites (Fig.1A),
were done in each case with DNA containing the respective mutations introduced intowild-type virus by site-directed mutagenesis (Fig. 2).The relative transcriptional activityin each region assayed wasnormalized for the number of C residues in each DNA fragment(Table 1).
DNA-binding assay. Thespecific DNA-binding activityof largeTantigenwith DNAfragmentscontainingmutantlarge T-antigen-bindingsiteswasdeterminedaccordingto a mod-ified
immunoprecipitation
assay(48)
asdescribedpreviously
(9, 64). A 670-nt DNA fragment
(nt
32 to702)
containing
B
on November 9, 2019 by guest
http://jvi.asm.org/
Virus Mutation Viability
167 Site C 130
I - - - I
WT
TAGAGGCTGAAATGAGG3CGGG3AAGAGGCGGTTGGGGCTT
C AA
AA
AA
AA
+111 Site B 84
WT
TGBtCCC~CACAGCGCCTCCAAGGCGCCAG
B TT TT TT +
69 Site A 49
TTGCCTCTGGAATGCCTCTc:
AA A A
T A AA
T
AA T
[image:3.612.132.491.83.263.2]TTGCCTC[11 nt
deletlon]TC
FIG. 2. Mutations introduced intolargeT-antigen-binding sites.
largeT-antigen-binding site A and wild-typeormutantlarge T-antigen-binding sites B and/or C was gel purified and
end-labeledwith 2P-labeled nucleosidetriphosphates by the Klenowfragment of DNA polymerase I. Duplicate samples of large T antigen-DNA complexes were
immunoprecipi-tated with a polyclonal antibody raised against large T
antigen.Theamountof DNAboundbylargeTantigenwas
quantitated by liquid scintillation counting, and binding levelswere corrected for nonspecific binding by using
nu-clearextractsprepared fromuninfected cells. RESULTS
Constructionofpolyomavirusmutants.Polyomavirus large Tantigen binds specificallytofoursitesdesignated C, B, A, and 1/2 in the intergenic region ofpolyomavirus DNA(20, 28, 33, 54) (Fig. 1A). Each site contains tandem repeatsof the pentanucleotide G(A/G)GGC known to be the target
sequencefor DNAbinding by largeTantigen (21, 53) (Fig. 1B). The guanine residues at positions 1, 3, and 4 in the pentanucleotide sequence andatposition5on theopposite
DNA strandarecriticalcontactpoints since methylation of
any of these nucleotides interferes with large T-antigen
binding(21).
Because ourgoalwasto generateviable mutantviruses,
we were restricted to mutating those binding sites not required for the initiation of viral DNA replication. Large T-antigen-bindingsites B andC havepreviously been shown to benonessential forviability (7). Double-point mutations replacing the final GC with AAwere therefore introduced
into the four G(A/G)GGC pentanucleotide sequences
present within site C and into the two pentanucleotide
sequencespresentwithinsite B,aswellasintoadegenerate
CTGGCpresentatoneend of site B(Fig.2).Viableviruses containing mutant site B or C or both B and C together
(mutantvirusBC)weregenerated.
Thepolyomavirus originofbidirectionalDNAreplication has beenmappedtoaregionlocated between sites1/2andA (37). Theminimalorigin ofviral DNAreplication,asdefined bydeletion mutants, containsbindingsite1/2and part ofsite A(23, 39, 55). Although site 1/2is absolutely required for DNAreplication, the role of site A remains unclear.
Essen-TABLE 1. EffectsofmutationsinlargeT-antigen-bindingsitesonRNApolymerasestalling
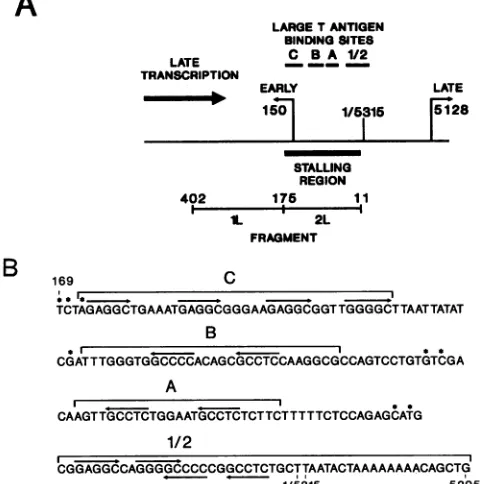
Virus Fragment No.of C Correction Avgcpm Transcriptional Stalling
residues
factor'
hybridizedb activity- ratiodWild typee 1 59 0.73 145 106 11
2 43 1.0 1,214 1,214
Bf 1 59 0.68 208 142 13
2 40 1.0 1,890 1,890
C!g 1 59 0.66 162 107 12
2 39 1.0 1,301 1,301
BCh 1 59 0.61 201 123 11
2 36 1.0 1,360 1,360
A1A2' 1 59 0.70 160 112 2.2
2 41 1.0 251 251
aNumber of Cresiduesinfragment2 dividedby the number ofC residues in thefragment used.
bAfter subtraction ofbackground.
cCounts perminutehybridizedxcorrection factor.
dRatiooftranscriptional activityinfragment2to that infragment1. 'Input, 25,000cpm.Backgroundbound in presenceofM13DNA,51cpm.
f Input, 26,400cpm.Background,69 cpm.
g Input, 22,800cpm.Background,60 cpm.
hInput, 35,400cpm.Background,81 cpm. iInput, 17,600cpm.Background,57 cpm.
WT A AO
Al A2 A1A2 A2dM
+
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.63.561.544.661.2]tial sequencesfor DNA
replication
arepresentwithinsiteA,ascertainmutationsreduceorabolish
replication. However,
large T-antigen
binding
to thisregion
may notbe essential(67).
Mutation of the GAGGCpentanucleotide
farther from site1/2
(nt
62to66)
allowed virusgrowth (mutant
Alvirus, GAGGC toGAGTT; Fig. 2).
However,
mutation of theGAGGC
pentanucleotide
closer to site1/2
(nt
51 to55)
to GATGTorGATGA(mutant
AandAOviruses,
respectively;
Fig. 2)
didnotallow virusgrowth.
Avirus(A2dl)
recoveredafter transfection of
baby
mousekidney
cells withmutantAO DNA hadundergone
a deletion of 11 nt(nt
51 to61)
that removed the mutated GATGApentanucleotide.
As aresultof this
deletion,
mutant virus A2dl has asingle
GAGGCpentanucleotide
in site A located inprecisely
the sameposition
relativetosite1/2
astheproximal
GAGGC(nt
51to55)
inwild-type
virus.Previous studies showed that mutation of the GAGGC
pentanucleotide proximal
to site1/2
to AAGGC does not affectvirusviability (46)
orviralDNAreplication (67),
eventhough
it interferes withlarge T-antigen binding
to site A(21).
Thismutationwasthereforeintroduced intothepenta-nucleotide
proximal
to site1/2,
and a viable virus wasgenerated
(mutant
A2virus; Fig. 2).
Wealso combinedthe A2mutation withtheAlmutationtogenerateamutantvirus in which both GAGGCpentanucleotides
in site A were mutated(mutant
A1A2virus; Fig. 2).
Transcriptional
run-on assay. The level of RNApoly-merase
stalling
in theintergenic region
ofpolyomavirus
was measuredby performing
atranscriptional
run-on assay(9,
62).
Inthis assay,viraltranscription complexes
areisolatedfrom infected cells and RNA
polymerases
are allowed to extendtheirnascentRNAchains forashorttime(2 min)
invitro in the presence of
[32P]CTP.
Radioactive RNA ishybridized
in solutiontosingle-stranded
recombinantDNA.Therelative levels of
hybridization,
corrected for the num-ber of C residues in each DNAfragment used, correspond
to the averagedensity
of active RNApolymerases
withintheregions
assayed.
RNApolymerases
thatstallininfected cells whiletraversing
thelarge T-antigen-binding region
onpoly-omavirus DNA accumulate there andcanresume
elongation
invitro under conditions used for therun-onassay
[150
mM(NH4)2SO4,
0.1%
Sarkosyl].
This results inahigher
level oftranscription
fromthestalling region (fragment
2L;
Fig. 1A)
than from
adjacent
upstream(fragment 1L)
ordownstreamregions (9).
Mutation of
large T-antigen-binding
siteAbutnotsiteBorC eliminates
stalling by
RNApolymerase
II. To determinewhether
binding
oflarge
Tantigen
toviralDNA isrequired
toblock
elongation
by
RNApolymerase II,
weexaminedthe level of RNApolymerase stalling
on thelate strand of viral DNAtemplates containing
alteredlarge T-antigen-binding
sites. Results from
representative experiments
areshown inTable 1 and
Fig.
3. Whentranscription
complexes
wereisolated from cells infected with
wild-type
virus(AT3-Mo-dori),
thedensity
of RNApolymerases
within thestalling
region (fragment
2L)
was 11-foldhigher
than that in theadjacent
upstreamregion (fragment 1L).
The ratios of RNApolymerases
infragment
2L to those infragment
1Lwerebetween 8 and 16 in
eight independent experiments using
wild-type
virus(data
notshown).
These levels of RNApolymerase
stalling
in theintergenic region
are similar to thosepreviously
observed forpolyomavirus
wild-type
strains AT3 and A3(9).
The AT3-Modoripolyomavirus
wild-type
strain usedin thisstudy
contains several mutationslocated
adjacent
tolarge T-antigen-binding
sitesC,
B,
andAQ 12. "1
WT
6- C 1A
VIRUS
FIG. 3. Mutation oflargeT-antigen-binding siteA, but not sites B andC,affectsstalling. Viral transcription complexes were isolated from cells infected for 30 h with wild-type (WT) virus or with mutant B, C, BC, or A1A2 virus. Transcriptional run-on assays were performedasdescribed in Materials and Methods, and thestalling ratio(relative density ofRNApolymerases infragment 2L to that in fragment 1L) for each virus was determined. Results are expressed asthe ratio of transcriptional activity in DNAfragmnent2L tothat in fragment1L. Data are taken from theexperimentshown in Table1.
(Fig. 1B); therefore, the presence of these mutations does notaffectstalling by RNA polymerase.
Wenextexaminedthe effects of mutating large
T-antigen-binding sites A, B, and C on stalling. In the experiment
shown, the ratios of RNA polymerases in fragment 2Lto those infragment 1Lwere 13 when site Bwasmutated, 12 when site Cwas mutated, and 11when both binding sites were mutated (Table 1 and Fig. 3). Since these ratios fall within thewild-typerange,we conclude that the mutations introducedinto theG(A/G)GGCmotifs present within
bind-ingsites B and C have no detectable effect onRNA poly-merase stalling. However, mutation of both GAGGCs in
binding site A (mutant AlA2 virus) essentially eliminated
stalling byRNA polymerases (Table 1 and Fig. 3). In this
experiment,theratio of RNApolymerasesinfragment 2L to that in
fragment
1L was 2.2, which represents a fivefold reduction in thelevel of stalled RNApolymerasesrelative to that observed with wild-type virus. In five independentexperiments, the stalling ratiowas between 2 and 3 when virusA1A2wasassayedfor RNApolymerase stallinglevels
(datanotshown).These ratios fall inthe rangeweobserved
previouslywhenlargeTantigenwasinactivatedbyusinga
temperature-sensitive
mutant virus(9).
We conclude thatstalling by RNA polymerases in the intergenic region of
polyomavirus
DNAis dependentonthe bindingoflarge Tantigentosite Abutnotsite BorC.
To rule out the possibility that the reduction in stalling levels observed with mutant A1A2 virus was due to an overallreductionin RNApolymeraselevelsonthe late DNA
strand,
we compared the level of RNA polymerases infragment 2L with those in fragments located just down-streamof the
major
latetranscriptionstartsite(fragments3L and 4L;Fig.
4A). The density of RNA polymerases de-creasesalong
the late DNA strand from amaximumat thetranscription
start site to the lowest leveljust prior to thestalling
site(9, 62).
Ontranscription complexesisolated from cells infected with wild-type virus, the density of RNApolymerases
in fragment 2Lwas greater than that in frag-mentslocatedjust downstream of themajor latetranscrip-tion start site
(Fig. 4B),
as waspreviously
observed (9). Whenbinding
site Awasmutated (mutant A1A2 virus;Fig.2),
theratio ofRNApolymerasesinfragment2Lrelativeto that infragment
1Ldecreased from 8(wild type)
to2(A1A2)in this
experiment,
while levels in the otherfragments
(1L,3L,
and4L)
remainedapproximately
the same(Fig.
4C).on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.369.513.87.186.2]A
LATE 1/5312[
1L 2L
I
F3L
4LA
I
I
I I I I402 175 11 5134 4968 4769
B
C
00 w
a
w0
>LO
Jz
:ic
[image:5.612.55.297.80.237.2]FRAGMENT
FIG. 4. Comparison of the level of stalled RNA polymerases withthe densityofpolymerasesdownstreamofthelatetranscription startsite.(A) Nucleotide numbers delimitingfragments 1L, 2L, 3L, and4Lareshown. The arrow shows thepositionof themajorlate transcriptionstartsite(nt 5128).Cells wereinfectedwithwild-type virus or with mutantA1A2virus for 40h, and viraltranscription complexeswereisolated. Transcriptional run-on assayswere per-formed, and the relative density of RNA polymerases in each fragmentwasdeterminedfor wild-type (WT)virus (B)and mutant A1A2 virus (C).
This result shows that the AlA2 mutation reduced the level ofstalledpolymerases in fragment2Lbuthad nodetectable
effectoninitiationby RNApolymeraseatthe late promoter, some200nt downstream.
We also examined whether both GAGGC
pentanucle-otides in binding site A were necessary for stalling. The
stallingratiosweredeterminedformutantAl and A2 viruses
(Fig. 2) and compared with those for wild-type virus and mutant A1A2 virus (Fig. SA). In the experiment shown, mutation of either of the two GAGGCs in binding site A reduced thestallingratio from 12(wild-typevirus)to4.5or 5.7 (mutant Al and A2 viruses, respectively). Mutation of both GAGGC pentanucleotides in site A (mutant AlA2 virus) further reduced the stalling ratioto 2.8. In twoother
experiments (data notshown), wealso observed intermedi-ate stalling ratios for mutant Al and A2 viruses relative to wild-type virus and mutant AlA2 virus. In addition, the
stalling ratio for mutant A2dl virus, which has only one GAGGCpentanucleotidein site A(Fig. 2),wasalso foundto be inthisintermediaterange(4.4; Fig. 5B).Weconclude that
stalling byRNApolymerasescan occurifonlyoneGAGGC
pentanucleotide is present insiteA, but that bothGAGGC
pentanucleotides are required for maximalaccumulation of RNApolymerases in the intergenic region.
Mutation of sites B and C interferes with large T-antigen binding in vitro. The introduction of mutations into the
G(A/G)GGC
pentanucleotidespresent withinbindingsites B andC hadnoeffectonstalling by RNA polymerases (Fig. 3). Onepossibleexplanationforthis result is that the mutationsintroduced into these binding sites did not interfere with largeT-antigenbinding. Since a previous study showed that a mutationwithin a G(A/G)GGCpentanucleotide sequence interferes with large T-antigen binding in vitro (21), we consider this explanation highlyunlikely. Nevertheless,we verified that the mutations introduced into binding sites B andC interfere with the binding of largeTantigento these sites in vitrobyperformingaDNA-binding assay(9, 64). In this assay, a radiolabeled DNA fragment containing large
T-antigen-binding sites A, B, and C was mixed with a
O
CDCIL
-1 15-,
co
.'A
AUl
A2dI A1A2
VIRUS
FIG. 5. BothGAGGCpentanucleotidesinlarge T-antigen-bind-ing site Aplay a role installing. Cellswereinfected with wild-type (WT)ormutantviruses for 30h,andtranscriptioncomplexeswere
isolated. Transcriptional run-on assays were performed, and the stalling ratio(relativedensityof RNApolymerasesinfragment2Lto
that infragment 1L)for each virus wasdetermined andcompared withthoseforwild-typevirus and mutant virusA1A2. (A) Results fromanexperiment with wild-type,Al,A2, andAlA2 viruses; (B) resultsfromaseparateexperiment with wild-type,A2dl,andAlA2 viruses.
nuclear extract containing large T antigen. The amount of DNAfragment immunoprecipitated with antibodies against large Tantigen isameasureof theaffinityoflargeTantigen to its binding sites. In the experiment shown (Fig. 6),
10-75
WO
>r
25
500
0
WT B C BC
DNA FRAGMENT
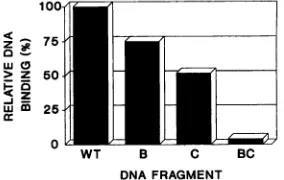
FIG. 6. Immunoprecipitation of DNAfragments containing
mu-tations in large T-antigen-binding sites B and C. The relative
amountsofimmunoprecipitated radiolabeled DNAfragments
con-tainingmutant large T-antigen-binding sites (see Fig. 2for
muta-tions) are shown. DNA fragments were incubated with nuclear
extracts containing largeTantigen, and the in vitroDNA-binding activity was determined as described in Materials and Methods. Under these conditions, approximately 10% of an input DNA fragmentcontainingbindingsitesA, B, and C(wildtype[WT])was
immunoprecipitated. In the experiment shown, this amount was
standardized to 100% forcomparisonwith other DNAfragments. DNAfragments assayed:WT (bindingsitesA,B,andCpresent);B (siteBmutant,sites A and Cpresent);C(siteC mutant, sites Aand Bpresent);BC(sitesBand C mutant,siteApresent).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.351.518.85.339.2] [image:5.612.365.507.516.606.2]A
DIRECTION OF EARLY
4
-TRANSCRIPTIONJ
LARGE T ANTIGEN BINDING SrrES
C B A 1/2
EARLY
1
I I I
1105 402 175 11
3E 2E 1E
B
U.
wo U
>nU
Z m
, 2
w 0
-Jz
O
3E de 1it
FRAGMENT
FIG. 7. Densities of RNApolymerasesontheearlyDNAstrand. (A) Map ofregion onpolyomavirus strain AT3 (62) analyzed by
transcriptional run-on assays. Large T-antigen-binding sites, the
early transcription start site, and nucleotide numbers delimiting fragments 1E, 2E,and3E,whichwereseparately cloned into M13 bacteriophagevectors,areshown.(B)Viraltranscription complexes wereisolatedfromcells incubatedat32°Cfor 3days after infection withpolyomavirusstrain AT3. Resultsareexpressedasthe ratio of transcriptional activity ineach DNAfragment to that infragment 3E.
mutation of site B or site C reduced the amount of DNA immunoprecipitated by25or50%, respectively. This result is consistent with previous findings showing that site C [containingfourG(AIG)GGCs]hasahigher affinityforlarge
T antigen than does site B or A [containing two G(A/ G)GGCseach inthis virusstrain] andtherefore contributes moststronglytothe interaction oflargeTantigenwiththis DNAfragment (20, 64).Mutation of bothbindingsites B and Cvirtuallyeliminatedbinding bylargeTantigentothis DNA fragment (96% reduction). It should be noted thatunder the conditions used, binding by large Tantigen to site Aalone could notbe detected (63); this requires higher concentra-tions of large T antigen than are available in the nuclear extract. We conclude that the mutations introduced into sites B andCstronglyreduceoreliminatespecific binding by
largeTantigento thesesites in vitro.
Large T antigen does not appear to block elongation by RNA polymerases transcribing the early DNA strand. We nextexamined whetherlargeTantigenblockselongation by RNA polymerases transcribing the early DNA strand of polyomavirus. RNA polymerases have been shown previ-ouslyby transcriptionalrun-onanalysistoaccumulateonthe
earlyDNA strand withina621-ntregionwhich includes the binding sites for polyomavirus large T antigen (62). We decided to map more precisely the distribution of RNA polymerases found within this region on the early DNA
strand. Three adjacent fragmentsfrom this regionof polyo-mavirus wild-type strain AT3 (62) were cloned into M13 vectors (Fig. 7A), and the density of RNA polymerases associated with eachfragmentwasdeterminedby transcrip-tional run-on analysis. In the experiment shown (Fig. 7B),
o10
8
< Z
UJ O 6
wO 6
I0
- z 2
CS
I. O
WT A1A2
[image:6.612.350.519.85.198.2]VIRUS
FIG. 8.
Late-to-early
transcription ratios. Cells were infected with wild-type (WT) or mutant A1A2 virus for 30 h, and viral transcription complexes were isolated. Transcriptional run-on as-sayswereperformed (RNA chains extended for 10min), and the corrected levels of hybridization to the early (nt 1498 to 2617) and late(nt 2991 to 3466) DNA strandsweredetermined. Results are expressed as ratio of RNA polymerases on the late DNA strand relative to the early DNA strand.and eightfold excess in RNApolymerase density was ob-served in fragment 2E (nt 175 to 402) relative to adjacent fragments 3E (nt 402 to 1100) and 1E (nt 11 to 175). Therefore, the accumulation of RNA polymerases previ-ously detected on the early DNAstrand by Skarnes et al.
(62) is located in fragment 2E, a 225-nt region which lies
immediately downstreamofthe earlytranscriptionstartsite
(Fig. 7A). The relative density ofRNApolymerases in the three DNAfragments examined further suggests that tran-scription is not blocked in the region which contains the
large T-antigen-binding sites (fragment 1E). Furthermore, inactivation of thermolabile large T antigen by using a temperature-sensitive virus had no effect on the relative density of RNA polymerases in fragment 1E or on the accumulation of RNApolymerasesinfragment2E(datanot shown). Therefore, largeTantigendoesnotappeartoaffect
elongation byRNApolymerases transcribingthe intergenic regiononthe earlyDNAstrand.
Effect of mutations inbinding site Aonearlytranscription and virusgrowth. ThebindingoflargeTantigentosite A has beenproposedtoplayarole in therepressionofearlygene
expression(23)and in the initiation ofviralDNAreplication
(55).
We were therefore interested indetermining
whetherthemutations introduced intolarge T-antigen-binding siteA
(mutantA1A2virus; Fig. 2) had any effect onthese func-tions.
To comparethe ratio of latetoearly transcriptionbetween wild-type virus and mutant A1A2virus, the levelsofRNA
polymerases transcribingtheearlyand late DNAstrandsfor each viruswere determinedbytranscriptional run-on
anal-ysis. The densities of RNA polymerases in the regions immediately precedingthepoly(A)sites of both theearly(nt
1498 to 2617) and late (nt 2991 to 3466) transcription units were measured. Analysis oftranscription complexes from wild-type virus revealed a9-to 10-fold ratio of RNA poly-merasestraversingthe latetranscriptionunitcomparedwith theearly transcriptionunit
(Fig.
8,WT). Analysis
ofmutant A1A2 virus transcription complexes also revealed a 9- to 10-foldhigherlevel of RNApolymerases onthe lateversus the early transcription unit(Fig.
8,A1A2). Therefore,
the mutations introduced intolargeT-antigen-binding
siteAhad noeffect onlevels ofearlytranscription.
Several stocks of A1A2 virus
generated by
transfection ofprimary babymousekidneycells with viral DNA hadtiters
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.98.256.87.313.2]A
6 h 30 h
WT AIA2 WT A1A2
WT A1A2 WT A1A2
48 h 72 h
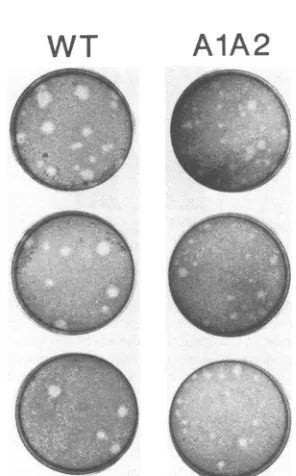
FIG. 9. Comparison of plaque size of wild-type and mutant
A1A2 viruses. Plaque assays were performed on primary baby
mouse kidneycells infectedwithwild-type (WT)ormutantA1A2
virus. Plaques were visualized by neutral red staining. Plaques formedbytwodifferentwild-type polyomavirusstrainsareshown;
the firsttwoplatesareAT3-Modori (63), andthirdplate is AT3(62).
(1 x 106to8 x 106PFU/ml) 50-to100-fold lower than those ofequivalentstocks ofwild-typevirus.A1A2virusalsogave
rise to smaller plaques than did wild-type virus (Fig. 9). Theseobservationsimpliedthat A1A2 virusgrowsless well
than wild-type virus. We therefore measured viral DNA replication and virus production after infection of baby
mouse kidney or 3T6 cells with either wild-type or A1A2
virus at an input multiplicity of 1 PFU per cell. Hirt extraction of infected monolayers revealed no detectable
differences betweenwild-type andA1A2virus in the
accu-mulation of viral DNAby 24, 30, 48,or72 h afterinfection
ofbabymousekidneycells(resultsfor6, 30, 48,and72 hare
shown in Fig. 10A). Similar results were obtained after infection of 3T6 cells(datanotshown). Yields of infectious
progenyvirus measuredbyplaqueassaysagainrevealedno
significant differences betweenwild-type andA1A2viruses (Fig. 10B). Restriction endonuclease digestion and DNA sequencing (data notshown) showed that the bulk of prog-eny DNAretained mutations in both GAGGC pentanucle-otides in the A1A2 virus in theseexperiments.Weconclude thatA1A2virusreplicatesits DNAgenomeandsynthesizes infectious progenyvirions at rates comparable to those of wild-type virus in bothbaby mouse kidney and 3T6 cells.
The reasons for small plaque size and low titers of virus
stocks generated by transfection remain obscure; these observations could beexplained bydelayedreleaseof A1A2 virus or by a small decrease in the rate of virus DNA replication (see Discussion).
DISCUSSION
We have examined the mechanism by which large T antigen blocks elongation by RNA polymerase II in vivo during transcription of the polyomavirus late DNAstrand. The results presented in this studysuggest that stalling by
0 24 48 72 96
[image:7.612.104.252.69.307.2]TIME POST-INFECTION(h)
FIG. 10. Growth properties of wild-type and mutant AiA2
vi-ruses. Primary babymousekidneycellswereinfectedwitheither wild-type (WT)ormutantA1A2virusatamultiplicity of 1 PFUper
cell. (A) Amountof viral DNA extractedfrom infected cells(38)at
6,30, 48, and 72hpostinfection. ViralDNAwasdigested with the
restriction endonucleaseHindIII, analyzed on a1%
agarose-Tris-borate-EDTAgel,andvisualizedby staining with ethidium bromide. Duplicate samples from each time point are shown. (B) Growth curves. At theindicated times, the amountofinfectious progeny
viruswasdeterminedby plaqueassay.Pointsrepresentaveragesof duplicate or triplicate samples of wild-type-infected cells (filled squares)andA1A2-infectedcells(open squares).
RNA polymerases in the intergenic region is mediated through the binding of large T antigen to the viral DNA template at siteA. Site A liesadjacenttotheorigin of viral DNA replication andconsists oftwo GAGGC pentanucle-otide sequence elements separatedbysixnucleotides (Fig. 1B). The interaction of large Tantigen withboth GAGGC pentanucleotides in site Aappearstobeessentialtoforman
effective blocktotranscriptionelongation,sincemutation of either of the GAGGC elements reduces stalling by RNA polymerase, andmutation ofboth elements eliminates stall-ing (Fig. 5). Althoughwe havenotperformed an extensive mutational analysis of large T-antigen-binding site A, the introduction ofmutations adjacenttothissitewasobserved to have little or no effect on stalling levels (AT3-Modori
sequence; Fig. 1B and Fig. 3). Therefore, the GAGGC pentanucleotides in site A appearto beessential sequence
elementsrequiredforlargeTantigentoblockelongation by RNApolymerase.
WT
A1A2
B
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.328.547.78.435.2]SeveralotherDNA-binding
proteins
have been shownto block elongation by RNA polymerases in anorientation-dependent manner(3, 18,25). In this study, an accumulation of RNA polymerases opposite site A on the early DNA strand was not observed (Fig. 7). This result implies that large T antigen may block only elongation by RNA poly-merases transcribing through the intergenic region on the late DNA strand. However, a moredetailedanalysiswill be necessarytodeterminewhetherlargeTantigenfunctions in an orientation-dependentmanner.
LargeTantigen also bindstothree othernearby sites
(B,
C, and1/2). Surprisingly,bindingtosites BandC, which lieupstreamof site A inthedirection of late transcription, has nomeasurable effect onRNApolymerase elongation, since
these sitescanbe inactivatedby mutationwithoutreducing stalling, andtheirpresencein the absenceofanintact siteA is not sufficientto induce stalling. Several explanations for these results canbe proposed. First, the GAGGC pentanu-cleotides in binding site C are oriented in the opposite direction to those in sites A and B
(see
Fig.1B).
If theorientationof boundlargeTantigen withrespectto elongat-ing RNA polymerases were important, RNA polymerases elongating in the latedirectionwouldnot beblocked atsite
C. However, this could not explain the
inability
oflarge
Tantigen boundat siteBtoblock
elongation. Second,
bound largeTantigenmaydissociaterapidlyfromsitesCandBin the infectedcell yet remain stablybound to site A. In thiscase, a largerproportion of DNA templates being
actively
transcribed may havelargeTantigenboundtositeAthanto site B or C. Little isknown about the occupancy of these sites in vivo. Third, the capacity of DNA-bound large TantigentoblockelongatingRNApolymeraseII maydepend on flanking sequences nearbyeach site, which could influ-encethetertiarystructureoflargeTantigenorits
dissocia-tionrate.Theseexplanationsare not
mutually
exclusive. We havenotbeen ableto testdirectlythedependenceofstalling ontheintegrity ofbinding site 1/2, asit isrequired
for DNAreplication and therefore forvirus
viability.
Itis knownthatlarge T antigen forms a complex double-hexamer structure aroundsite 1/2 and that this double hexamerenables DNA
replication to begin by unwinding the DNA helix and by attracting cellularproteinsto this site (10,
71).
Clearly, site1/2 by itself is not sufficient to block RNA polymerase elongation efficiently in the absence of an intact site A.
However, it may be that blockage
requires
site 1/2 in additionto site A.LargeTantigenboundto siteA mayblock
elongation
on the late DNA strand by physicallypreventing
RNApoly-merases from transcribing through the DNA-protein com-plex. The lac repressor protein has been shown to block
elongation byE. coliRNA
polymerase
andeukaryotic
RNApolymerasesI andII
(24,
25,42,
61), suggesting
that block-age in these cases occurby
simple
steric interference. Alternatively, large T antigen may interact with aspecific
domain of RNApolymerase IIwhichleads to
stalling.
Themurine TTFI
protein,
which terminatestranscription by
RNApolymerase I (5), doesnotterminate transcription byRNApolymerase IIorprokaryotic RNApolymerases (42).
In addition, the termination and DNA-binding functions of
TTFI
appeartoreside indifferentproteindomains(5).These results imply that a specific protein-protein interaction be-tween theblockingprotein and thetranscriptionelongation
complex is necessary fortermination tooccur. Itwill be of interesttodetermine whetherlargeTantigenboundtosite A is abletoblockelongation byother
eukaryotic
and prokary-otic RNApolymerases.RNApolymerasesmayrespondinanumber of waysto a
blocking protein. First,
RNApolymerases
may stall butretain the
ability
to continueelongationfollowing
the disso-ciation of the blockingprotein (52). Alternatively,
termina-tion oftranscription
may occur,resulting
in thedissociation of RNApolymerase
and its nascent RNA chain from the DNAtemplate (24,42).
The characteristics of theblocking
proteins
that determine theirability
to stall or terminatetranscription by
RNApolymerases
arenotyetunderstood.Theaccumulationof RNA
polymerases
in vivonearby large
T-antigen-binding
sites and theirsubsequent
reactivation invitro onviral
transcription complexes
indicatethatlarge
Tantigen
blockselongation by
astalling
mechanism(9, 62;
thisstudy).
However, it ispossible
that stalled RNApoly-merases
eventually
terminatetranscription
andarereleasedfrom the DNA template. An accumulation of stalled or paused RNA
polymerases
has been observed within the terminationregions
of several genes transcribedby
RNApolymerase
11(30,
47,69,
73).
Transcription by
RNApolymerases
through
prokaryotic
promoters(2, 34, 35)
andeukaryotic
promoters(6, 19, 22, 36,
50, 56,
68)
has been shown todisrupt
the formation of initiationcomplexes,
causing
promoter occlusion. RNApolymerase
II hasrecently
been observedto stall or termi-natetranscription
in the5'
flanking regions
ofseveralviral and cellular genes(3, 13, 17,
62).
Interestingly,
specific
DNA-binding
proteins
areresponsible,
at least in part, formediating
theseblockstotranscription
elongation
(3, 9, 11,
18,
58).
It has therefore beenproposed
that certainDNA-binding proteins
mayfunctiontopreventRNApolymerases
from readingthrough
downstream promoterregions
anddisplacing
proteins
that are necessary for the initiation oftranscription
(3, 11, 17,
18).
DNA-binding proteins
that bind upstreamof RNApolymerase
Ipromotershave been shown toprevent such interference fromoccurring (36, 49). Large
T-antigen-binding
site A is locatedsome200ntupstreamof thepolyomavirus
late promoter,suggesting
thatlarge
Tantigen
may function to prevent RNApolymerases
tran-scribing
the late DNA strand fromdisrupting
protein
binding
atthe late promoter.
However,
mutationoflarge
T-antigen-binding
site A had littleor noeffectonlatepromoterfunction(Fig. 4).
Two otherregulatory
elements are also locateddownstream of
binding
site A: theorigin
of viral DNAreplication
and the enhancerregion,
which augments viral DNAreplication
andtranscription
from theearly
promoter(26, 27, 51, 65). Perhaps large
Tantigen
bound to site Afunctionstopreventthe
displacement
ofproteins
assembled at theseregulatory
elementsby
readthrough
transcription.
The
binding
oflarge
Tantigen
tositeA,
besidesblocking
elongation by
RNApolymerases,
has beenproposed
to augmentthe initiation of viral DNAreplication
atadjacent
site1/2
(55)
andmayplay
arole inrepressing
transcription
initiationat the
early
promoter(23).
Inthisstudy,
mutationof both GAGGC
pentanucleotides
insite A hadnoapparenteffect on the
repression
ofearly
transcription
(Fig.
8).
However,
several nucleotides within the GAGGC pentanu-cleotideproximal
to site1/2
were found to beabsolutely
required
for virusgrowth
(mutations
AandAO;
Fig.
2).
Inaddition,
mutant A1A2 virusproduced
smallplaques,
sug-gesting
someimpairment
of virusgrowth
(Fig.
9).
Unexpect-edly,
furthercharacterizationof the A1A2mutantvirus did not show any reduction in the levels of viral DNAsynthe-sized
(Fig.
10A)
orintheproduction
of progeny virions(Fig.
lOB). Perhaps
the reduction inplaque
size that we have observed with the A1A2mutantvirus is duetoonly
asmallreduction in viral DNA
synthesis
that is not detectableby
on November 9, 2019 by guest
http://jvi.asm.org/
the assay used in this
study.
Severalpolyomavirus
mutantshaving small-plaque phenotypes
have been isolated and characterized(7, 8, 46, 65).
Some of these mutantviruses showonly
a two- tothreefold reduction in thelevel of DNAsynthesis
relative towild-type
virus.Alternatively,
the re-lease of infectiousprogenyvirions from infectedcells may be affectedby
mutations in site A. Thisimpairment
would account for thesmall-plaque phenotype (Fig. 9)
and ourinability
toobtainhigh-titer
virusstocks,
infour attempts,bytransfection of cells withmutantA1A2virus DNA
(data
notshown).
Inboth of thesesituations, repeated cycles
ofvirus infection and release arerequired, thereby revealing
anyimpairment
in virus release. Since all cells were initiallyinfected
during
the DNAsynthesis
andgrowthcurve exper-imentsperformedinthisstudy,noeffectonthe level of virus DNAsynthesizedor onvirusyieldwould beexpected,as we have observed (Fig. 10).CP1,MAZ,andTBF1areotherexamplesofDNA-binding
proteinswhich havebeen shownto stall or terminate tran-scription by RNA polymerase II (3, 11, 17, 18, 58). In addition to blocking elongation, CP1 and MAZ proteins functionasactivators oftranscriptioninitiation(4, 14). The TBF1
protein,
which bindsto asite within the P2 promoter of c-myc (58), may also participate in the initiation oftranscription. Displacement
of theseproteins
by elongatingRNA
polymerases
might
therefore be expectedto interfere withtranscription
initiation. Thebinding
oflargeT antigen tositeA,
besidesblocking elongation by
RNApolymerases,
also appears to berequired
for normal virusgrowth,
assuggested by
the smallplaque
size of the A1A2mutantvirus(Fig. 9). Perhaps
thestalling
of RNApolymerases
by largeTantigen
functionsprimarily
to prevent thedisplacement
oflarge
Tantigen
from siteA, thereby retaining
animportant
protein-DNA
interactionrequired
for the normal growth ofpolyomavirus.
ACKNOWLEDGMENTS
Wethank Lynn Kellyand SusanSillaots for excellent technical assistance and Peter Liston forhelpfulcomments.
J.B. was supported by studentships from the J. W. McConnell MemorialFund, the McGillFacultyofMedicine, andthe Medical Research Council of Canada.N.-A.S.wassupported bya student-shipfromTheCancer ResearchSociety, Inc.,andbytheMedical Research CouncilofCanada. Thisresearchwas supported bythe Medical ResearchCouncilofCanada(grant MT-7281).
REFERENCES
1. Acheson,N. H.1978.Polyomavirus giantRNAscontain tandem repeatsofthe nucleotide sequence of theentire viral genome. Proc. Natl. Acad. Sci. USA75:4745-4748.
2. Adhya, S.,and M.Gottesman. 1982. Promoterocclusion:
tran-scription through a promoter may inhibit its activity. Cell 29:939-944.
3. Ashfield,R.,P.Enriquez-Harris,and N.Proudfoot.1991. Tran-scriptionaltermination between closelylinked human
comple-mentgenesC2 and factor B:commonterminationfactor for C2 and c-myc. EMBO J.10:4197-4207.
4. Asselin, C.,A.Nepveu,andK. Marcu. 1989. Molecular
require-ments fortranscriptionalinitiation of the murine c-myc gene. Oncogene4:549-558.
5. Bartsch,I.,C.Schonenberg,and I.Grummt. 1988.Purification andcharacterizationofTTFI,afactor that mediates termination ofmouseribosomalDNAtranscription.Mol.Cell.Biol. 8:3891-3897.
6. Bateman, E.,and M. R.Paule. 1988.Promoter occlusionduring ribosomal RNAtranscription.Cell54:985-992.
7. Bendig, M. M., and W. R. Folk 1979. Deletion mutants of polyomavirusdefining a nonessentialregionbetween the
ori-gin of replication and the initiation codon for early proteins. J. Virol. 32:530-535.
8. Bendig, M. M., T. Thomas, and W. R. Folk 1980. Regulatory
mutantsofpolyomavirusdefectivein DNAreplication and the synthesis of early proteins. Cell 20:401-409.
9. Bertin, J., N.-A. Sunstrom, P. Jain,and N. H.Acheson. 1992. Stalling by RNA polymerase IIin thepolyomavirus intergenic region is dependent on functional large T antigen. Virology 189:715-724.
10. Borowiec, J. A.,F. B.Dean,P. A.Bullock,andJ.Hurwitz.1990. Binding and unwinding-how T antigenengagesthe SV40 origin of DNAreplication.Cell 60:181-184.
11. Bossone,S.A.,C.Asselin,A.J. Patel,and K. B.Marcu.1992. MAZ, a zinc finger protein, binds to c-myc and C2 gene sequencesregulating transcriptional initiation andtermination. Proc. Natl. Acad. Sci. USA 89:7452-7456.
12. Briggs, D.,D.Jackson,E.Whitelaw,and N.J. Proudfoot. 1989. Direct demonstration of termination signals for RNA
poly-merase II from the seaurchinH2Agene.Nucleic Acids Res. 17:8061-8071.
13. Chodchoy, N., N. B. Pandey, and W. F. Marzluff. 1991. An intact histone3' endprocessing site is required for transcription termination in a mouse histone H2A gene. Mol. Cell. Biol. 11:497-509.
14. Chodosh,L.A.,A. S.Baldwin,R. W.Carthew,andP. A.Sharp. 1988.HumanCCAAT-binding proteins have heterologous sub-units. Cell 53:11-24.
15. Cogen,B. 1978.Virus-specific earlyRNAin 3T6cellsinfected byatsAmutantofpolyoma virus. Virology 85:222-230. 16. Connelly, S., and J. L. Manley. 1988. A functional mRNA
polyadenylation signalisrequiredfortranscription termination byRNApolymeraseII.Genes Dev.2:440-452.
17. Connelly,S.,andJ.L.Manley.1989. ACCAATbox sequence in the adenovirusmajorlate promoterfunctionsaspartofan
RNApolymeraseIIterminationsignal. Cell 57:561-571. 18. Connelly, S., and J. L. Manley. 1989. RNA polymerase II
transcriptionterminationsignalis mediatedspecificallyby pro-tein binding to a CCAAT box sequence. Mol. Cell. Biol. 9:5254-5259.
19. Corbin, V., and T. Maniatis. 1989. Role of transcriptional interference in the Drosophila melanogaster Adh promoter switch. Nature(London)337:279-282.
20. Cowie, A., and R. Kamen. 1984. Multiple binding sites for polyomavirus large T antigen within regulatory sequences of polyomavirusDNA. J. Virol. 52:750-760.
21. Cowie,A., and R.Kamen. 1986. Guaninenucleotide contacts within viral DNA sequencesboundby largeTantigen. J. Virol. 57:505-514.
22. Cullen,B.R.,P.T.Lomedico,andG.Ju.1984.Transcriptional interference in avian retroviruses-implications for the
pro-moter insertion model ofleukaemogenesis. Nature (London) 307:241-245.
23. Dailey,L.,and C. Basilico.1985.Sequences inthepolyomavirus regulatory region involvedin viral DNAreplicationand early geneexpression.J. Virol. 54:739-749.
24. Deuschle, U., R. Gentz, andH. Bujard. 1986. Lac repressor blockstranscribingRNApolymeraseandterminates transcrip-tion.Proc. Natl. Acad.Sci.USA 83:4134-4137.
25. Deuschle, U., R. A. Hipskind, and H. Bujard. 1990. RNA polymerase II transcription blocked by Escherichia coli lac repressor. Science248:480-483.
26. deVilliers, J., and W. Schaffner. 1981. A small segment of polyomavirusDNAenhances theexpressionofacloned 3-glo-bin geneoveradistance of 1400 basepairs.Nucleic Acids Res. 9:6251-6264.
27. deVilliers, J., W. Schaffner, C. Tyndall, S. Lupton, and R. Kamen. 1984. PolyomavirusDNAreplication requires an
en-hancer.Nature(London)312:242-246.
28. Dilworth, S. M.,A. Cowie, R. Kamen, and B. Griffin. 1984. DNA binding activity of polyomavirus large tumor antigen. Proc.Natl. Acad. Sci.USA 81:1941-1945.
29. Eckhart, W. 1990. Polyomavirinae and their replication, p. 1593-1607. In B. N. Fields and D. M. Knipe (ed.), Fields
on November 9, 2019 by guest
http://jvi.asm.org/
virology, 2nd ed. RavenPress, New York.
30. Enriquez-Harris, P.,N.Levitt, D. Briggs, and N. J. Proudfoot. 1991. A pause site for RNApolymeraseII is associated with termination of transcription. EMBO J. 10:1833-1842.
31. Farmerie, W. G., and W. R. Folk. 1984. Regulation of poly-omavirus transcription by large tumor antigen. Proc. Natl. Acad. Sci. USA81:6919-6923.
32. Fenton, R. G., and C. Basilico. 1982.Regulation of polyomavi-rusearlytranscription intransformed cells by large T antigen. Virology 121:384-392.
33. Gaudray, P., C.Tyndall, R. Kamen, and F. Cuzin. 1981. The high affinity binding site on polyomavirus DNA for the viral large T antigen. NucleicAcidsRes. 9:5697-5710.
34. Gay, N. J., V. L. Tybulewicz, and J. E.Walker. 1986. Insertion of transposon Tn7into theEscherichia coliglmStranscriptional terminator. Biochem. J.234:111-117.
35. Hausler, B., and R. L. Somerville. 1979. Interaction in vivo between strong closely spaced constitutive promoters. J. Mol. Biol. 127:353-356.
36. Henderson, S. L., K. Ryan, and B. Sollner-Webb. 1989. The promoter-proximal rDNA terminator augments initiation by preventingdisruption of stable transcription complexescaused bypolymerase read-in. Genes Dev. 3:212-223.
37. Hendrickson, E. A., C. E. Fritze, W. R. Folk, and M. L. DePamphilis. 1987. The origin ofbidirectional DNAreplication inpolyomavirus. EMBO J. 6:2011-2018.
38. Hirt, B. 1967. Selective extraction of polyomavirus DNA from infectedmouse cellcultures. J. Mol. Biol. 26:365-369. 39. Katinka, M., and M. Yaniv. 1983. DNA replication origin of
polyomavirus: early proximal boundary. J. Virol. 47:244-248. 40. Kern, F. G., S. Pellegrini, A. Cowie, and C. Basilico. 1986.
Regulation ofpolyomavirus late promoter activity by viral early proteins. J. Virol. 60:275-285.
41. Kruse, B., N.Narasimhan, and G. Attardi. 1989.Termination of transcription in human mitochondria: identification and purifi-cation of a DNA binding protein factor thatpromotes termina-tion. Cell 58:391-397.
42. Kuhn, A.,I.Bartsch, andI.Grummt.1990.Specificinteraction of themurinetranscriptionterminator factorTTFIwithclass-I RNApolymerases. Nature (London) 344:559-562.
43. Kunkel, T. A. 1985. Rapid and efficientsite-specificmutagenesis without phenotypic selection. Proc. Natl. Acad. Sci. USA 82:488-492.
44. Lanoix, J., and N. H. Acheson. 1988. A rabbit1-globin polyad-enylation signal directs efficient termination of polyomavirus DNA.EMBO J.7:2515-2522.
45. Logan, J., E.Falck-Pederson, J. E.Darnell, Jr., and T.Shenk. 1987. A poly(A) addition site and a downstream termination region are required for efficient cessation of transcription by RNApolymerase II in the mouse c major-globin gene. Proc. Natl.Acad.Sci. USA84:8306-8310.
46. Luthman, H., M. Osterlund, and G. Magnusson. 1984. Inhibition ofpolyomavirusDNA synthesis by base pairsubstitutions at the replicationorigin. Nucleic Acids Res. 12:7503-7515.
47. Maa, M., J. M. Chinsky, V. Ramamurthy, B. D. Martin, and R. E.Kellems. 1990.Identification of transcription start sites at the 5' and 3' ends of the murine adenosine deaminase gene. J. Biol. Chem.265:12513-12519.
48. McKay, R. D. G. 1981. Binding of simian virus 40 large T antigen to DNA. J. Mol. Biol. 145:471-488.
49. McStay, B., and R.H.Reeder. 1990. ADNA-binding protein is required fortermination of transcription by RNA polymerase I in Xenopuslaevis. Mol. Cell. Biol. 10:2793-2800.
50. McStay, B., and R. H. Reeder. 1990. An RNA polymerase I terminator sitecan stimulate the adjacent ribosomal gene pro-moter by two distinct mechanisms in Xenopus laevis. Genes Dev.4:1240-1252.
51. Muller, W. J., C. R. Mueller, A. M. Mes, and J. Hassell. 1983. Polyomavirus origin for DNA replication comprises multiple geneticelements. J. Virol. 47:586-599.
52. Pavco, P. A., and D. A. Steege. 1990. Elongation by Escherichia
coli RNA polymerase is blocked in vitro by a site-specific DNA binding protein. J. Biol. Chem. 265:9960-9969.
53. Pomerantz, B. J., and J. A. Hassell. 1984. Polyomavirus and simian virus 40 large T antigen bind to common DNA se-quences. J. Virol. 49:925-937.
54. Pomerantz, B. J., C. R. Mueller, and J. A. Hassell. 1983. Polyomavirus large T antigen binds independently to multiple, unique regions on the viral genome. J. Virol. 47:600-610. 55. Prives, C., Y. Murakami, G. Kern, W. R. Folk, C. Basilico, and
J.Hurwitz. 1987. DNA sequence requirements for replication of polyomavirus DNA in vivo and in vitro. Mol. Cell. Biol. 7:3694-3704.
56. Proudfoot, N. J. 1986. Transcriptional interference and termina-tion between duplicated a-globulin gene constructs suggests a novel mechanism for gene regulation. Nature (London) 322: 562-565.
57. Proudfoot, N. J. 1989. How RNA polymerase II terminates transcription in higher eukaryotes. Trends Biochem. Sci. 14: 105-110.
58. Roberts, S., T. Purton, and D. L. Bentley. 1992. A protein-binding sitein the c-myc promoter functions as a terminator of RNA polymerase II transcription. Genes Dev. 6:1562-1574. 59. Sancar, G. B., A. Sancar, J. W.Little, and W. D. Rupp. 1982.
TheuvrBgeneofEscherichia coli has both lexA-repressed and LexA-independent promoters. Cell 28:523-530.
60. Sato, K.,R.Ito, K.-H. Baek, andK Agarwal. 1986. A specific DNA sequence controls termination of transcription in the gastringene. Mol. Cell. Biol. 6:1032-1043.
61. Sellitti, M. A., P. A. Pavco, and D. A. Steege. 1987. Lac repressor blocks in vivo transcription of lac control region DNA.Proc. Natl.Acad. Sci. USA 84:3199-3203.
62. Skarnes,W. C., D. C. Tessier, and N. H. Acheson. 1988. RNA polymerases stall and/or prematurely terminate nearby both early and late promoters on polyomavirus DNA. J. Mol. Biol. 203:153-171.
63. Sunstrom, N.-A. 1991. Ph.D. thesis. McGill University, Mon-treal, Canada.
64. Sunstrom, N.-A., N. H. Acheson, and J. A. Hassell. 1991. Determination of the origin-specific DNA-binding domain of polyomavirus large Tantigen. J. Virol. 65:6998-7003.
65. Tang,W.-J., S. L. Berger, S. J. Triezenberg, and W. R. Folk. 1987. Nucleotides in the polyomavirus enhancer that control viral transcription and DNA replication. Mol. Cell. Biol. 7:1681-1690.
66. Tantravahi,J.,M. Alvira, and E. Falk-Pederson. 1993. Charac-terization of the mouse ,B-major globin transcription region: a spacing sequence is required between the poly(A) signal se-quence and multiple downstream termination elements. Mol. Cell. Biol. 13:578-587.
67. Triezenberg, S. J., andW. R.Folk. 1984. Essential nucleotides in thepolyomavirus origin region. J. Virol. 51:437-444. 68. Vales, L. D., andJ. E. Darneli, Jr. 1989. Promoter occlusion
prevents transcription of adenovirus polypeptide IX mRNA until after DNAreplication. Genes Dev. 3:49-50.
69. Vandenbergh, D. J., M. James-Peterson, and R. C. Hardison. 1991. An apparent pause site in the transcription unit of the rabbit a-globin gene. J. Mol. Biol.220:225-271.
70. Wahle, E., and W. Keller. 1992. The biochemistry of 3' end cleavage and polyadenylation of messenger RNA precursors. Annu. Rev. Biochem. 61:419-440.
71. Wang, E. H., and C.Prives. 1991. ATP induces the assembly of polyomavirus large T antigeninto hexamers. Virology 184:399-403.
72. Whitelaw, E., and N. J. Proudfoot. 1986. Alpha thalassaemia caused by poly(A) site mutation reveals that transcription is linked to 3' endprocessing in the human a-2 globingene. EMBO J. 5:2915-2922.
73. Xu, M., M. B. Barnard, S. M. Rose, P. N. Cockerill, S.-Y. Huang, and W. T. Garrard. 1986. Transcriptional termination and chromatin structure of the active immunoglobulin K gene locus. J. Biol. Chem.261:3838-3845.