JOURNAL OFVIROLOGY, Oct. 2009, p. 10299–10304 Vol. 83, No. 19 0022-538X/09/$08.00⫹0 doi:10.1128/JVI.00217-09
Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Mapping of Functional Domains in Herpesvirus Saimiri Complement
Control Protein Homolog: Complement Control Protein Domain 2
Is the Smallest Structural Unit Displaying Cofactor and
Decay-Accelerating Activities
䌤
Akhilesh K. Singh,† Viveka Nand Yadav, Kalyani Pyaram, Jayati Mullick, and Arvind Sahu*
National Centre for Cell Science, Pune University Campus, Ganeshkhind, Pune 411007, India
Received 30 January 2009/Accepted 17 July 2009
Herpesvirus saimiri encodes a functional homolog of human regulator-of-complement-activation proteins named CCPH that inactivates complement by accelerating the decay of C3 convertases and by serving as a cofactor in factor I-mediated inactivation of their subunits C3b and C4b. Here, we map the functional domains of CCPH. We demonstrate that short consensus repeat 2 (SCR2) is the minimum domain essential for classical/lectin pathway C3 convertase decay-accelerating activity as well as for factor I cofactor activity for C3b and C4b. Thus, CCPH is the first example wherein a single SCR domain has been shown to display complement regulatory functions.
The complement system is an ancient and yet highly evolved effector mechanism of immune defense that forms an imper-ative branch of innate immunity (23, 46). In addition, recent findings have clearly revealed its role as a vital viaduct between the innate and acquired immune systems (6, 18). Thus, it is not surprising that the system helps in purging a wide array of invaders, including viruses. Consequently, for their successful survival, many viruses have developed mechanisms to subvert the host complement system (7, 24, 26, 29, 39, 45). Herpesvi-ruses and poxviHerpesvi-ruses, in particular, subvert host complement by encoding structural and/or functional homologs of human complement regulators belonging to the regulator-of-comple-ment-activation (RCA) family, by capturing host membrane complement regulators and by using cellular receptors for en-tering cells (1, 8, 15, 23).
The RCA proteins are formed by multiple tandem repeats of bead-like complement control protein (CCP) domains or short consensus repeats (SCRs) separated by short linkers. It has been suggested that the sequence variations enforced upon these SCR domain folds and the interdomain dynamics dictate the functionality of the complement regulators (17, 19, 44, 49). Because sequence similarity in herpesviral complement regu-lators varies between 43% and 89% and in poxviral comple-ment regulators exceeds 91%, it is likely that the structural diversity in herpesviral complement regulators may have re-sulted in functional differences in these proteins and/or have resulted in variation in structural requirements for comple-ment regulation. In the herpesviridae family, detailed func-tional characterization has been performed for complement regulators of Kaposi’s sarcoma-associated herpesvirus
(Ka-posica/KCP) (28, 42), herpesvirus saimiri (HVS) (CCPH) (10, 38), and rhesus rhadinovirus (RCP) (31). All these proteins showed conservation of complement regulatory activities, in-dicating thereby that structural diversity has not resulted in loss of complement regulatory functions in these proteins. How-ever, it is not clear whether sequence variations within the herpesviral complement regulators have resulted in differences
* Corresponding author. Mailing address: National Centre for Cell Science, Pune University Campus, Ganeshkhind, Pune 411007, India. Phone: 91-20-2570-8083. Fax: 91-20-2569-2259. E-mail: arvindsahu @nccs.res.in.
† Present address: Department of Molecular Biophysics and Bio-chemistry, School of Medicine, Yale University, New Haven, CT 06520.
[image:1.585.311.530.410.605.2]䌤Published ahead of print on 29 July 2009.
FIG. 1. Schematic illustration of sCCPH and SDS-PAGE analysis of purified recombinant sCCPH and its deletion mutants. (Top) Sche-matic representation of the structure of the soluble form of CCPH (sCCPH), which is composed of four SCRs. The domains are num-bered, and the minimum domains shown to be important for C3b and C4b cofactor activities (CFA) and CP DAA are identified. (Bottom) Expressed and purified sCCPH and its deletion mutants were analyzed by 12% (left) and 13% (right) SDS-PAGE under reducing conditions and stained with Coomassie blue. Molecular weights as determined by SDS-PAGE: for sCCPH, 32,000; for SCR1-3, 26,000; for SCR2-4, 27,500; for SCR1-2, 17,000; for SCR2-3, 17,500; for SCR3-4, 16,500; for SCR1, 9,500; for SCR2, 7,000; for SCR3, 8,000; and for SCR4, 8,000. Molecular mass is expressed as kilodaltons in the figure.
10299
on November 8, 2019 by guest
http://jvi.asm.org/
in the domain requirements for complement regulatory activ-ities, since mapping of functional domains has been performed only for Kaposica (30, 43). In the present study, we therefore have mapped the complement regulatory domains of HVS CCPH to get further insight into diversity in domain require-ments for functional activities.
HVS is a classical prototype of the gamma 2-herpesviruses or rhadinoviruses. It causes rapidly progressing fulminant lym-phoma, lymphosarcoma, and leukemia of T-cell origin in mar-mosets, owl monkeys, and other species of New World pri-mates but not in its natural host, the squirrel monkey (9, 16). Unlike other herpesviruses, it encodes two complement regu-lators: an RCA homolog (ORF 4; CCPH) that regulates the early steps of complement activation (2, 10) and a CD59 ho-molog (ORF 15) that inhibits the late steps of complement activation (4, 36). The RCA homolog is formed of four SCR modules (Fig. 1). As a result of alternative splicing, the protein is expressed as a full-length membrane-bound form (mCCPH) containing the transmembrane region as well as a spliced se-cretory form (sCCPH) lacking the transmembrane region (2). Earlier, we showed that sCCPH inhibits complement by tar-geting C3 convertases: (i) it supports serine protease factor I-mediated inactivation of C3b and C4b, the subunits of C3 convertases (cofactor activity), and (ii) it accelerates the irre-versible decay of the classical pathway (CP)/lectin pathway and to a limited extent the alternative pathway (AP) C3 converta-ses (decay-accelerating activity [DAA]) (38).
(This work was done in partial fulfillment of the Ph.D. thesis requirements of A.K.S., University of Pune, Pune, India.)
In order to map the functional domains of sCCPH, we have generated a series of soluble triple, double, and single SCR deletion mutants. In brief, the deletion mutants of sCCPH comprising SCR1-3, -2-4, -1-2, -2-3, and -3-4 as well as SCR1, -2, -3, and -4 were constructed from the full-length HVS sCCPH clone (38) by PCR amplification and cloning into the bacterial expression vector pET29. The authenticity of each of the clones was confirmed by DNA sequencing, and then they
were transformed into the Escherichia coli BL21 strain for
expression. The mutants carried the histidine tag at the C terminus and hence were purified to homogeneity by using histidine affinity chromatography. Refolding of the purified proteins was performed by using the rapid dilution method as previously described (38, 47, 48), and the refolded proteins were loaded onto a Superose 12 gel filtration column (Phar-macia) to obtain monodisperse populations of the expressed mutants (38, 48). The preservation of various functions in mutants (see below) suggests that the mutants have maintained their proper conformation. The expressed proteins were
⬎95% pure as judged by sodium dodecyl
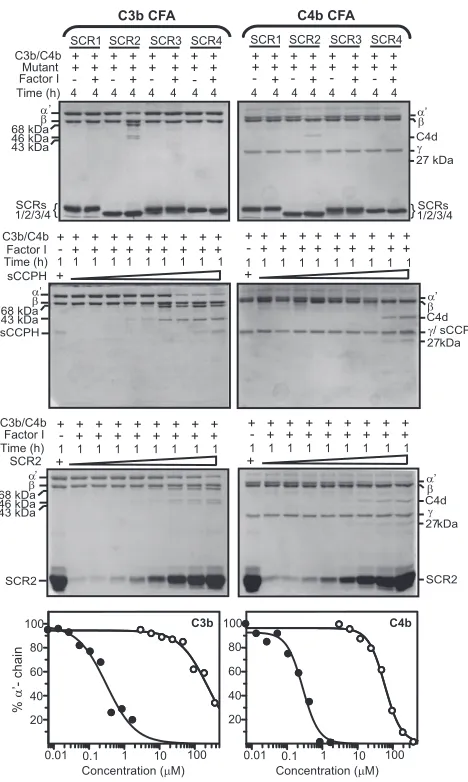
sulfate-polyacryl-amide gel electrophoresis (SDS-PAGE) analysis (Fig. 1). To identify the domains required for cofactor activities of sCCPH against C3b and C4b, we utilized a fluid phase assay wherein C3b or C4b was incubated with each of the deletion mutants and factor I, and inactivation of C3b/C4b (cleavage of
FIG. 2. Analysis of factor I cofactor activity of sCCPH and its deletion mutants for human complement proteins C3b and C4b. Cofactor activity was assessed by incubating 3.0g of human C3b (upper panels) or C4b (lower panels) with sCCPH/SCR1-3/SCR2-4 (4.0M) or SCR1-2/2-3/3-4 (24M) in the presence or absence of factor I (100 ng) for the indicated time periods at 37°C in 10 mM sodium phosphate, pH 7.4, containing 145 mM NaCl. The reactions were stopped by addition of sample buffer containing dithiothreitol, and the amount of C3b or C4b cleaved was visualized by subjecting the samples to SDS-PAGE analysis on 10% or 11.5% gel, respectively, and staining with Coomassie blue. During C3b cleavage, the␣⬘-chain is cleaved into N-terminal 68-kDa and C-terminal 46-kDa fragments. The 46-kDa fragment is then cleaved into a 43-kDa fragment. These cleavages indicate inactivation of C3b. In the case of C4b, the␣⬘-chain is cleaved into N-terminal 27-kDa, C-terminal 16-kDa (not visible in the gel), and central C4d fragments. These cleavages indicate the inactivation of C4b.
on November 8, 2019 by guest
http://jvi.asm.org/
the␣⬘-chain) was determined by running the samples on SDS-PAGE gels. It is clear from the data presented in Fig. 2 that sCCPH and the mutants SCR1-3, -2-4, and -1-2 supported the
cleavage of the␣⬘-chain of C3b. A very weak cleavage was also
supported by SCR2-3 and -3-4. The cleavage of the␣⬘-chain of
C4b, however, was supported by sCCPH and the mutants SCR1-3, -2-4, -1-2, and -2-3 but not by SCR3-4 (Fig. 2). To-gether, these data point out that SCR1 and -2 considerably contribute to the C3b and C4b cofactor activities of sCCPH but that SCR3 and SCR4 in the case of C3b cofactor activity and SCR3 in the case of C4b cofactor activity contribute to its optimal activity. These results, however, did not elucidate whether a single domain(s) could impart the cofactor activities. We therefore expressed the single-domain mutants (SCR1, SCR2, SCR3, and SCR4) and analyzed their cofactor activities. The results presented in Fig. 3 indicate that SCR2, by itself, possesses the ability to support factor I-mediated inactivation of C3b and C4b; SCR3 also displayed very weak cofactor activity against C3b when used at higher concentrations (88
M; data not shown). These results suggest that structural
elements involved in the interaction of sCCPH with factor I are primarily located within SCR2 and -3. Admittedly, the single-domain mutants possess very weak cofactor activities and other domains too contribute to the optimal activity; the cofactor activities of SCR2 for C3b and C4b were 781- and 212-fold lower than that for sCCPH (Fig. 3). It should be mentioned here that earlier observations on mapping of the human RCA proteins (factor H, C4b-binding protein, membrane cofactor protein, and complement receptor 1) (3, 11–13, 21), Kaposica (30), and vaccinia virus CCP (VCP) (27) indicated that a min-imum of two (in Kaposica) or three (in all other RCA proteins) successive SCR domains are necessary for factor I cofactor activities. Thus, sCCPH is the first complement regulator in which a single SCR domain has been shown to display the factor I cofactor function.
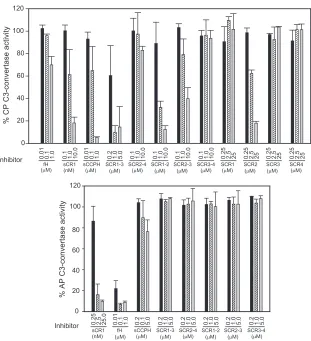
As discussed above, in addition to the inactivation of sub-units of C3 convertases (C3b and C4b), sCCPH also regulates C3 convertases by accelerating their decay. It possesses con-siderable DAA for the CP/lectin pathway C3 convertase (C4b,2a) and a poor decay activity for the AP C3 convertase (C3b,Bb). Thus, we next examined the DAAs of the various sCCPH mutants to map the domains required for this function. To measure the CP C3 convertase decay activity, the C4b,2a enzyme was formed on sheep erythrocytes and allowed to decay in the presence of various mutants. The remaining en-zyme activity was then measured by incubating the reaction mixture with EDTA sera (a source of C3 to C9) and measuring hemolysis. Apart from sCCPH, mutants SCR1-3, -1-2, and -2-3 showed substantial DAA for the CP C3 convertase (Fig. 4). These data suggested that SCR1-3 is primarily responsible for this activity. On a molar basis, SCR1-3 was 1.6-fold less effi-cient than sCCPH. Because both SCR1-2 and SCR2-3 pos-sessed the decay activity, it was likely that similar to the cofac-tor activities, a single SCR domain of sCCPH might also possess the DAA for the CP C3 convertase. Hence, we also assessed the DAAs of the single-domain mutants. Interestingly again, SCR2 was the only single domain that distinctly dis-played CP DAA (Fig. 4); however, on a molar basis, it was 26-fold less active than sCCPH. Previous data on the involve-ment of SCR domains in decay acceleration of CP C3
conver-tase in human RCA proteins (decay-accelerating factor, com-plement receptor 1, and C4b-binding protein) (3, 5, 20) and viral RCA homologs (Kaposica and VCP) (27, 30) have shown that a minimum of two or three consecutive domains are nec-essary for the activity. Thus, sCCPH is the only prototype to date in which a single SCR is adequate to impart the CP DAA. Although sCCPH is known to possess limited AP C3
con-FIG. 3. Analysis of factor I cofactor activity (CFA) of single SCR mutants of sCCPH for human complement proteins C3b and C4b. (Upper panels) Cofactor activity was assessed by incubating 3.0g of human C3b or C4b with the single SCR mutants (44 M) in the presence or absence of factor I (100 ng) for 4 h at 37°C in PBS (10 mM sodium phosphate, pH 7.4, containing 145 mM NaCl). The reactions were stopped by addition of sample buffer containing dithiothreitol, and the amount of C3b or C4b cleaved was visualized by subjecting the samples to 13% SDS-PAGE and stained with Coomassie blue. Cleav-age of the␣⬘-chain of C3b and C4b and generation of cleavage prod-ucts indicate the inactivation of these proteins. (Middle panels) Hu-man C3b (3.0g) or C4b (3.0g) and factor I (100 ng) were incubated in PBS with increasing concentrations of sCCPH or the SCR2 mutant at 37°C for 1 h, and the cleavage products were analyzed as described above. (Lower panels) The intensity of the␣⬘-chains of C3b and C4b in the middle panels was determined densitometrically and is repre-sented graphically. The closed and open circles represent sCCPH and the SCR2 mutant, respectively.
VOL. 83, 2009 NOTES 10301
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.585.302.536.65.456.2]vertase DAA, we sought to determine whether this limited activity is localized in a specific region or the full-length pro-tein. To measure the AP DAA, the C3 convertase C3b,Bb was formed on the sheep erythrocytes and incubated with sCCPH or with each of its deletion mutants. The decay of the AP C3 convertase was assessed by adding EDTA sera and measuring hemolysis. Although the full-length protein displayed a limited AP C3 convertase, none of the deletion mutants exhibited any activity (Fig. 4).
Inactivation of C3 convertases by the RCA proteins, owing to their cofactor and decay activities, requires interaction of these proteins with C3b and C4b. The ligand binding activity of the RCA proteins, however, does not always correlate with their cofactor and decay activities (12, 34), as apart from ligand binding, cofactor activity involves interaction of the RCA pro-tein with factor I (40), and decay activity involves interaction of the RCA protein with C2a or Bb (22, 25). In order to deter-mine whether cofactor and decay activity data of sCCPH and the various mutants correlate with the ligand binding data, we
[image:4.585.137.448.65.406.2]measured binding of these proteins to C3b and C4b by using a surface plasmon resonance-based assay (38). As observed ear-lier (38), sCCPH displayed higher affinity for C4b than for C3b (Fig. 5 and Table 1). When we measured binding of various deletion mutants to C3b and C4b, only SCR2-4 showed bind-ing to C3b, and SCR1-3 showed bindbind-ing to C4b (Fig. 5). How-ever, there were reductions of about 16- and 14-fold in the affinities of these deletion mutants for C3b and C4b, respec-tively, compared to that for sCCPH (Table 1), suggesting that all the four domains contribute to binding to C3b and C4b. Because most of the deletion mutants that displayed com-plement regulatory activities possessed negligible binding to C3b and C4b, it is clear that binding of the mutants does not correlate with their cofactor and decay activities. It is likely that during cofactor activity, interaction of the mutants with C3b and C4b is stabilized by the interaction of factor I with C3b/C4b and the mutants. Similarly, during DAAs, the mu-tants may possess better affinity for the convertases than their subunits C3b and C4b. Consistent with this argument,
FIG. 4. Analysis of CP and AP C3 convertase DAAs of sCCPH and its mutants. (Upper panel) The CP C3 convertase C4b,2a was formed on antibody-coated sheep erythrocytes (EA) by sequentially incubating them with human C1, C4, and C2 (Calbiochem). The C3 convertase on the cells was then allowed to decay by incubating EA-C4b,2a with various concentrations of sCCPH or its mutants for 5 min at 22°C, and the activity of the remaining enzyme was assessed by measuring the cell lysis following incubation for 30 min at 37°C with Guinea pig sera containing 40 mM EDTA (27, 32). (Lower panel) The AP C3 convertase C3b,Bb was formed on sheep erythrocytes (ES) by incubating them with human C3 (Calbiochem) and factors B and D in the presence of NiCl2. The C3 convertase on the cells was then allowed to decay by incubating ES-C3b,Bb
with various concentrations of sCCPH or its mutants for 10 min at 37°C, and the activity of the remaining enzyme was assessed by measuring the cell lysis following incubation with EDTA-sera for 30 min at 37°C (35, 37). The data obtained were normalized by considering the lysis that occurred in the absence of an inhibitor as 100% lysis.
on November 8, 2019 by guest
http://jvi.asm.org/
decay-accelerating factor has previously been shown to bind to CP C3 convertase with 1,000-fold higher affinity than to C4b (33).
The presence of SCR domains is not restricted to com-plement regulators, as SCR domains are also present in other complement proteins (e.g., C1r, C1s, MASP-1, MASP-2, factor B, C2, C6, and C7) and noncomplement
proteins (e.g., 2-GPI, interleukin-2 and -15 receptors,
GABAB receptor type 1a, E-selectin, brevican, CSMD-1, and polydom) (41). The SCR domains are always present as a pair or more, and the presence of a single SCR domain in proteins is rare (e.g., interleukin-15R and brevican). Fur-ther, data obtained thus far from domain mapping studies indicate that a minimum of two successive SCR domains are required for imparting any function. Together, these find-ings led to a paradigm: a two-SCR structure is the smallest
basic structural unit required for exhibiting any function (44). In the present study, data obtained for HVS sCCPH elucidate for the first time that a single SCR domain (SCR2) is able to impart factor I cofactor activities as well as DAA. Therefore, clearly, the current belief regarding the require-ment of multiple domains for displaying any functional ac-tivity requires revision. We would like to point out here that though earlier studies of viral complement regulators have used comparable molar excess of regulators for domain mapping studies, similar studies performed for human com-plement regulators utilized 5- to 50-fold less molar excess of regulators than the present study. Thus, it is likely that single domains in human complement regulators too may possess the complement regulatory activities.
In summary, our findings demonstrate that though three SCR domains of HVS CCPH are necessary for displaying the optimum complement regulatory activities, a single domain is sufficient to impart the various complement regulatory activi-ties. These data therefore point out that sequence variations in herpesviral complement regulators have resulted in a notable difference in domain requirements for the functional activities in these proteins.
We thank John D. Lambris (Department of Pathology and Labora-tory Medicine, University of Pennsylvania, Philadelphia, PA) and Mi-chael K. Pangburn (Department of Biochemistry, University of Texas Health Center, Tyler, TX) for their continuous support. We also thank John Bernet for site-specific labeling of C3b and C4b with biotin and express appreciation to Yogesh Panse and Sarang Satoor for their excellent technical assistance.
[image:5.585.81.504.70.252.2]This work was supported by the Wellcome Trust Senior Research Fellowship in Biomedical Science in India and a project grant from the Department of Biotechnology, New Delhi, India, to A.S. We also FIG. 5. Binding of sCCPH and its mutants to C3b and C4b. Binding was determined by a surface plasmon resonance-based assay (38). Sensograms were generated by immobilizing biotinylated C3b (1,200 response units [RUs]) and C4b (940 RUs) on streptavidin chips (Sensor Chip SA; Biacore AB; additional RUs of C3b [⬃6,000 RUs] were deposited by forming AP C3 convertase on the chip and flowing native C3 [14]) and injecting sCCPH or its mutants in PBS-T (10 mM sodium phosphate and 145 mM NaCl, pH 7.4, containing 0.05% Tween 20) over the chip. Flow cells immobilized with bovine serum albumin-biotin (Sigma) served as control flow cells. (Left panels) Binding of sCCPH and its various mutants to C3b (top) and C4b (bottom). The sensograms were generated by injecting 500 nM and 2M of sCCPH and its various mutants over C3b and C4b chips, respectively. (Middle panels) Sensogram overlay for the interaction between sCCPH and C3b (top) or sCCPH and C4b (bottom). (Right panels) Sensogram overlay for the interaction between SCR2-4 and C3b (top) and SCR1-3 and C4b (bottom). The concentrations of proteins injected are indicated at the right of the sensograms. The solid lines in the top middle and top right panels represent the global fitting of the data to a 1:1 Langmuir binding model with a drifting baseline (A⫹B7AB; Biaevaluation 4.1). The small arrows in the bottom middle and right panels indicate the time points used for evaluating the steady-state affinity data.
TABLE 1. Kinetic and affinity data for the interactions of sCCPH and the deletion mutants with human complement
proteins C3b and C4ba
Ligand Analyte kd(1/s)/ka
(1/m 䡠 s) SE (kd/ka) KD(m) 2
C3b sCCPH 4.6⫻10⫺3/206 4.41⫻10⫺5/11.3 2.23⫻10⫺5 2.02b
C4b sCCPH NA NA 3.51⫻10⫺7 1.69c
C3b SCR2-4 0.0542/156 4.74⫻10⫺4/12.4 3.48⫻10⫺4 1.63b
C4b SCR1-3 NA NA 4.9⫻10⫺6 3.27c
a
NA, not applicable;ka, association rate constant;kd, dissociation rate
con-stant;KD, equilibrium rate constant; SE, standard error. b
Data were calculated by global fitting to a 1:1 Langmuir binding model with a drifting baseline (BIAevaluation 4.1).
c
Data did not fit the 1:1 model and were calculated by steady-state analysis (BIAevaluation 4.1).
VOL. 83, 2009 NOTES 10303
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.43.284.611.675.2]acknowledge the financial assistance to A.K.S., K.P., and V.N.Y. from the Council of Scientific and Industrial Research, New Delhi, India, and to J.M. from the Department of Science and Technology, New Delhi, India.
REFERENCES
1.Ahmad, M., K. Pyaram, J. Mullick, and A. Sahu.2007. Viral complement regulators: the expert mimicking swindlers. Indian J. Biochem. Biophys.
44:331–343.
2.Albrecht, J. C., and B. Fleckenstein.1992. New member of the multigene family of complement control proteins in herpesvirus saimiri. J. Virol.66:
3937–3940.
3.Blom, A. M., L. Kask, and B. Dahlback.2001. Structural requirements for the complement regulatory activities of C4BP. J. Biol. Chem.276:27136– 27144.
4.Bramley, J. C., A. Davies, and P. J. Lachmann.1997. Herpesvirus saimiri CD59—baculovirus expression and characterisation of complement inhibi-tory activity. Biochem. Soc. Trans.25:354S.
5.Brodbeck, W. G., D. Liu, J. Sperry, C. Mold, and M. E. Medof.1996. Localization of classical and alternative pathway regulatory activity within the decay-accelerating factor. J. Immunol.156:2528–2533.
6.Carroll, M. C. 2004. The complement system in regulation of adaptive immunity. Nat. Immunol.5:981–986.
7.Cooper, N. R.1998. Complement and viruses, p. 393–407.InJ. E. Volanakis and M. M. Frank (ed.), The human complement system in health and disease. Marcel Dekker, Inc., New York, NY.
8.Finlay, B. B., and G. McFadden.2006. Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell124:767–782. 9.Fleckenstein, B., and R. C. Desrosiers.1982. Herpesvirus saimiri and
her-pesvirus ateles, p. 253–332.InB. Roizman (ed.), The herpesviruses. Plenum Publishing Corporation, New York, NY.
10.Fodor, W. L., S. A. Rollins, S. Biancocaron, R. P. Rother, E. R. Guilmette, W. V. Burton, J. C. Albrecht, B. Fleckenstein, and S. P. Squinto.1995. The complement control protein homolog of herpesvirus saimiri regulates serum complement by inhibiting C3 convertase activity. J. Virol.69:3889–3892. 11.Gordon, D. L., R. M. Kaufman, T. K. Blackmore, J. Kwong, and D. M.
Lublin.1995. Identification of complement regulatory domains in human factor H. J. Immunol.155:348–356.
12.Hourcade, D., M. K. Liszewski, M. Krych-Goldberg, and J. P. Atkinson.
2000. Functional domains, structural variations and pathogen interactions of MCP, DAF and CR1. Immunopharmacology49:103–116.
13.Iwata, K., T. Seya, Y. Yanagi, J. M. Pesando, P. M. Johnson, M. Okabe, S. Ueda, H. Ariga, and S. Nagasawa.1995. Diversity of sites for measles virus binding and for inactivation of complement C3b and C4b on membrane cofactor protein CD46. J. Biol. Chem.270:15148–15152.
14.Jokiranta, T. S., J. Hellwage, V. Koistinen, P. F. Zipfel, and S. Meri.2000. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J. Biol. Chem.275:27657–27662.
15.Judson, K. A., J. M. Lubinski, M. Jiang, Y. Chang, R. J. Eisenberg, G. H. Cohen, and H. M. Friedman.2003. Blocking immune evasion as a novel approach for prevention and treatment of herpes simplex virus infection. J. Virol.77:12639–12645.
16.Jung, J. U., and R. C. Desrosiers.1994. Herpesvirus saimiri and ateles, p. 614–622.InR. Webster and A. Granoff (ed.), Encyclopedia of virology. Saunders Scientific Publications, Inc., Philadelphia, PA.
17.Kirkitadze, M. D., and P. N. Barlow.2001. Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunol. Rev.180:146–161.
18.Kohl, J.2006. The role of complement in danger sensing and transmission. Immunol. Res.34:157–176.
19.Krych-Goldberg, M., and J. P. Atkinson.2001. Structure-function relation-ships of complement receptor type 1. Immunol. Rev.180:112–122. 20.Krych-Goldberg, M., R. E. Hauhart, V. B. Subramanian, B. M. Yurcisin,
D. L. Crimmins, D. E. Hourcade, and J. P. Atkinson.1999. Decay acceler-ating activity of complement receptor type 1 (CD35). Two active sites are required for dissociating C5 convertases. J. Biol. Chem.274:31160–31168. 21.Kuhn, S., C. Skerka, and P. F. Zipfel.1995. Mapping of the complement
regulatory doamins in the human factor H-like protein 1 and in factor H. J. Immunol.155:5663–5670.
22.Kuttner-Kondo, L. A., M. P. Dybvig, L. M. Mitchell, N. Muqim, J. P. Atkinson, M. E. Medof, and D. E. Hourcade.2003. A corresponding tyrosine residue in the C2/factor B type A domain is a hot spot in the decay accel-eration of the complement C3 convertases. J. Biol. Chem.278:52386–52391. 23.Lachmann, P. J.2002. Microbial subversion of the immune response. Proc.
Natl. Acad. Sci. USA99:8461–8462.
24.Lambris, J. D., D. Ricklin, and B. V. Geisbrecht.2008. Complement evasion by human pathogens. Nat. Rev. Microbiol.6:132–142.
25.Lukacik, P., P. Roversi, J. White, D. Esser, G. P. Smith, J. Billington, P. A. Williams, P. M. Rudd, M. R. Wormald, D. J. Harvey, M. D. Crispin, C. M. Radcliffe, R. A. Dwek, D. J. Evans, B. P. Morgan, R. A. Smith, and S. M. Lea.
2004. Complement regulation at the molecular level: the structure of decay-accelerating factor. Proc. Natl. Acad. Sci. USA101:1279–1284.
26.Means, R. E., J. K. Choi, H. Nakamura, Y. H. Chung, S. Ishido, and J. U. Jung.2002. Immune evasion strategies of Kaposi’s sarcoma-associated her-pesvirus. Curr. Top. Microbiol. Immunol.269:187–201.
27.Mullick, J., J. Bernet, Y. Panse, S. Hallihosur, A. K. Singh, and A. Sahu.
2005. Identification of complement regulatory domains in vaccinia virus complement control protein. J. Virol.79:12382–12393.
28.Mullick, J., J. Bernet, A. K. Singh, J. D. Lambris, and A. Sahu.2003. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) open read-ing frame 4 protein (kaposica) is a functional homolog of complement control proteins. J. Virol.77:3878–3881.
29.Mullick, J., A. Kadam, and A. Sahu.2003. Herpes and pox viral complement control proteins: ‘the mask of self.’ Trends Immunol.24:500–507. 30.Mullick, J., A. K. Singh, Y. Panse, V. Yadav, J. Bernet, and A. Sahu.2005.
Identification of functional domains in kaposica, the complement control protein homolog of Kaposi’s sarcoma-associated herpesvirus (human her-pesvirus 8). J. Virol.79:5850–5856.
31.Okroj, M., L. Mark, A. Stokowska, S. W. Wong, N. Rose, D. J. Blackbourn, B. O. Villoutreix, O. B. Spiller, and A. M. Blom.2009. Characterization of the complement inhibitory function of rhesus rhadinovirus complement con-trol protein (RCP). J. Biol. Chem.284:505–514.
32.Pan, Q., R. O. Ebanks, and D. E. Isenman.2000. Two clusters of acidic amino acids near the NH2 terminus of complement component C4 alpha⬘ -chain are important for C2 binding. J. Immunol.165:2518–2527. 33.Pangburn, M. K.1986. Differences between the binding sites of the
com-plement regulatory proteins DAF, CR1, and factor H on C3 convertases. J. Immunol.136:2216–2221.
34.Pangburn, M. K., K. L. Pangburn, V. Koistinen, S. Meri, and A. K. Sharma.
2000. Molecular mechanisms of target recognition in an innate immune system: interactions among factor H, C3b, and target in the alternative pathway of human complement. J. Immunol.164:4742–4751.
35.Pryzdial, E. L., and D. E. Isenman.1986. A reexamination of the role of magnesium in the human alternative pathway of complement. Mol. Immu-nol.23:87–96.
36.Rother, R. P., S. A. Rollins, W. L. Fodor, J. C. Albrecht, E. Setter, B. Fleckenstein, and S. P. Squinto.1994. Inhibition of complement-mediated cytolysis by the terminal complement inhibitor of herpesvirus saimiri. J. Vi-rol.68:730–737.
37.Sahu, A., T. R. Kozel, and M. K. Pangburn.1994. Specificity of the thioester-containing reactive site of human C3 and its significance to complement activation. Biochem. J.302:429–436.
38.Singh, A. K., J. Mullick, J. Bernet, and A. Sahu.2006. Functional charac-terization of the complement control protein homolog of herpesvirus saimiri: R118 is critical for factor I cofactor activities. J. Biol. Chem.281:23119– 23128.
39.Smith, G. L., J. A. Symons, A. Khanna, A. Vanderplasschen, and A. Alcami.
1997. Vaccinia virus immune evasion. Immunol. Rev.159:137–154. 40.Soames, C. J., and R. B. Sim.1997. Interactions between human
comple-ment components factor H, factor I and C3b. Biochem. J.326:553–561. 41.Soares, D. C., and P. N. Barlow.2005. Complement control protein modules
in the regulators of complement activation, p. 19–62.InD. Morikis and J. D. Lambris (ed.), Structural biology of the complement system. Taylor & Fran-cis, New York, NY.
42.Spiller, O. B., D. J. Blackbourn, L. Mark, D. G. Proctor, and A. M. Blom.
2003. Functional activity of the complement regulator encoded by Kaposi’s sarcoma-associated herpesvirus. J. Biol. Chem.278:9283–9289.
43.Spiller, O. B., L. Mark, C. E. Blue, D. G. Proctor, J. A. Aitken, A. M. Blom, and D. J. Blackbourn. 2006. Dissecting the regions of virion-associated Kaposi’s sarcoma-associated herpesvirus complement control protein re-quired for complement regulation and cell binding. J. Virol.80:4068–4078. 44.Stehle, T., and M. Larvie.2003. Structure of complement control proteins, p. 231–253.InR. A. B. Ezekowitz and J. A. Hoffmann (ed.), Innate immunity. Humana Press, Totowa, NJ.
45.Stoiber, H., M. Pruenster, C. G. Ammann, and M. P. Dierich.2005. Com-plement-opsonized HIV: the free rider on its way to infection. Mol. Immu-nol.42:153–160.
46.Volanakis, J. E., and M. M. Frank.1998. The human complement system in health and disease. Marcel Dekker, Inc., New York, NY.
47.White, J., P. Lukacik, D. Esser, M. Steward, N. Giddings, J. R. Bright, S. J. Fritchley, B. P. Morgan, S. M. Lea, G. P. Smith, and R. A. Smith.2004. Biological activity, membrane-targeting modification, and crystallization of soluble human decay accelerating factor expressed in E. coli. Protein Sci.
13:2406–2415.
48.Yadav, V. N., K. Pyaram, J. Mullick, and A. Sahu.2008. Identification of hot spots in the variola virus complement inhibitor (SPICE) for human comple-ment regulation. J. Virol.82:3283–3294.
49.Zhang, L., and D. Morikis.2006. Immunophysical properties and prediction of activities for VCP and SPICE using molecular dynamics and electrostatics. Biophys. J.90:3106–3119.

![FIG. 5. Binding of sCCPH and its mutants to C3b and C4b. Binding was determined by a surface plasmon resonance-based assay (38).Sensograms were generated by immobilizing biotinylated C3b (1,200 response units [RUs]) and C4b (940 RUs) on streptavidin chips (Sensor Chip](https://thumb-us.123doks.com/thumbv2/123dok_us/158536.35680/5.585.43.284.611.675/determined-resonance-sensograms-generated-immobilizing-biotinylated-response-streptavidin.webp)
