0022-538X/80/09-0682/12$02.00/0
Transcriptional
Map
for Newcastle Disease Virus
PETER L. COLLINS,* LAWRENCE E. HIGHTOWER, ANDL. ANDREWBALLt
MicrobiologySection U-44, Biological Sciences Group, University of Connecticut, Storrs, Connecticut06268
Atranscriptionalmap ofNewcastledisease virus wasdeterminedbymeasuring
the kinetics ofUV inactivation of the transcription ofindividualgenes and of viral infectivity.The inactivation of single genes wasmonitoredbymeasuring the reduction in theaccumulation of viralgeneproductsin vivo and invitro.Invivo, the accumulation of viral polypeptides in infected cells was measured after reversal ofacycloheximidetreatmentdesignedtoinhibit secondarytranscription. Actinomycin Danda hypertonic mediumwere usedtodecreaseselectively the synthesis of hostcellpolypeptides in infected cells.Invitro, mRNA'ssynthesized by irradiated viruses were analyzed by translation in cell-free systems under conditions in which the amount of each polypeptide synthesized reflected the relative abundanceof the corresponding mRNA. UV targetsizeswereobtained for thegenescoding for the HN, Fo, NP, M, L,and Ppolypeptides; the
47,000-dalton protein was notdetected. Acomparison ofthe UV targetsizes with the
corresponding genesizessuggested thattranscription of thesegenesinitiated at a single promotor and proceeded in the order NP, P, (Fo, M), HN, L. These experimentswereperformed with Newcastle disease virus strains Australia-Vic-toria andB1-Hitchner; for both strains, two forms ofthe P polypeptide which differed in electrophoretic mobilitywere detected. Proof that the P protein is
virus specific was obtained. Inaddition, infection of chicken embryo cells with avirulent strainB1-Hitchner enhanced the accumulation ofatleast fourtpolypep-tides thatappeared tobespecified by the host cell rather than by the infecting virus.
Measurement of the sensitivity ofa gene to
inactivation by UV irradiation can be used to
determine the proximity of its promotor site (18). Whenseveralgenes aretranscribed froma
single promotor, the order oftranscription can also be determined. For several rhabdoviruses andparamyxoviruses, thesingle-strandedRNA
genomes appear to contain a single promotor
site (2, 4-7, 15-17, 19), in contrast to the
seg-mentedgenomesof orthomyxoviruses (1, 39).A
completetranscriptionalmap has beenobtained
for the rhabdovirus vesicular stomatitis virus (VSV) (2,4, 6, 7,14).
Thedetermination of the VSVtranscriptional order, in conjunction with physical mapping
studies (20), provided considerable insight into the mechanism oftranscription bystandard and
defectiveparticles.The orderprovidedan
expla-nation for the relative amounts of viral gene products which accumulate in vitro and in vivo
(45). Measurements of the UV target sizes for
transcription of individual genes provided the basis forinterpretation oftarget sizes obtained for viral activities, such as cell killing (33) and inhibition of host cell protein (34) and RNA
tPresent address: Biophysics Laboratory, University of Wisconsin,Madison,WI 53706.
syntheses (47).Finally, the VSV transcriptional order was found to correspond to the 3'-to-5'
physical order of the viralgenes (20).
The UVmapping studies reportedinthis pa-per were conducted to investigate the
mecha-nism oftranscription, the organization, and the
coding capacity of thegenomeoftheavian
par-amyxovirus Newcastle disease virus (NDV) (9). The genome of this virus is a single negative
strand of RNA with a molecular weight esti-matedbyelectron microscopytobe 5.1x 106to 5.7 x 106 (27). Transcription of the genome in
vitro orextraction of RNA from infected cells
yields at least six electrophoretically distinct
viralmRNAspecies(5, 12, 13, 26, 42, 44). Trans-lation ofthe mRNA's in vitro or extraction of
polypeptidesfrominfectedcells yields polypep-tides corresponding tosix or seven viralgenes (5, 11, 13, 22, 35;G. W.Smith, J. C. Schwalbe, and L. E.Hightower, in C. F. Fox, ed., Biological
Assembly,inpress).Thefollowingviral
polypep-tides are known: HN, the hemagglutinin-neur-aminidaseglycoprotein; F, the fusion
glycopro-tein; Fo, a glycosylated precursor of F (36, 40);
NP, the most abundant protein of the nucleo-capsid; P,second in abundanceamong the core-associatedproteins; L,thelargest proteinof the
nucleocapsid; and a 47-kilodalton minor
poly-682
on November 10, 2019 by guest
http://jvi.asm.org/
VOL. 35, 1980
peptide. The P proteins of NDV have been identified and characterized by G. W. Smith and L. E. Hightower (manuscript in preparation) and by T. G. Morrison (manuscript in preparation). The X polypeptide described by Madansky and Bratt (32) is also a P-related protein (personal communication).
In this work, transcriptional map positions were obtained for the genes coding for proteins NP, P, Fo, M, HN, and L, which together rep-resent most orall ofthe genome coding capacity. For transcriptional mapping in vivo, a cyclohex-imidetreatment was used toinhibit secondary transcription, which otherwise obscured the po-lar effects of UV irradiation. In infected cells, viral polypeptides translated from primary tran-scripts could be detected clearly only by using the avirulent strain Bl-Hitchner (Bl) rather than the virulent strain Australia-Victoria(A V).
Since strainBlhasnotbeen wellcharacterized, its polypeptides were identified by comparison with thoseof the previously studied strain AV.
In the course of this comparison, it was observed
that at least two electrophoretically distinct forms of the NDV P polypeptide accumulated during infections by both strains. Furthermore, infection by the avirulent strain led to an
in-creaseintheaccumulation ofatleast four poly-peptides thatappear tobecellspecific, namely,
p88, p72,p7l, andp23.
(Some of these resultswere presentedatthe 79thAnnual Meeting of the American Society for Microbiology, 1979, andatthe Third Cam-bridge ConferenceonVirology, 1977.)
MATERIALS AND METHODS
Virus preparation, assay, and irradiation. NDV strains AV and Blweregrowninembryonated eggsandpurified by centrifugationasdescribed pre-viously (13, 22). Some of the virus stocks used in this workweregifts from C. H.Madanskyand M. A. Bratt. Plaque assays of virulent (32, 46) strain AVwere
performedonsecondary cultures of chicken embryo (CE) cells with an overlay of 0.8% agarose in NCl medium(GIBCO Laboratories) containing 2%calf se-rum. Avirulent (32, 46) strain BI was assayed on
monolayers of MDBK cells with an overlay of 0.8% agaroseinNCl mediumcontaining9jgoftrypsin per ml.Preparations of NDVwereexposedtoUV irradia-tionasdescribedpreviously (6). The doserate was6.5
ergs/mm'pers.
Cellculture. Secondarycultures of CE cellswere
preparedas described previously (13; Smithetal.,in
press). MDBK cells were maintained in Dulbecco modified Eagle medium containing 10% fetal bovine
serum.Plaque assaysandinfections of CEcell cultures
wereperformedat40°C; MDBK cultureswere incu-batedat37.5°C.
Conditions for infection, incubation, and la-beling of cell cultures. Secondary cultures of CE
cells were infected at a multiplicity of 5 PFU/cell
NDV TRANSCRIPTIONAL MAP 683
(standard inoculum) or >300 PFU/cell (high-multi-plicity inoculum). Beginning with an adsorption period of 45 min, thecultures were incubated at 40°C in NCl medium or Eagle minimal essential medium contain-ing 2% calf serum. In the case of infections with high-multiplicity inocula, virus adsorption and infection before the labeling period were performed in the pres-ence of 50 ug of cycloheximide per ml. For some experiments, cells were also incubated in the presence of 0.5 yg of actinomycin D per ml beginning 4 h before infection and continuing throughout infection before thelabeling period.
The labeling period beganat 6hpostinfection for cultures infected with standard inocula and at 5 h when high-multiplicity inocula were used. For labeling under standardconditions, cultures were washedwell with minimal essential medium containing 10 mM HEPES-hydrochloride (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid hydrochloride) (pH 7.6) and 2.5% (0.38 mg/liter) of thenormalmethionine content. Cellswerethen incubatedfor 30 min at 40°C in the
samemediumcontaining[35S]methionine(Amersham Corp.) at a concentration of 30 to 50MCi/ml. Cells were washed quickly with cold phosphate-buffered saline (pH 7.6) and solubilized in gel sample buffer (28). The extracts were boiled immediately for 2 min. For labeling under hypertonic conditions, cellswere washedwellwith minimal essential medium contain-ing 10 mM HEPES-hydrochloride (pH 7.6), 2.5% of the normal methionine content, and 126 mM addi-tional NaCl. Cultures were incubated in the same medium for 10 min at 40°C. The medium was then replaced by identical mediumcontaining30 to 50t,Ci of[3S]methionineperml and incubatedfor30minat
40°C. The cells were washed and solubilized as de-scribed above.
Preparation of NDV AVmRNA's in vitro. Ir-radiated and unirIr-radiatedpreparationsof NDVwere
usedtosynthesize mRNA in vitro in the presence of L-cell cytoplasmic extracts and 0.4% Triton N-101 (13).mRNAwaspurified bychromatographyon
col-umns of oligodeoxythymidylic acid-cellulose as de-scribedpreviously (13). Forcomparison,mRNA was
preparedfrom reaction mixtureslackingNDV. Translation of NDV AV mRNA's in vitro. Translation of AV mRNA's in cell-free systems pre-pared frommouseL-cellswasperformedasdescribed previously(13), except that the mRNA concentration
was 0.5 ,ug/ml or less. Reticulocyte lysate reaction mixtures contained 20 mM HEPES-hydrochloride (pH 7.6), 80 mM KCI, 1 mM magnesium acetate, 1
mMATP,0.2mMGTP,5.5mMcreatinephosphate,
100,ugof creatine kinase perml,50,uMamino acids
except methionine, 1 ,uM methionine (including
[355]methionineat aconcentration of250,Ci/ml),0.5
mM dithiothreitol, 60
MLg
of tRNA prepared frommouseL-cells(3) perml,25MuMhemin,and70%(vol/ vol) reticulocyte lysatecontaining0.05% ,8-mercapto-ethanol.Reticulocyteswereobtainedfrom phenylhy-drazine-treated rabbits, and lysates were prepared (37).Immediately beforethereticulocyte lysateswere
addedtothereactionmixtures, theywereadjustedto 1 mMCaCl2,incubated for 10min at20°Cwith 125 U
of Staphylococcus aureus nuclease (Boehringer Mannheim) perml,and thenadjustedto 2mM
on November 10, 2019 by guest
http://jvi.asm.org/
684 COLLINS, HIGHTOWER, AND BALL
yleneglycol-bis(2-aminoethyl ether)-N,N'-tetraacetic acid byusinganeutralized stock solution (37). NDV mRNA's made in vitro were added to a final concen-tration of0.5ytg/mlorless.
SDS-PAGE. Sodium dodecyl sulfate-polyacryl-amide gel electrophoresis (SDS-PAGE) was per-formed indiscontinuous slab gels by the method of Laemmli (28).Electrophoresiswas at20 mA(constant current)for16 hfor 11.5% gels and for9h for 7%gels, exceptasnoted below. Forautoradiography, gelswere
acidfixed, dried underavacuum,andexposedto
XR-5 X-ray film. Forfluorography, gelswere acidfixed, treatedfor fluorography (30), dried, and exposed to XR-5 X-rayfllm that had been presensitizedto linear-ize response (31). Quantitation of radioactive bands was bydensitometry of the corresponding autoradi-ogram orfluorogram.
Peptide mapping. Polypeptide bands resolved by SDS-PAGE were excised from the dried gels and analyzed bypartial digest peptide mapping according
to the procedure of Cleveland et al. (10), using S.
aureusprotease(Boehringer Mannheim). The proce-durewas asdescribedpreviously(21, 23), except that thebuffers used for treating thegelslices contained 1%(wt/vol) dithiothreitol (29) and SDS-PAGE of the digestfragmentswas at 20 mAfor8h.
RESULTS
Comparison of the polypeptides
ex-tracted from CE cells infected with strains A V and B1.Secondarycultures ofCEcellswere infected withstandard inocula of strainsAVand
Bl, incubated for 6 h, exposed to
[35S]methio-nine for30min, andsolubilized. The radioactive polypeptideswereanalyzed by SDS-PAGE(Fig. 1). InAV-infected cells (Fig. 1, channel b), the mostabundant radioactively labeled species in-cluded thefollowingviral polypeptides: L, HN,
Fo,NP,M,
F1
(thelarger subunit of the F protein [40]), andtwoelectrophoreticallydistinct forms ofan additional viral protein, P, that has notbeen reportedpreviously (Smith and Hightower, manuscript in preparation; Morrison,
manu-script in preparation). The P polypeptides of
strain AV were designated according to their
apparent molecularweights of55,000 (P55) and
53,000 (P53), which were obtained by SDS-PAGE in 11.5% gels (Smith and Hightower,
manuscript inpreparation).The NP,P53, and
F1
species comigrated in the 7% gels shown in Fig. 1.The47,000-dalton protein (theremaining re-ported viral protein [11, 22, 42]) was not identi-fied inthis work.
The SDS-PAGE pattern of the radioactive polypeptides extracted from Bl-infected cells
(Fig. 1, channel c) resembled the pattern
de-scribed above for strain AV. However, three differences were noted. First, the NP, P, and M
proteins displayed small strain-specific differ-ences in electrophoretic mobility. Second,
Bl-a b c
_ -L
.: :.. Mly
pas.---H
p71,72
....
p68-
_-pZ-l HN
}ye- A
9P
53'F,
-NP5F'n
_0
-MFIG. 1. SDS-PAGE ofpolypeptidesextractedfrom secondary culturesofCE cells thatwereuninfected (channel a)orinfectedwith standard inoculaofNDV
strains AV(channel b)andBl(channelc).Infected andmock-infected cultureswere incubatedfor6h, exposedto
[CS]methionine
for 30 min, solubilized, andanalyzedby electrophoresis ina 7%1 SDS-poly-acrylamide gel. Anautoradiogramofthefixed,dried gel is shown. Thepositionsofthemajor virus-speci-fiedandvirus-stimulatedpolypeptides (p88, p72,p71) aremarked.infected cells accumulated a relatively smaller
amount ofFO and appeared to accumulate no
polypeptide corresponding in sizein theF1
gly-coprotein of strain AV. However, the SDS-PAGEpatterncontainedaheterodisperseband
ofpolypeptides (designated FA) whichwere in-termediate inelectrophoretic mobility between theFOandF1glycoproteins of strainA V.FAwas
foundtobe relatedtoFOonthe basis ofpartial digest peptide mapping by the procedure of Cleveland et al. (10) (data not shown). This resultconfirmed earlier observations of Madan-sky andBratt(32; C. H. Madansky,Ph.D.thesis, Harvard University, Cambridge, Mass., 1979). Third, Bl-infected cells accumulated increased amounts of at least four polypeptides, which were designated p88, p72, p71, and p23 on the basis of their estimated molecularweights. p23 wasnotretainedby the gelrepresentedby the autoradiogram in Fig. 1. p72 and p71 could be
separated in 11.5% gels (data not shown).
Smalleramountsofpolypeptideswith the same
electrophoretic mobilities were detected in un-infected cells (Fig. 1, channel a) and in AV-infectedcells(channelb).
Identification ofmultiple forms of the P
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.510.285.429.64.277.2]VOL. 35, 1980
Fo
a b c
IW
4.i
.w.
NDV-AV
P55
a b c
P P NP 53A
d d d
'aw
VW
NDV -Bi
Fo P59 P57 NP
a b c a b c a b c a b c
qwmm
~ ~
:.: .:
[image:4.510.59.252.82.445.2]Ay&
FIG. 2. Identification of the P polypeptides of
strains AVand BI by peptide mapping, using the methodofCleveland etal.(10). Thefollowing
/;S]-methionine-labeledpolypeptides were analyzed for
strain AV:Fo,P53,and NPfrom infectedcells and P made in vitro, allobtainedbySDS-PAGE in 11.5%
gelsasdescribedin thelegendstoFig.5 and6;and
P55 from infected cells,obtained bySDS-PAGE ina
7%gelasdescribedin thelegendtoFig. 1. TheB1 polypeptides wereFo, Pa,P57, andNP,allfrom
in-fectedcellsand obtainedbySDS-PAGE ina7%gel
asdescribedin thelegendtoFig.1. The viral poly-peptides,whichwereexcisedfromthe driedgels,were
treated withthefollowingconcentrationsofS.aureus
protease: 0.1 pg/ml (channels labeled a), 10 ptg/ml (channels b), 100pg/ml (channels c), and 25pg/ml (channels d). Thepartial digest fragmentswere
re-solved bySDS-PAGE in 15%gels. Fluorograms of fixed, treated, dried gels are shown. Some ofthe characteristicdigest fragmentsaremarked with ar-rows.
NDV TRANSCRIPTIONAL MAP 685
protein. Recently, the NDV Pprotein has been
identified and characterizedby analysis of strain
AV virions (Smith and Hightower, manuscript in preparation). As Fig. 2 shows, partial digest peptide mapping (10) was used to identify the
cellular forms of the P proteins of strains AV
and Bl and the AV P protein synthesized in
vitro. The
P,%
polypeptideof strain A V and theP57 andP59polypeptides of strainBl were
ob-tained by SDS-PAGE of infected cell extracts
by usinga7% gel (Fig. 1).An 11.5%gelwas used (see Fig. 5) to obtain the AV
P53
polypeptidefrom infected cellsand the 53,000-dalton AV P
protein thatwas synthesized in a mouse L-cell cell-freesystem by usingmRNA made in vitro
by detergent-activated AVvirions. The partial
digest mapping patterns obtained for the AV and Bl P polypeptideswere very similar (Fig. 2), confirming the identifications. Similar
pat-terns were obtained by partial digest peptide
mapping of virion P polypeptides (Smith and
Hightower, manuscript in preparation).In con-trast, the patterns were different than those
obtained for the viral proteins Foand NP (Fig.
2), the CE cell polypeptides contained in the same gel region, and theother major viral
pro-teins (datanotshown).
Transcriptional mapping ofNDV B1 in
vivo.PreparationsofNDV Bl were exposed to
UV irradiation and used to infect secondary
cultures of CE cells at high multiplicities of
infection; cycloheximide was present toinhibit
thereplication of undamagedviral genomes. To measurethetranscripts synthesizedunder these
conditions, the cycloheximidewasremoved, the
cultureswereincubated for30min in the
pres-ence of [35S]methionine, and the radioactively
labeled polypeptides were extracted and
ana-lyzed by SDS-PAGE.
Preliminary experiments (data not shown)
showed that the accumulation of viral polypep-tides under these conditions was severalfold
greater incells infected withthe avirulent strain
Blthan incellsinfectedwith the virulent strain AV. This was consistent with the observation
thatavirulent strains synthesize relatively larger
amounts of RNA during primary transcription
thanvirulent strains (Madansky,manuscript in
preparation). Cycloheximide treatment did not
alter the relative abundancesorelectrophoretic
mobilities of most of theviral polypeptides syn-thesizedafterremoval ofthe drug.Anexception wasthe FA polypeptide of the avirulentstrain,
whichcouldnot be detected under these condi-tions.
Other preliminary experiments (data not
shown)wereperformedtoobtain conditions that
*W,
I
qo* Aft: 4mkk
on November 10, 2019 by guest
http://jvi.asm.org/
facilitated the detection of the viral polypep-tides. In cultured cellsinfected by a variety of
viruses, the accumulation of host cell polypep-tidescanbe reducedby increasing the
osmolar-ity of the medium, whereas the accumulation of viralpolypeptides is affectedto a lesserextent
(16,29).The accumulation of host cell polypep-tideswasfurther reduced when actinomycinD waspresentthroughoutinfectionbefore the
ad-dition ofhypertonic labeling medium. Thus, for transcriptional mapping, infected cells were
treated withactinomycin D,cycloheximide, and hypertonic labeling mediumtorestrict viral
ac-tivity to primary transcription and selectively reduce the host cell biosynthetic background (Fig. 3).
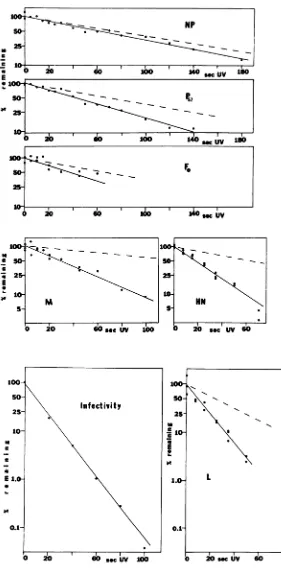
TheratesofUV inactivation of the individual viral genes were determined by monitoring
changes in viral polypeptide accumulation, as
measuredbydensitometry offluorograms such
asthose showninFig.3 (Fig. 4).Inactivation of
infectivitywas measured by plaque assay (Fig. 4). The targetfor UV inactivation ofinfectivity
was assumed to be the entire viral genome. Basedon thisassumption, a comparison ofthe
inactivation ratefor eachgenewith the inacti-vationrate forinfectivity provided an estimate
of themolecularweightofaUVtargetfor tran-scription of an individual gene (Table 1; see
below). The dashed lines inFig. 4 show hypo-thetical inactivationkinetics,whichwere
calcu-lated by assumingthat the UVtarget for each
gene was equal to its individual weight. These molecular weightswere calculated onthe basis
of the electrophoretic mobilities of the
corre-sponding protein products (22; unpublished data) and RNA products (12, 26, 42, 49). The measured UV target sizes and estimated gene sizes were compared (Table 1).The UV target size obtained for NPapproached thepredicted gene size,but for the othergenesthe measured target sizes were two to five times largerthan the sizes predicted and increased in the order NP, P, Fo, M, HN, L.The target size obtained for the L genewas thesame as thetarget size
forinfectivity.
Transcriptional mapping of NDV AV in vitro.Itwaspossible that thechangesin poly-peptide accumulation in vivo did not reflect reliably the inactivation of the viralgenes.
Mea-surementsof the viralmRNA's, the immediate products oftranscription, would have afforded
a more direct assay. However, the NDV
mRNA'shave beenonly partiallyidentified (35, 42), and single-stranded RNAs are difficult to
resolve completely. Nevertheless, it is possible
to determine relative changesinthe accumula-tion of mRNA's which have been isolated by
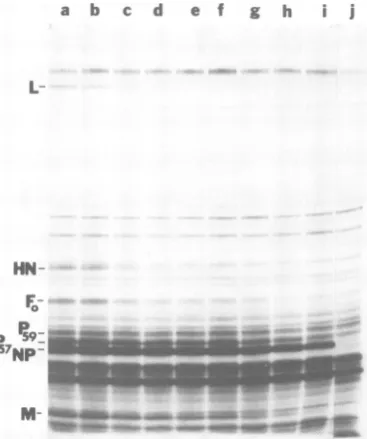
a b c d e f g h i i
1-
HN-
Fo-q59
NP
[image:5.510.271.455.83.303.2]M-Ii
FIG. 3. SDS-PAGE ofpolypeptides extracted from cells infected with UV-irradiated NDV Bl under conditions limiting viral activity to primary tran-scription and inhibitory to synthesis of host cell poly-peptides. Actinomycin D-pretreated secondary cul-turesof CE cells were infected with high-multiplicity inoculaof NDV Bl (channels a through i) or mock infected (channelj). Viral adsorption and infection
wereinthe presenceof cycloheximide and actinomy-cinD. Before infection, the virus preparations were exposed to UVirradiation for the following times: channel a, 0 s; channel b, 10 s; channel c, 20 s; channel d, 30 s; channel e, 45 s; channel f, 60 s; channel g, 80s; channel h, 100 s; channeli, 180 s. The37%osurvival pointfor infectivity was 13 s.
Cul-tures wereexposedto(35S]methionineunder hyper-tonic conditions, solubilized, and analyzed by elec-tropho&esis ina 7%oSDS-polyacrylamide gel. A fluo-rogram ofa fixed, treated, dried gel is shown. To simplify the figure, some irradiation time points and duplicatepoints analyzed in the same experiment
wereomitted. Thepositions of themajorviral poly-peptidesaremarked.
using oligodeoxythymidylic acid-cellulose and translated incell-freesystems byquantifying the
polypeptide products (13).
Preparationsof NDV AV wereirradiatedand
used to synthesize mRNA in vitro. The
tran-scripts were isolated by oligodeoxythymidylic
acid-cellulose column chromatography and
as-sayedfor theirabilitytodirectthe synthesis of viralpolypeptidesincell-free systems prepared from mouse L-cells (Fig. 5) or rabbit reticulo-cytes (Fig. 6). The concentrations of added mRNA in these experimentswere 0.5yg/ml or less (at least 10-fold lower than the levels that
on November 10, 2019 by guest
http://jvi.asm.org/
100-
50-E @11
E
be
n l- I ~I I
b 20 100
NO4
UKw
UV ISO10 D20 D 60 100 140 s.UV 160
100 _
*~~~~~~~~~
_- _P.57
25-10- 1I Iv
IA~~~I T
20 100 S UV
V 20 60 SWCUV 100 0 20mc UV 60
FIG. 4. Kineticsofreductionofboth NDVBIinfectivityand theaccumulationof viralpolypeptidesin vivo
asfunctions ofincreasingUV irradiationofthe viruspreparation. Infectivitywasdeterminedbytheplaque assay.Theamountofeachpolypeptidewasdeterminedby densitometry of fluorograms,suchasthose shown
inFig.3. Forcomparison,the dashed linesrepresenthypotheticalinactivationkinetics, basedonestimations
ofgenesizesfromthecorresponding transcriptsorpolypeptidesizes(Table 1)andassumingthat transcription
ofeachgeneinitiatesataseparate,adjacentpromotor. 687
*-__-~- *~~0 ~ NP
_~0
4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.510.133.414.57.625.2]688 COLLINS, HIGHTOWER, AND
TABLE 1. Comparison of the target sizes for infectivity and individual genes of NDVBl
Gene size
(molwt) Gene size Viralactiv- D Targetsize estimated
(mol wt)
ity" (ergs/ (mol wt,byepoly
by tran-mm) pep6moltide script molmol wt wt(x10)' Infectivity 91 5.10-5.70
Lprotein 91 5.10-5.70 1.98
HN protein 156 2.97-3.32 0.60 0.83 Mprotein 267 1.74-1.95 0.36 0.42 Fprotein 351 1.32-1.48 -f 0.77 P protein 429 1.08-1.21 0.51 0.49 NPprotein 585 0.79-0.89 0.50 0.70
IInactivation ofinfectivity and ofindividual viral genes wasmonitored as described in thelegend to Fig. 4 and in the text.
bD"),Amountof UV irradiationrequiredtoreduce activity to 37% survival. The valuesobtained for four experiments varied over a range of 10 to 15% of the values shown. Similar results were obtained in two experiments with strain A V and oneexperiment with the avirulent strain N. J. LaSota (data notshown).
'The target size forinfectivity was assumed to be the molecularweight of the genome,5.1 x 106to5.7 x106(27). For individual genes, the target sizeequalled(D"'for infectiv-ity/D"forindividual gene)xgenomemolecularweight.
'dMinimumgenesizes basedonestimates of the molecular weights ofRNAs sufficient to code for thepolypeptide gene products,assumingonenucleotidetripletperamino acid. The polypeptide molecular weightswerebasedonthe electropho-reticmobilities of the Blpolypeptides.For the HN glycopro-tein, the molecularweightoftheproduct made in vitro (un-publisheddata)wasassumedtocorrespondtothepolypeptide portion of theglycoprotein.Thefollowing polypeptide molec-ularweightswereused:L,220,000;HN,67,000;NP, 55,000; P, 57,000;andM, 40,000.
eGene sizes basedonthemolecularweightsof the corre-sponding mRNA's were estimated by using the data of Thomas et al.(42) for strain California.Acontribution of 130 nucleotide residueswasassumedfor thepolyadenylicacid tail (48)andwassubtracted. Direct identifications of the L mRNA (35) and M mRNA (42) have been made. Identifications of the other mRNA'saretentative(6,42).Band2of the poly-acrylamide gel pattern of NDV mRNA's was assumed to contain the Pprotein mRNA. The other tentativeassignments are asfollows:band 3,NPmRNA; band 4,FmRNA; band 5, HN mRNA.
fAmolecularweightestimate for thepolypeptideportion ofthe Fglycoproteinwas notavailable.
approachsaturation of the translationalcapacity
of the cell-freesystems). Therefore, changesin mRNA abundances could be detected reliably
byquantitationoftheproteinproducts.
The viralpolypeptides synthesizedbymouse L-cell cell-free systems in responsetoA VmRNA
(Fig. 5) includedproductscorresponding tothe
following four viral genes: HN, NP, P, and M. Theproductsof the cell-free system have been
designatedHN67, NP59,NP,P,andM,according
topeptidemappingidentifications (Fig. 2) (13).
Forthe invitroproductsthat differed in
electro-phoretic mobility from the corresponding
au-thenticproteins, subscriptshavebeen addedto
a b c d e f g h
HN~~~~~~N
NP
~~~~~N
Ps
invitro M
FIG. 5. SDS-PAGEofpolypeptidessynthesizedin L-cell cell-free systems containing mRNA made in vitro by UV-irradiated NDV AV. Reaction mixtures for transcription included L-cell extracts and no
NDV (channel h) or detergent-activated NDV AV that had been exposed to UV irradiation for 0 s (channel c), 25s (channeld), 50s(channel e), 100s (channel/%, and160s(channelg).The 37% survival pointfor infectivitywas25s.The mRNAwaspurified
and addedtocell-freesystemsprepared frommouse
L-cells. The [35S]methionine-labeled polypeptides
synthesizedin vitrowereanalyzedinan11.5%1 SDS-polyacrylamidegelrun at20mAfor13h. For
com-parison, secondary culturesofCE cells were mock-infected (channel a)orinfectedwithastandard in-oculumofNDV AV(channelb), incubated, exposed
to/55S]methioninefor30min solubilized,andrunin parallel.Anautoradiogramofthefixeddriedgelis shown. Thepositions ofthemajorviralpolypeptides synthesizedin vitro and in vivoareshown. For A V polypeptidesextractedfrom infectedcells(channelb),
theP53andF,proteinscomigratedinasingleband under these conditions, with P53 in considerably greater abundance.
indicate their apparent molecular
weights
(inthousands), based on migration in 11.5% gels
(Fig.
6) (13). HN67, previously'designated
P67,
(13), appeared to represent an
unglycosylated
formn of the HNprotein. NP.59,previously
desig-nated
P59,
(13), wasrelatedto the56,000-daltonauthentic NP. Because NP59was ofhigher
ap-parent molecular weight, the suggestion was
madepreviouslythat itmightbeaprecursor of
the56,000-daltonform(13, 23).NPand M made
in vitro corresponded to authentic NP and M. The existence of the NDV P protein had not
beenappreciated previously. The53,000-dalton
A V Pprotein made invitro exhibited the same
electrophoretic mobility as theF, glycoprotein
and
previously
wasthoughttobeanunglycosy-lated, uncleaved form of the precursor Fo,
al-thoughthiscouldnotbeconfirmed byour
pep-VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.510.68.257.100.226.2]NDV TRANSCRIPTIONAL MAP 689
a b c d e f
_____
L
HN17
mP-NP
-M
FIG. 6. SDS-PAGE of [35S]methionine-labeled polypeptides synthesized inareticulocytelysate
con-taining mRNA made in vitro byUV-irradiatedNDV
AV. Twoexposures areshownforasingle gelthat
had been acidfixed, treatedforfluorography, and
dried. Reaction mixtures fortranscription included L-cellextractsandnoNDV (channela)orNDVAV
thathadbeen exposedtoUVirradiationOs(channel
b),25s(channel c),50s(channeld), 100s(channele), and 160s(channel f). Thesameseries oftranscription
reactionmixturesprovided the mRNA for the
exper-imentsshownhereandinFig.5.The purifiedmRNA
wasanalyzedby translation inareticulocytelysate,
andtheproteinproductswereresolved by
electropho-resis inan11.5%SDS-polyacrylamidegel.The posi-tions ofviral polypeptides synthesized in vitro are
shown.
tidemapping studies (13).Subsequent mapping studies (Fig. 2) (Smith and Hightower,
manu-script in preparation) have established that this 53,000-dalton protein made in vitro is the P protein and is not related to Fo. A cell-free product related to F0 has yet to be identified. The 53,000-dalton protein electrophoretic
spe-cies from AV-infected cell extracts was
previ-ously assumed to contain only F1, but is now
knowntobeamixture ofP53 andFl,withP53 in
considerably greater abundance (Smith and
Hightower,manuscript in preparation). The virus-specified polypeptides synthesized by rabbitreticulocyte lysates (Fig. 6) inresponse
toAVmRNAwerethesame asthepolypeptides
described aboveexceptfortwodifferences.First,
nodistinctNP59 bandwasobserved, although in some cases (datanotshown) itwaspossible to detect aheterogeneousseries ofvirus-specified
bandsresembling NP59 in electrophoretic mobil-ity.Second,apolypeptide ofrelativelylow
abun-dance was detected and tentatively designated
L on the basis ofthe following three criteria:
comigration with authentic L during SDS-PAGE (data not shown), synthesis in cell-free
systemsonlyinresponse toNDV mRNAmade invitro (Fig.6) or extracted frominfected cells (data notshown), and a UV sensitivity appro-priate for a product ofthe L gene (Fig. 6). In summary,thepolypeptides synthesized in vitro corresponded to five NDV genes, namely, NP, P, M, HN, andL.
Analyses of experiments such asthoseshown
inFig.5 and 6provided rates of inactivation for
these fivegenesandfor viral infectivity. Overall,
the relative sensitivity to UV inactivation for
eachNDV genewasthe same whether transcrip-tionwasassayed invitro or in vivo. The orderof increasing sensitivity calculated from
experi-mentsperformedin vitro was as follows: NP,
P,
M, HN,L.
DISCUSSION
Recently,the Pprotein has been shown to be distinct from other viral proteinsonthe basisof
peptide mapping byseveral methods (Smithand
Hightower, manuscript in preparation). Proof that the P protein is virus specificwasobtained
by synthesis in cell-free systems directed by NDV mRNA made in vitro (Fig. 5 and 6) (13)
and by identification of the protein productsby peptidemapping (Fig. 2) (Smith andHightower,
manuscript in preparation). Both of thestrains analyzed specified the synthesis in vivo of at leasttwoforms ofP, but the significance of the multiple forms is not known.
The identification of theFApolypeptideofthe avirulent strainwasbased ona closesimilarity to Fo by partial digest peptide mapping (data not shown) and is in agreement withprevious observations (32). FO appears to be processed into FA within 30minofsynthesis(unpublished
data; MadanskyandBratt, manuscriptin
prep-aration). It is not known whether the shift in electrophoretic mobility reflectsachangein
car-bohydrateorpolypeptidecontent.Interestingly,
processing ofFointo FA didnotoccurduringa
labeling periodimmediatelyaftercycloheximide treatment (Fig. 3). (Similar experiments [data notshown] withoutthe additional treatmentsof
actinomycin D and hypertonic medium
pro-duced the same results.) This might reflect a
lack of the processing enzyme(s) under these conditions.Alternatively,since FAbut notFO is incorporated into virions (32; Madansky and Bratt, manuscriptinpreparation)theconversion ofFOand FA maybe closely coupled to virion morphogenesis, which is blocked by cyclohexi-mide (24).
UV irradiationof NDVpreparationsresulted inthe loss of bothinfectivityand theexpression of individualgenesin accordance withsingle-hit a b c d e f
VOL. 35, 1980
V,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.510.62.248.75.251.2]690 COLLINS, HIGHTOWER, AND
kinetics. Theinactivatinghits presumably rep-resent theformation of transcription-terminat-inguracil dimers. Targetsize analysiswas
per-formed as describedpreviously (5, 6).The rate of inactivation of each genewasdeterminedby measuring the reductionintheaccumulationof thecorrespondingRNAorprotein product.This wasexpressed as aphysicaltarget sizeby
com-parisonwith the kinetics of reduction of
infectiv-ity, as measured by plaque assay. For animal
virusespossessinga smallnumberof genes,itis likely that expression of the entire genome is
requiredtoinitiate themultiplerounds of
infec-tionnecessary toforma plaque.Therefore, the rateof loss ofinfectivitycorrespondstoatarget
equivalent to the molecular weight of the ge-nome. Alowerrateof inactivationwould
corre-spondto aproportionatelysmaller target. In the caseofindividual viralgenes, the targetsize is thelengthof nucleic acid thatmustbe traversed
by RNA polymerasesinorderforexpression of aparticular viralactivityorsynthesisofagene
product (18). For comparison, the molecular
weights of the NDV genes were estimated
ac-cording tothe molecular weights of the
corre-sponding proteinand mRNAgeneproducts
(Ta-ble 1). Thepredictionsbasedontranscriptsize
probably providedmore accurateestimates,
al-though onlythe L and M mRNA's have been
directlyidentified (35, 42),andaccurate
molec-ular weight determinations forL mRNAwere not available. Therefore, gene sizes were also estimatedaccordingtothe molecularweightsof
the protein products, assuming one nucleotide
triplet peramino acid. These presumably rep-resentminiimumestimates,since mRNA's
char-acteristically contain noncoding sequences at
both ends.
UVtargetsizeswereobtained for theNP,P,
F, M, HN, and L genes. The combined gene
molecularweight, calculatedbyusingthe tran-script sizes estimated by gel electrophoresis
whenavailable(Table 1),isabout5.2x106.The
genome molecularweight is 5.1 x 106 to5.7 x 106 (27). Thetarget sizes obtained for the NP gene wereconsistently10to50%largerthan the twovalues forthepredictedgenesize(Table1). The physical target forNP gene transcription mightbelargerthanpredicted dueto contribu-tionsfromsequences traversedbyRNA
polym-erasesbutnotrepresentedinthe NPtranscript
orpolypeptide.Forexample, transcription might
bedependentuponreadthroughfromagenome
regioncoding foraleader RNA.Banerjee etal.
found that NDV transcription yielded a small RNA resembling the VSV leader RNA. The genome regioncodingfor thissmall NDV RNA
presumably precedes the NP gene in the
tran-scriptional order,as isthecaseforVSV (8, 14). Furthermore, the observed UV target size for the NPgenewouldappear unexpectedly largeif the NPgene (andleader RNAtemplateregion) contained proportionately more sites for UV
damage, such as adjacent uracilresidues, than the overall genome. Insupport ofthis, theNP mRNA isreportedtocontainadenylate-rich
re-gions, which perhaps code for series of basic
amino acids (42).The NDV leaderRNAmight
be similar in composition to the VSV leader,
which contains nearly 50% adenylate residues
(8).
Theobservedtargetsizes for theP,F, M, HN, andL genes were two tofive timeslarger than the predictedgenesizes(Table 1). This showed
that an extensiveportion of thetargetforeach
gene consisted of sequences notrepresented in
thetranscript.Furthermore, itwasevident that
the UV targets for the individual genes must
overlap extensively, because the combined tar-getsize molecularweightwasmore thantwice the genome molecular weight. The simplest model sufficient to explain these results (6, 18)
predicts that the NDVgenes arecontained ina
singletranscriptional unit equal to the genome
molecular weight. The cumulative increasesin
the UVtargetsizes suggest thattranscriptionof
eachgeneisdependentuponinitiationand
read-through fromasinglepromotor site.
Transcriptional readthroughmay explain the
existence of several unusual NDV mRNA's
made both invivo and invitro. Two of the major
electrophoretic classes of viral mRNA,
desig-natedRNAs6and 7, contain bothaggregatesof smaller18S mRNA speciesandlarge 22S to 24S
molecules thatresist denaturingconditions (43, 44, 49; G. W. Wertz and P. L. Collins,
unpub-lished data). Other less abundantlarge
nondis-sociable mRNA's also exist (unpublisheddata).
These large RNAs probably do not represent additionalgenes,sincethat would greatly exceed
thegenomecoding capacity.Instead, these large
RNAs might be generated by transcriptional readthrough in conjunctionwith incomplete
nu-cleolytic processing. The same possibility has
beensuggested byVarich et al. on the basis of
hybridization-competition experiments (43,44). Thetarget size analysis shown in Fig. 4 and
Table 1indicates that the order oftranscription
offive ofthe viralgenes is asfollows:NP, P, M,
HN, L. The relative position of the F gene,
although clearlyintermediate in the
transcrip-tionalorder,wasnot as
firmly
established.This was because the F-relatedpolypeptideproduct made in vitro has not been identified and be-causeduring SDS-PAGEofinfectedcellextracts the Fo glycoprotein comigrated with a CE cellJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
NDV TRANSCRIPTIONAL MAP 691
polypeptide, interfering with measurements of
changes in abundance. Our datasuggestthat the position of the Fgenelies between the P and M
genes.
The UVtargetforageneconsists ofasingle
promotor at the 3' target end, anyintervening
genes, anda coding regionatthe 5'targetend. Ifthe NDVgenomeconsists ofcontiguous,
non-overlapping genes,then each UVtarget molec-ularweight shouldapproximatethecumulative molecularweights of the inclusivegenes.Inthis regard there are two apparent discrepancies in the UVtargetmolecularweightsshown in Table
1. First, the UV target molecular weights esti-mated for thePandF genes appearinsufficient
to contain the expected coding regions. Since the NP, P, and F polypeptides are notrelated (Fig. 2) (Smith and Hightower, manuscript in
preparation), these data raisethepossibility that
geneoverlapoccursinthisgenomeregion.Gene
overlap appears to occurfor fowlplague virus
(25). Alternatively,thisapparent overlapcould reflect inaccuracies in the target size analysis.
Second, the difference between the M and HN
targets was about 1.30 x 10 daltons, which is
considerably largerthan theHNgenemolecular
weight (0.60x
10'
to0.82x106).Thismightalso reflect inaccuracies in the target size analysis.An alternative possibility isthat this apparent gap might contain an additional gene, which
precedes HN in the transcriptional order. It is
possible that the 47-kilodalton NDV structural
protein (11, 22, 42) isa product of thisgenome
region. Otherparamyxovirusesappear todirect
synthesis ofa smallnonstructural polypeptide
(15, 29), whichmightalsocorrespondto agene
placed between M and HN in thetranscriptional
order.
The failuretodetect the 47-kilodaltonprotein
inthis workmight be duetosubstantialchanges made in conditions for cell culture and SDS-PAGE since the original report (22). The
47-kilodaltonproteinisnotabundant,anddetection
mightbe obscuredbythepresenceof NP
frag-ments and two abundant cellular polypeptides
in the correspondinggel region. The identifica-tion of viralgeneproducts iscomplicatedby the
ability ofparamyxoviruses toinduce some
cel-lularpolypeptidesandtoincorporateothers into virions.
Glazieretal.(17)obtainedevidenceof asingle
promotor site for transcription of the Sendai viral genome. However, the gene order which was predicted for Sendai virus (NP, Fo, M, P,
HN, L) onthe basis of tentativeidentifications
of the mRNA's is different than
the
orderre-portedhere forNDV,primarilyintheplacement
of the P gene, assuming that the NDV and
Sendai Pproteinsareanalogous.The transcrip-tional orders of NDV(NP, P,Fo,M,HN, L) and VSV(N,NS, M, G, L)arealsoverysimilarapart fromtheFgene,which hasnoVSVcounterpart.
Both orders begin with the major nucleocapsid protein NP and end withthelargest viral protein (L), which is core-associated for both viruses. The second position in the orders corresponds
toanothernucleocapsid protein, whichmaybe
part of the polymerase complexes. The
enve-lope-associated M proteins and the glycopro-teinsinvolved in viral attachmentoccupy
com-parablepositions intermediate in the transcrip-tional orders.
Infection of CE cell cultures with avirulent strainBl resulted in elevated accumulation of
atleast fourcellular polypeptides, namelyp88, p72,p71, andp23.Thesamespecieswere present
inuninfected andAV-infected cells, althoughat
lower levels. An analysis of the mRNA's
ex-tracted from NDV-infected CE cells indicated that infection resulted inlargeincreases in the
amounts oftranslatable mRNA's for p88, p72,
p71,p23,andseveral othercellular polypeptides (unpublished data). The less dramatic increases
inpolypeptideamountsshown inFig.1
presum-ably reflect inhibitionatthelevel of synthesis of cellularpolypeptides.
The four most abundant NDV-stimulated polypeptides (p88, p72, p71, and p23) are iden-ticaltothefourmostabundant CE cell
polypep-tides induced by theincorporation of amino acid analogs intoproteins (21,23;unpublished data).
Recently,infection of CE cellsbythe
paramyxo-viruses Sendai virus and simian virus 5 was
found to stimulate the syntheses of p88 and polypeptides correspondingtothe two glucose-regulated inducible CE cellgenes(38,41). These observations indicate the existence ofanumber of inducible cellulargeneswhich code for abun-dant but unidentifiedpolypeptidesthatcan
re-spondtoinfectionbyparamyxoviruses.
ACKNOWLEDGMENTS
We thank M. A. Bratt and C. H. Madansky for providing someof theB)and N.J. LaSota stocks used in this work, C. H.Madansky for useful discussions of the avirulent strains, L. Jean and J. Winters forhelp in preparing the manuscript, and AllenPhillipsforphotography ofthefigures.
Thisworkwassupported by PublicHealth Service grants HL23588from theNational Heart and Lung Institute and CA 14733fromtheNationalCancer Institute and by grant PCM 78-08088 from the National ScienceFoundation. We benefited from the use of acell culture facility supported by thegrant from theNational CancerInstitute. P.L.C. was a National Science FoundationGraduate Fellow, andL.A.B. was arecipientof aNational InstitutesofHealth Research CareerDevelopmentAward.
LITERATURE CITED
1. Abraham, G.1979.Theeffect ofultravioletradiationon
theprimarytranscriptionofinfluenza virus messenger
VOL. 35, 1980
on November 10, 2019 by guest
http://jvi.asm.org/
692 COLLINS, HIGHTOWER, AND BALL RNAs.Virology 97:177-182.
2. Abraham, G., and A. K. Banerjee. 1976. Sequential transcriptionof thegenesof vesicular stomatitisvirus. Proc. Natl. Acad. Sci. U.S.A. 73:1504-1508.
3. Aviv, H.,I. Boime, and P. Leder.1971.Protein synthesis directed by encephalomyocarditis virus RNA:
proper-ties of a transfer-RNA-dependent system. Proc.Natl. Acad.Sci. U.S.A. 68:2303-2307.
4. Ball, L. A. 1977. Transcriptional mapping of vesicular stomatitis virusin vivo. J. Virol.21:411-414. 5. Ball, L. A., P. L. Collins, and L. E. Hightower. 1978.
Transcription,translation and mapping of thegenesof
Newcastle diseasevirus,p.367-382. In B. W. J.Mahy and R. D. Barry (ed.), Negative strandvirusesand the host cell.Academic Press, Inc., London.
6. Ball, L. A.,and C. N.White.1976.Order of transcription of genes of vesicular stomatitis virus. Proc. Natl. Acad. Sci.U.S.A. 73:442-446.
7. Ball, L. A., C. N. White, and P. L. Collins. 1976. A transcriptionalmapofvesicular stomatitis virus,p.
419-438. In D. Baltimore,A.S. Huang, and C.F. Fox (ed.), Animal virology. ICN-UCLA Symposiaon Molecular
and Cellular Biology,vol.4.Academic Press, Inc., New York.
8. Banerjee,A. K., R. C. Colonno, D. Testa, and M. T. Franze-Fernandez.1978.Mechanism of RNA synthe-sis in vitro byvesicular stomatitis virus,p.249-259.In
B. W. J. Mahyand R. D. Barry(ed.), Negative strand viruses andthehostcell. AcademicPress, Inc.,London. 9. Choppin,P. W., and R. W. Compans. 1975. Reproduc-tion of paramyxoviruses, p. 98-178. In H.
Fraenkel-Conrat and R. R. Wagner (ed.), Comprehensive virol-ogy. PlenumPublishing Corp.,New York.
10. Cleveland, D. W., S. G. Fischer, W. M. Kirschner, and U. K. Laemmli.1977.Peptidemapping bylimited proteolysis in sodiumdodecyl sulfate and analysisby gel electrophoresis.J.Biol. Chem.252:1102-1106. 11. Clinkscales,C.W.,M. A. Bratt, and T. G.Morrison.
1977. SynthesisofNewcastle disease virus polypeptides in a wheatgermcell-freesystem.J. Virol. 22:97-101. 12. Collins,B. S., and M. A. Bratt. 1973.Separation of the
messenger RNAs of Newcastle disease virus by gel electrophoresis.Proc. Natl. Acad. Sci. U.S.A.
70:2544-2548.
13. Collins,P. L., L. E. Hightower, and L. A. Ball. 1978. Transcription and translation of Newcastle disease
vi-rus mRNA'sin vitro. J. Virol.28:324-336.
14. Colonno, R.J.,and A. K. Banerjee. 1977. Mapping and inhibitionstudies ontheleaderRNA of vesicular
sto-matitis virus.Virology 77:260-268.
15. Etkind, P.R., R. K. Cross,R. A. Lamb, D. C. Merz, and P. W.Choppin. 1980. In vitro synthesis of
struc-tural and nonstructural proteins of Sendai and SV5 viruses.Virology100:22-23.
16. Flamand, A., and J. F. Delagneau. 1978. Transcrip-tional mappingof rabies virus in vivo. J. Virol.
28:518-523.
17. Glazier,K.,R. Raghow, and D. W. Kingsbury. 1977.
Regulationof Sendai virus transcription:evidencefora
singlepromotorin vivo. J. Virol. 21:863-871.
18. Hackett,P.B., and W. Sauerbier. 1975. The
transcrip-tional organization of the ribosomal RNA genes in
mouseL-cells.J. Mol. Biol.91:234-256.
19. Hall, W.W.,W. R.Kiessling,andV.terMeulen. 1978.
Biochemicalcomparison of measles and subacute
scle-rosingpanencephalitis (SSPE) viruses, p. 146-156. In
B. W. J.Mahyand R. D. Barry (ed.),Negative strand
viruses andthehostcell. Academic Press,Inc.,London.
20. Herman, R. C., S. Adler, R. A. Lazzarini, R. J.
Co-lonno,A.K. Banerjee, and H. Westphal. 1978.
In-terveningpolyadenylate sequencesin RNA transcripts
of vesicularstomatitisvirus.Cell15:587-596.
J. VIROL.
21. Hightower, L. E. 1980. Cultured animal cells exposed to amino acid analogues or puromycin rapidly synthesize several polypeptides. J. Cell.Physiol. 102:407-427. 22. Hightower, L. E., T. G. Morrison, and M. A.Bratt.
1975. Relationships among the polypeptides of Newcas-tle disease virus. J. Virol. 16:1599-1607.
23. Hightower, L. E., and M. D. Smith. 1978. Effects of canavanine on proteintnetabolismin Newcastle disease virus-infected and uninfected chicken embryo cells, p. 395405. In B. W. J. Mahy and R. D. Barry (ed.), Negative strand viruses and the host cell. Academic Press, Inc., London.
24. linuma,M., Y. Nagai, K. Maeno, T. Yoshida, and T. Matsumoto. 1971. Studies on the assembly of Newcas-tle disease virus: incorporation of structural proteins into virus particles. J. Gen. Virol. 12:239-247. 25. Inglis, S. C., T. Barrett, C. M. Brown, and J. W.
Almond. 1979. The smallest genome RNA segment of influenza virus contains two genes that may overlap. Proc. Natl. Acad. Sci. U.S.A. 76:3790-3794.
26. Kaverin, N. W., and N. L. Varich. 1974. Newcastle disease virus-specific RNA: polyacrylamide gel analysis of single-stranded RNA and hybrid duplexes. J. Virol. 13:253-260.
27. Kolakofsky, D., E. Boy de la Tour, and H.Delius. 1974. Molecular weight determination of Sendai and Newcastle disease virus RNA. J. Virol. 13:261-268. 28. Laemmli, U. K. 1970. Cleavage of structural proteins
during the assembly of the head of bacteriophage T4. Nature (London) 277:680-685.
29. Lamb, R. A., R. W. Peluso, and P. W. Choppin. 1978. SV5 and Sendai virus polypeptides: aspects of compo-sition, synthesis and phosphorylation, p. 195-203. InB. W. J. Mahy and R. D. Barry (ed.), Negative strand viruses and the hostcell.Academic Press, Inc.,London. 30. Laskey, R. A., and A. D. Mills. 1975. Quantitative film detection of'H and"4Cin polyacrylamide gels by fluo-rography. Eur. J. Biochem. 56:335-341.
31. Laskey, R. A., and A. D. Mills. 1977. Enhanced auto-radiographic detection of:"Pand12'I using intensifying screens and hypersensitive film. FEBS Lett. 82:314-316.
32. Madansky, C. H., and M. A. Bratt. 1978.Noncytopathic mutants of Newcastle disease virus: comparison with naturally occurring avirulent strains, p. 709-720. In B. W. J. Mahy and R. D. Barry (ed.), Negative strand viruses and the host cell. Academic Press, Inc.,London. 33. Marcus, P. I., and M. J. Sekellick. 1975. Cell killingby viruses. II. Cell killing by vesicular stomatitis virus: a requirement for virion-derived transcription. Virology 63:176-190.
34. Marvaldi, J. L., M. J. Skellick, P.1.Marcus, and J. Lucas-Lenard. 1978. Inhibition of mouse L cellprotein synthesis by ultraviolet-irradiated vesicular stomatitis virus requires viral transcription. Virology84:127-133. 35. Morrison,T. G., S. Weiss, L. Hightower, B.
Spanier-Collins,
and M. A. Bratt. 1975. Newcastle diseasevirus protein synthesis, p.281-290. In A. L. Haenniand
G. Beaud (ed.), In vitro transcription andtranslation of viral genomes. Institut National de laSante et de la RechercheMedicale,Paris.
36. Nagai, I., and H.-D. Klenk. 1977. Activation of
precur-sors to both glycoproteins of Newcastle disease virusby proteolytic cleavage. Virology 77:125-134.
37. Pelham, H. R. B., and R. J. Jackson. 1976. Anefficient mRNA-dependent translation system from rabbit retic-ulocyte lysates. Eur. J. Biochem. 67:247-256. 38. Peluso, R. W., R. A. Lamb, and P. W. Choppin. 1978.
Infection with paramyxoviruses stimulates synthesisof cellular polypeptides that arealso stimulated in cells
transformed by Rous sarcoma virus or deprived of
glucose. Proc. Natl. Acad. Sci. U.S.A. 75:6120-6124.
on November 10, 2019 by guest
http://jvi.asm.org/
NDV TRANSCRIPTIONAL MAP 693
39. Pons, M. W., and0.M.Rochovansky.1979.Ultraviolet
inactivation ofinfluenza virus RNA in vitro and in vivo. Virology 97:183-189.
40. Scheid, A., and P. W. Choppin. 1977. Two disulfide-linkedpolypeptide chains constitute the active F
pro-tein ofparamyxoviruses.Virology 80:54-66.
41. Shiu, R. P. C., J. Pouyssegur, and I Pastan. 1977. Glucose depletion accounts for the induction oftwo
transformation-sensitive membrane proteins in Rous
sarcoma virus-transformed chickembryo fibroblasts. Proc.Natl.Acad. Sci. U.S.A.74:3840-3844.
42. Thomas, G. P., R. D. Barry, P. Feilner, and J. Smith. 1978.Newcastle disease virusmessengerRNAs,p.
381-394.In B.W.J.Mahy, and R. D. Barry (ed.), Negative strand virusesand the host cell. AcademicPress, Inc., London.
43. Varich, N. L., I. S. Lukashevich, and N. V. Kaverin. 1979.Newcastlediseasevirus-specific RNA:an analysis of24S and 35S RNA transcripts. Acta Virol. 23:273-283.
44. Varich, N. L., I. S. Lukashevich, and N. V. Kaverin.
1979.Newcastle disease virus-specificRNA: hybridiza-tion-competition of the non-dissociable358RNA with individual18S RNA species. Acta Virol. 23:341-343. 45. Villarreal, L P., M.Breindl, and J. J. Holland. 1976.
Determination of molar ratios of vesicular stomatitis virusinduced RNA species inBHK2,cells.
Biochemis-try15:1663-1667.
46. Waterson,A.P.,T. H.Pennington, and W. H. Allan. 1967.Virulence in Newcastle disease virus. A
prelimi-narystudy. Br. Med. Bull. 23:128-143.
47. Weck, P.K., A. R. Carroll,0.M.Shattuck, and R. R.
Wagner. 1979. Use of UVirradiationtoidentify the genetic information of vesicular stomatitis virus
respon-siblefor shutting off cellular RNA synthesis. J. Virol. 30:746-753.
48. Weiss, S. R., and M. A. Bratt. 1974. Polyadenylate
sequences onNewcastle diseasevirus mRNA synthe-sized invivo and in vitro. J. Virol.13:1220-1270. 49. Weiss, S.R.,and M. A. Bratt.1976.Comparative
elec-trophoresis of the 18-22S RNAs of Newcastle disease virus.J. Virol. 18:316-323.
VOL. 35, 1980
on November 10, 2019 by guest
http://jvi.asm.org/