for Haplotype Inference by Pure Parsimony
using Delayed Haplotype Selection
Jo˜ao Marques-Silva1, Inˆes Lynce2, Ana Grac¸a2, and Arlindo L. Oliveira2 1 School of Electronics and Computer Science, University of Southampton, UK
jpms@ecs.soton.ac.uk
2 IST/INESC-ID, Technical University of Lisbon, Portugal
{ines,assg}@sat.inesc-id.pt,aml@inesc-id.pt
Abstract. Haplotype inference from genotype data is a key step towards a better
understanding of the role played by genetic variations on inherited diseases. One of the most promising approaches uses the pure parsimony criterion. This ap-proach is called Haplotype Inference by Pure Parsimony (HIPP) and is NP-hard as it aims at minimising the number of haplotypes required to explain a given set of genotypes. The HIPP problem is often solved using constraint satisfaction techniques, for which the upper bound on the number of required haplotypes is a key issue. Another very well-known approach is Clark’s method, which resolves genotypes by greedily selecting an explaining pair of haplotypes. In this work, we combine the basic idea of Clark’s method with a more sophisticated method for the selection of explaining haplotypes, in order to explicitly introduce a bias towards parsimonious explanations. This new algorithm can be used either to ob-tain an approximated solution to the HIPP problem or to obob-tain an upper bound on the size of the pure parsimony solution. This upper bound can then used to efficiently encode the problem as a constraint satisfaction problem. The experi-mental evaluation, conducted using a large set of real and artificially generated examples, shows that the new method is much more effective than Clark’s method at obtaining parsimonious solutions, while keeping the advantages of simplicity and speed of Clark’s method.
1
Introduction
For a number of reasons, these studies have focused on the tracking of the inher-itance of Single Nucleotide Polymorphisms (SNPs), point mutations found with only two common values in the population. This process is made more difficult because of technological limitations. Current methods can directly sequence only short lengths of DNA at a time. Since the sequences of the chromosomes inherited from the parents are very similar over long stretches of DNA, it is not possible to reconstruct accurately the sequence of each chromosome. Therefore, at a genomic site for which an individual inherited two different values, it is currently difficult to identify from which parent each value was inherited. Instead, currently available sequencing methods can only deter-mine that the individual is ambiguous at that site.
Most diseases are due to very complex processes, where the values of many SNPs affect, directly and indirectly, the risk. Due to a phenomenon known as linkage dise-quilibrium, the values of SNPs in the same chromosome are correlated with each other. This leads to the conservation, through generations, of large haplotype blocks. These blocks have a fundamental role in the risk of any particular individual for a given dis-ease. If we could identify maternal and paternal inheritance precisely, it would be pos-sible to trace the structure of the human population more accurately and improve our ability to map disease genes. This process of going from genotypes (which may be am-biguous at specific sites) to haplotypes (where we know from which parent each SNP is inherited) is called haplotype inference.
This paper introduces a greedy algorithm for the haplotype inference problem called Delayed Haplotype Selection (DS) that extends and improves the well-known Clark’s method[5]. We should note that recent work on Clark’s method studied a number of variations and improvements, none similar to DS, and all performing similarly to Clark’s method. This new algorithm takes advantage of new ideas that have appeared recently, such as those of pure parsimony[10]. A solution to the haplotype inference by pure parsimony (HIPP) problem provides the smallest number of haplotypes required to ex-plain a set of genotypes. This algorithm can then be used in two different ways: (1) as a standalone procedure for giving an approximate solution to the HIPP problem or (2) as an upper bound to the HIPP solution to be subsequently used by pure parsimony algo-rithms which use upper bounds on their formulation. Experimental results, obtained on a comprehensive set of examples, show that, for the vast majority of the examples, the new approach provides a very accurate approximation to the pure parsimony solution.
2
Problem Formulation and Related Work
2.1 Haplotype Inference
A haplotype represents the genetic constitution of an individual chromosome. The un-derlying data that forms a haplotype is generally viewed as the set of SNPs in a given region of a chromosome. Normal cells of diploid organisms contain two haplotypes, one inherited from each parent. The genotype represents the conflated data of the two haplotypes. The value of a particular SNP is usually represented by X, Y or X/Y, de-pending on whether the organism is homozygous with value X, homozygous with value Y or heterozygous. The particular base that the symbols X and Y represent varies with the SNP in question. For instance, the most common value in a particular location may be the guanine (G) and the less common variation cytosine (C). In this case, X will mean that both parents have guanine in this particular site, Y that both parents have cytosine at this particular site, and X/Y that the parents have different bases at this particular site. Since mutations are relatively rare, the assumption that at a particular site only two bases are possible does not represent a strong restriction. This assumption is supported by the so called infinite sites model[14], that states that only one mutation has occurred in each site, for the population of a given species.
Starting from a set of genotypes, the haplotype inference task consists in finding the set of haplotypes that gave rise to that set of genotypes. In computational terms, the problem may be formulated as follows.
Definition 1. (Haplotype Inference) Given a setGofngenotypes, each of lengthm, the haplotype inference problem consists in finding a setHof 2·n haplotypes, not necessarily different, such that for each genotypegi ∈ Gthere is at least one pair of
haplotypes(hj, hk), withhjandhk ∈ Hsuch that the pair(hj, hk)explainsgi. The
variablendenotes the number of individuals in the sample, andmdenotes the number of SNP sites. A specific genotype is denoted bygi, with1 ≤ i ≤n. Furthermore,gij
denotes a specific sitejin genotypegi, with1≤j≤m. The objective is to find the set Hof haplotypes that is most likely to have originated the set of genotypes inG.
Without loss of generality, we may assume that the two values of each SNP are either 0 or 1. Value 0 represents the wild type and value 1 represents the mutant. A haplotype is then a string over the alphabet{0,1}. Genotypes may be represented by extending the alphabet used for representing haplotypes to{0,1,2}. We say that a geno-typegican be explained by haplotypeshjandhkiff for each position wherehjhas the same value ashk (homozygous sites),giis equal to hj, and, for each position where hj andhk have different values (heterozygous sites), gi has value 2. In general, if a genotypegihasrheterozygous sites, there are2r−1pairs that can explaingi.
Example 1. (Haplotype Inference) Consider genotype 02122 having 5 SNPs, of which
We may distinguish between a number of approaches that are usually used for solv-ing the haplotype inference problem: the statistical, the heuristic and the combinato-rial approaches. The statistical approaches[19, 22] use specific assumptions about the underlying haplotype distribution to approximate different genetic models, and may obtain highly accurate results. The heuristic approaches include, among others, Clark’s method[5]. Finally, most combinatorial approaches are based on the pure parsimony cri-terion[10]. The later has shown to be one of the most promising alternative approaches to statistical models[3, 17].
2.2 Clark’s Method
Clark’s method is a well-known algorithm that has been proposed to solve the haplotype inference problem[5]. Clark’s algorithm has been widely used and is still useful today. This method considers both haplotypes and genotypes as vectors. The method starts by identifying genotype vectors with zero or one ambiguous sites. These vectors can be resolved in only one way, and they define the initially resolved haplotypes. Then the method attempts to resolve the remaining genotypes by starting with the resolved haplotypes. The following step infers a new resolved vector NR from an ambiguous vector A and an already resolved genotype vector R.
Suppose A is an ambiguous genotype vector withrambiguous sites and R is a re-solved vector that is a haplotype in one of the2r−1potential resolutions of vector A. Then the method infers that A is the conflation of the resolved vector R and another unique vector NR. All of the ambiguous positions in A are set in NR to the opposite value of the position in R. Once inferred, this vector is added to the set of known re-solved vectors, and vector A is removed from the set of unrere-solved vectors.
The key point to note is that there are many ways to apply the resolution rule, since for an ambiguous vector A there may be many choices for vector R. A wrong choice may lead to different solutions, or even leave orphan vectors, in the future, i.e., vectors that cannot be resolved with any already resolved vector R.
The Maximum Resolution (MR) problem[9] aims at finding the solution of the Clark’s algorithm with the fewest orphans, i.e. with the maximum number of genotypes resolved. Despite being an accurate criterion for using the Clark’s method, this problem is NP-hard as shown by Gusfield[9], who also proposed an integer linear programming approach to the MR problem.
2.3 Pure Parsimony
Chromosomes in the child genome are formed by combination of the corresponding chromosomes from the parents. Long stretches of DNA are copied from each parent, spliced together at recombination points. Since recombination is relatively infrequent, large segments of DNA are passed intact from parent to child. This leads to the well known fact that the actual number of haplotypes in a given population is much smaller than the number of observed different genotypes.
Definition 2. (Haplotype Inference by Pure Parsimony) The haplotype inference by
the total number of distinct haplotypes used. The problem of finding such a parsimo-nious solution is APX-hard (and, therefore, NP-hard)[16].
Example 2. (Haplotype Inference by Pure Parsimony) Consider the following
exam-ple, taken from a recent survey on the topic[11], where the set of genotypes is: 02120, 22110, and 20120. There are solutions that use five different haplotypes1, but the solu-tion (00100, 01110), (01110, 10110), (00100, 10110) uses only three different haplo-types.
It is known that the most accurate solutions based on Clark’s method are those that infer a small number of distinct haplotypes[10, 20]. Although Clark’s method has sometimes been described as using the pure parsimony criterion[19, 1, 22], this criterion is not explicitly used and an arbitrary choice of the resolving haplotype does not lead to a pure parsimony solution. The present paper proposes a method that, while still based on Clark’s method, explicitly uses the pure parsimony criterion, leading to more precise results.
Several approaches, have been proposed to solve the HIPP problem. The first algo-rithms are based on integer linear programming[10, 2, 24], whereas the most recent and competitive encode the HIPP problem as a constraint satisfaction problem (either using propositional satisfiability[17, 18] or pseudo-Boolean optimization[4]).
One should note that the implementation of exact algorithms for the HIPP problem often requires computing either lower or upper bounds on the value of the HIPP so-lution[24, 18]. Clearly, Clark’s method can be used for providing upper bounds on the solution of the HIPP problem. Besides Clark’s method, which is efficient but in general not accurate, existing approaches for computing upper bounds to the HIPP problem require worst-case exponential space, due to the enumeration of candidate pairs of hap-lotypes[12, 24]. Albeit impractical for large examples, one of these approaches is used in Hapar[24], a fairly competitive HIPP solver when the number of possible haplotype pairs is manageable.
The lack of approaches both accurate and efficient for computing upper bounds, prevented their utilization in recent HIPP solvers, for instance, in SHIPs[18]. Algo-rithm 1 summarizes the top-level operation of SHIPs. This SAT-based algoAlgo-rithm iter-atively models whether there exists a setHof distinct haplotypes, withr =|H|such that each genotypeg ∈ G is explained by a pair of haplotypes in H. The algorithm considers increasing sizes forH, from a lower boundlbto an upper boundub. Trivial lower and upper bounds are, respectively,1and2·n. For each value ofrconsidered, a CNF formulaϕris created, and a SAT solver is invoked (identified by the function call SAT(ϕr)). The algorithm terminates for a size ofHfor which there existr=|H| haplotypes such that every genotype inGis explained by a pair of haplotypes inH, i.e. when the constraint problem is satisfiable. (Observe that an alternative would be to use binary search.)
This paper develops an efficient and accurate approach for haplotype inference, inspired by pure parsimony, and which can be used to compute tight upper bounds
1
Algorithm 1 Top-level SHIPs algorithm
SHIPS(G, lb)
1 r←lb
2 while (true)
3 do GenerateϕrgivenGandr
4 if SAT(ϕr
) =true
5 then returnr
6 else r←r+ 1
to the HIPP problem. Hence, the proposed approach can be integrated in any HIPP approach, including Hapar[24] and SHIPs[18].
3
Delayed Haplotype Selection
A key drawback of haplotype inference algorithms based on Clark’s method is that these algorithms are often too greedy, at each step seeking to explain each non-explained genotype with the most recently chosen haplotype. As a result, given a newly selected haplotypeha, which can explain a genotypegt, a new haplotypehb is generated that only serves to explaingt. If the objective is to minimize the number of haplotypes, then the selection ofhbmay often be inadequate.
This section develops an alternative algorithm which addresses the main drawback of Clark’s method. The main motivation is to avoid the excessive greediness of Clark’s method in selecting new haplotypes, and therefore a delayed greedy algorithm for hap-lotype selection (DS) is used instead.
In contrast to Clark’s method, where identified haplotypes are included in the set of chosen haplotypes, the DS algorithm maintains two sets of haplotypes. The first set, the selected haplotypes, represents haplotypes which have been chosen to be included in the target solution. A second set, the candidate haplotypes, represents haplotypes which can explain one or more genotypes not yet explained by a pair of selected haplotypes.
The initial set of selected haplotypes corresponds to all haplotypes which are re-quired to explain the genotypes with no more than one heterozygous sites, i.e. geno-types which are explained with either one or exactly two haplogeno-types. Clearly, all these haplotypes must be included in the final solution.
At each step, the DS algorithm chooses the candidate haplotypehc which can ex-plain the largest number of genotypes. The chosen haplotypehc is then used for iden-tifying additional candidate haplotypes. Moreover,hc is added to the set of selected haplotypes, and all genotypes which can be explained by a pair of selected haplotypes are removed from the set of unexplained genotypes. The algorithm terminates when all genotypes have been explained.
Algorithm 2 Delayed Haplotype Selection
DELAYEDHAPLOTYPESELECTION(G)
1 HSis the set of selected haplotypes;HCis the set of candidate haplotypes
2 HS←CALCINITIALHAPLOTYPES(G)
3 G ←REMOVEEXPLAINEDGENOTYPES(G,HS)
4 for eachh∈ HS
5 do
6 for eachg∈ G
7 do if CANEXPLAIN(h, g)
8 thenhc←CALCEXPLAINPAIR(h, g)
9 HC← HC∪ {hc}
10 Associatehcwithg
11 while (G 6=∅) 12 do if (HC=∅)
13 then
14 hc←PICKCANDHAPLOTYPE(G)
15 HC← {hc}
16 h←hc∈ HCassociated with largest number of genotypes
17 HC← HC− {h}
18 HS← HS∪ {h}
19 G ←REMOVEEXPLAINEDGENOTYPES(G,HS)
20 for eachg∈ G
21 do if CANEXPLAIN(h, g)
22 thenhc←CALCEXPLAINPAIR(h, g)
23 HC← HC∪ {hc}
24 Associatehcwithg
25 HS←REMOVENONUSEDHAPLOTYPES(HS)
26 returnHS
not yet explained. Clearly, other alternatives could be considered, but the experimental differences, obtained on a large set of examples, were not significant.
Observe that the proposed organization allows selecting haplotypes which will not be used in the final solution. As a result, the last step of the algorithm is to remove from the set of selected haplotypes all haplotypes which are not used for explaining any genotypes.
Table 1. Classes of problem instances evaluated
Class #Instances minSNPs maxSNPs minGENs maxGENs
uniform 245 10 100 30 100
nonuniform 135 10 100 30 100
hapmap 24 30 75 7 68
biological 450 13 103 5 50
Total 854 10 103 5 100
In practice, the delayed haplotype selection algorithm is executed multiple times, as in other recent implementations of Clark’s method[20]. At each step, ties in picking the next candidate haplotype (see line 16) are randomly broken. The run producing the smallest number of haplotypes is selected.
Results in the next section suggest that delayed haplotype selection is a very effec-tive approach. Nonetheless, it is straightforward to conclude that there are instances for which delayed haplotype selection will yield the same solution as Clark’s method. In fact, it is possible for DS to yield solutions with more haplotypes than Clark’s method. The results in the next section show that this happens very rarely. Indeed, for most ex-amples considered (out of a comprehensive set of exex-amples) DS is extremely unlikely to compute a larger number of haplotypes than Clark’s method, and most often computes solutions with a significantly smaller number of haplotypes.
4
Experimental Results
This section compares the delayed haplotype selection (DS) algorithm described in the previous section with a recent implementation of Clark’s method (CM)[8]. In addition, the section also compares the HIPP solutions, computed with a recent tool[18], with the results of DS and CM. As motivated earlier, the objectives of the DS algorithm are twofold: first to replace Clark’s method as an approximation of the HIPP solution, and second to provide tight upper bounds to HIPP algorithms.
Recent HIPP algorithms are iterative[18], at each step solving a Boolean Satisfiabil-ity problem instance. The objective of using tight upper bounds is to reduce the number of iterations of these algorithms. As a result, the main focus of this section is to ana-lyze the absolute difference, in the number of haplotypes, between the computed upper bound and the HIPP solution.
4.1 Experimental setup
The instances used for evaluating the two algorithms have been obtained from a number of sources[18], and can be organized into four classes shown in Table 1. For each class, Table 1 gives the number of instances, and the minimum and maximum number of SNPs and genotypes, respectively2. The uniform and nonuniform classes of instances
2Table 1 shows data for the original non-simplified instances. However, all instances were
are the ones used by other authors[3], but extended with additional, more complex, problem instances. The hapmap class of instances is also used by the same other au-thors[3]. Finally, the instances for the biological class are generated from data publicly available[13, 21, 7, 6, 15]. To the best of our knowledge, this is the most comprehensive set of examples used for evaluating haplotype inference solutions.
All results shown were obtained on a 1.9 GHz AMD Athlon XP with 1GB of RAM running RedHat Linux. The run times of both algorithms (CM and DS) were always a few seconds at most, and no significant differences in run times were observed between CM and DS. As a result, no run time information is included below.
4.2 Experimental evaluation
The experimental evaluation of the delayed haplotype selection (DS) algorithm is or-ganized in two parts. The first part compares DS with a publicly available recent im-plementation of Clark’s method (CM)[8], whereas the second part compares DS with an exact solution to the Haplotype Inference by Pure Parsimony (HIPP) problem[18]. In all cases, for both CM and DS, we select the best solution out of 10 runs. Other implementations of Clark’s method could have been considered[20]. However, no sig-nificant differences were observed among these implementations when the objective is to minimize the number of computed haplotypes.
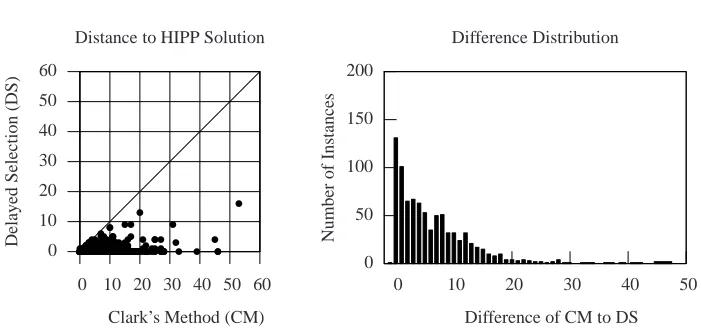
The results for the first part are shown in Figure 1. The scatter plot shows the differ-ence of CM and of DS with respect to the exact HIPP solution for the examples consid-ered. The results are conclusive. DS is often quite close to the HIPP solution, whereas the difference of CM with respect to the HIPP solution can be significant. While the dis-tance of DS to the HIPP solution never exceeds 16 haplotypes, the disdis-tance of CM can exceed 50 haplotypes. Moreover, for a large number of examples, the distance of DS to the HIPP solution is 0, and for the vast majority of the examples the distance does not exceed 5 haplotypes. In contrast, the distance of CM to the HIPP solution often exceeds 10 haplotypes.
The second plot in Figure 1 provides the distribution of the difference between the number of haplotypes computed with DS and with CM. A bar associated with a valuekrepresents the number of examples for which CM exceeds DS bykhaplotypes. With one exception, DS always computes a number of haplotypes no larger than the number of haplotypes computed with CM. For the single exception, DS exceeds CM in 1 haplotype (hence -1 is shown in the plot). Observe that for 85% of the examples, DS outperforms CM. Moreover, observe that for a reasonable number of examples (40.1%, or 347 out of 854) the number of haplotypes computed with CM exceeds DS in more than 5 haplotypes. Finally, for a few examples (3 out of 854), CM can exceed DS by more than 40 haplotypes, the largest value being 46 haplotypes.
It should also be noted that, if the objective is to use either DS or CM as an up-per bound for an exact HIPP algorithm, then a larger number of computed haplotypes represents a less tight, and therefore less effective, upper bound. Hence, DS is clearly preferable as an upper bound solution.
0 10 20 30 40 50 60
0 10 20 30 40 50 60
D el ay ed S el ec ti o n (D S )
Clark’s Method (CM) Distance to HIPP Solution
0 50 100 150 200
0 10 20 30 40 50
N u m b er o f In st an ce s
[image:10.595.141.492.113.280.2]Difference of CM to DS Difference Distribution
Fig. 1. Comparison of Clark’s Method (CM) with Delayed Haplotype Selection (DS)
as an upper bound for recent HIPP algorithms[18]. For examples where DS computes the HIPP solution, exact HIPP algorithms are only required to prove the solution to be optimum. For a negligible number of examples (0.9%, or 8 out of 854) the difference of DS to the HIPP solution exceeds 5 haplotypes. Hence, for the vast majority of examples considered, DS provides a tight upper bound to the HIPP solution.
The results allow drawing the following conclusions. First, DS is a very effective alternative to CM when the objective is to minimize the total number of computed hap-lotypes. Second, DS is extremely effective as an upper bound for exact HIPP algorithms. For most examples (99.1%, or 846 out of 854) the number of haplotypes identified by DS is within 5 haplotypes of the target HIPP solution.
5
Conclusions and Future Work
0 100 200 300 400 500 600 700
0 5 10 15 20
N
u
m
b
er
o
f
In
st
an
ce
s
[image:11.595.196.384.121.276.2]Difference of DS to HIPP Solution
Fig. 2. Comparison of Delayed Haplotype Selection (DS) with HIPP solution
As mentioned earlier, recent approaches for the HIPP problem iterate through creasingly higher lower bounds[18]. This implies that solutions to the haplotype in-ference problem are only obtained when the actual solution to the HIPP problem is identified. Thus, these recent approaches to the HIPP problem[18] cannot be used for computing approximate HIPP solutions. The work described in this paper provides an efficient and remarkably tight approach for computing upper bounds. This allows recent HIPP based algorithms[18] to compute the exact solution by iterating through decreas-ing upper bounds. Hence, at each step a solution to the haplotype inference problem is identified, and, therefore, these methods can be used for approximating the exact HIPP solution. The integration of the DS algorithm in recent solutions to the HIPP problem is the next natural step of this work.
Acknowledgments This work is partially supported by Fundac¸˜ao para a Ciˆencia e Tecnologia under research project POSC/EIA/61852/2004 and PhD grant SFRH/BD/ 28599/2006, by INESC-ID under research project SHIPs, and by Microsoft under con-tract 2007-017 of the Microsoft Research PhD Scholarship Programme.
References
1. R.M. Adkins. Comparison of the accuracy of methods of computational haplotype inference using a large empirical dataset. BMC Genet, 5(1):22, 2004.
2. D. Brown and I. Harrower. A new integer programming formulation for the pure parsimony problem in haplotype analysis. In Workshop on Algorithms in Bioinformatics (WABI’04), 2004.
4. A. Grac¸a, J. Marques-Silva, I. Lynce, and A. Oliveira. Efficient haplotype inference with pseudo-Boolean optimization. In Algebraic Biology 2007, pages 125–139, July 2007. 5. A. G. Clark. Inference of haplotypes from pcr-amplified samples of diploid populations.
Molecular Biology and Evolution, 7(2):111–122, 1990.
6. M. J. Daly, J. D. Rioux, S. F. Schaffner, T. J. Hudson, and E. S. Lander. High-resolution haplotype structure in the human genome. Nature Genetics, 29:229–232, 2001.
7. C. M. Drysdale, D. W. McGraw, C. B. Stack, J. C. Stephens, R. S. Judson, K. Nandabalan, K. Arnold, G. Ruano, and S. B. Liggett. Complex promoter and coding regionβ2-adrenergic receptor haplotypes alter receptor expression and predict in vivo responsiveness. In National
Academy of Sciences, volume 97, pages 10483–10488, September 2000.
8. G. Greenspan and D. Geiger. High density linkage disequilibrium mapping using models of haplotype block variation. Bioinformatics, 20, Supp. 1, 2004.
9. D. Gusfield. Inference of haplotypes from samples of diploid populations: complexity and algorithms. Journal of Computational Biology, 8(3):305–324, August 2001.
10. D. Gusfield. Haplotype inference by pure parsimony. In 14th Annual Symposium on
Combi-natorial Pattern Matching (CPM’03), pages 144–155, 2003.
11. D. Gusfield and S.H. Orzach. Handbook on Computational Molecular Biology, volume 9 of Chapman and Hall/CRC Computer and Information Science Series, chapter Haplotype Inference. CRC Press, December 2005.
12. Y-T. Huang, K-M. Chao, and T. Chen. An approximation algorithm for haplotype inference by maximum parsimony. Journal of Computational Biology, 12(10):1261–1274, December 2005.
13. B. Kerem, J. Rommens, J. Buchanan, D. Markiewicz, T. Cox, A. Chakravarti, M. Buch-wald, and L. C. Tsui. Identification of the cystic fibrosis gene: Genetic analysis. Science, 245:1073–1080, 1989.
14. M. Kimura and J.F. Crow. The number of alleles that can be maintained in a finite population.
Genetics, 49(4):725–738, 1964.
15. D. L. Kroetz, C. Pauli-Magnus, L. M. Hodges, C. C. Huang, M. Kawamoto, S. J. Johns, D. Stryke, T. E. Ferrin, J. DeYoung, T. Taylor, E. J. Carlson, I. Herskowitz, K. M. Giacomini, and A. G. Clark. Sequence diversity and haplotype structure in the human abcd1 (mdr1, multidrug resistance transporter). Pharmacogenetics, 13:481–494, 2003.
16. G. Lancia, C. M. Pinotti, and R. Rizzi. Haplotyping populations by pure parsimony: complexity of exact and approximation algorithms. INFORMS Journal on Computing,
16(4):348–359, 2004.
17. I. Lynce and J. Marques-Silva. Efficient haplotype inference with Boolean satisfiability. In
National Conference on Artificial Intelligence (AAAI), July 2006.
18. I. Lynce and J. Marques-Silva. SAT in bioinformatics: Making the case with haplotype inference. In International Conference on Theory and Applications of Satisfiability Testing
(SAT), 2006.
19. T. Niu, Z. Qin, X. Xu, and J. Liu. Bayesian haplotype inference for multiple linked single-nucleotide polymorphisms. American Journal of Human Genetics, 70:157–169, 2002. 20. S. H. Orzack, D. Gusfield, J. Olson, S. Nesbitt, L. Subrahmanyan, and Jr. V. P. Stanton.
Analysis and exploration of the use of rule-based algorithms and consensus methods for the inferral of haplotypes. Genetics, 165:915–928, October 2003.
21. M. J. Rieder, S. T. Taylor, A. G. Clark, and D. A. Nickerson. Sequence variation in the human angiotensin converting enzyme. Nature Genetics, 22:481–494, 2001.
22. M. Stephens, N. Smith, and P. Donelly. A new statistical method for haplotype reconstruc-tion. American Journal of Human Genetics, 68:978–989, 2001.
24. L. Wang and Y. Xu. Haplotype inference by maximum parsimony. Bioinformatics,