CopyrightC 1984, American Societyfor Microbiology
Selective Cloning of
a
DNA
Single-Strand Initiation Determinant
from
XX174
Replicative-Form DNA
MICHAEL D. STRATHEARN,' ROBERT L. LOW,2tAND DAN S.
RAY'*
Molecular Biology Institute, University of California, Los Angeles, California 90024,1 and Department of Biochemistry,
Stanford University School of Medicine, Stanford, California94305 Received 28 July 1983/Accepted 27 September 1983
An M13 phage deletion mutant, M13AE101, developed as a vector for selecting DNA sequences that
direct DNA strand initiation on asingle-stranded template, has been used for cloningrestriction enzyme
digests ofXX174 replicative-form DNA. Initiation determinants, detected on the basis of clear-plaque formationbythechimeric phage,werefound onlyinrestriction fragments containing the unique effector site
inXX174 DNA fortheEscherichia coli proteinn' dATPase (ATPase). Furthermore,thesesequences were
functional only when cloned in the orientation in whichthe4)X174viral strandwasjoinedtothe M13 viral strand. A181-nucleotide viral strand fragment containing this initiationdeterminant confersa XX174-type
complementary-strand replication mechanism on M13 chimeras. The chimeric phage isconverted to the
parental replicative form in vivo by a mechanism resistant to rifampin, a specific inhibitor of the normal RNApolymerase-dependent mechanism of M13. In vitro, the chimeric single-strandedDNApromotesthe
assembly ofa functional multiprotein priming complex, orprimosome, identical to that utilized by intact XX174 viral strand DNA. Chimeric phage containing the sequence complementary to the 181-nucleotide
viral strand sequence shows noinitiation capability, either in vivo orin vitro.
The single-stranded (SS) viral DNA of
4X174
is distin-guished from that of the SS phages G4 and M13 by the presence of a specific DNA sequence that serves as an effector for the Escherichia coli protein n' (Y factor) dATPase (ATPase) activity. This interaction between pro-teinn' and the effector sequence initiatesthe assembly ofamultiprotein priming complex, the primosome, that once formedmovesalong the DNAlayingdown RNAprimersfor complementary strand synthesis (1, 3). In vitro analysis of SSfragments of
4fX174
viral DNA haslocalized theprotein n' effector activity to a 55-nucleotide sequence within the intergenicspace between the4X174 genes FandG (22).Onepurposeofthis work was todetermine whether such sequences might also be present in the XX174 complemen-tary strand to allow a discontinuous mode of viral strand synthesis. Continuous synthesis of the XX174 viral strand has beenobservedinanin vitro system withpurifiedgeneA
protein, rep helicase, single-strand-binding protein (SSB), andpolIIIholoenzyme (4, 6).
Synthesis
is initiatedbygene A protein nicking at a specific site on the viral strand, followedby continuouselongation byarolling-circle
mecha-nism. In contrast,recentinvivoexperiments
have indicated that viral strand synthesiscanalsooccurbyadiscontinuous processwithinitiationsatsites other than the gene Anicking
site (14). Such internal initiations might be directed by n'-type effector sequences in the complementary strand. We have, therefore, analyzed the entire4X174
genome for the presence of such initiation determinants, using the M13 deletionmutantAE101as a vector toallowselectionofDNAsequences that direct initiation on an SS template (15, 19). The M13AE101 vector contains a 105-nucleotide deletion spanning the normal M13 complementary strand origin and consequentlygivesa 20-fold reducedphageyieldandforms very turbidplaques (13). The insertion ofaDNA sequence capable of directing DNA initiation on an SS template
*Correspondingauthor.
tPresentaddress:DepartmentofPathology,Washington Univer-sity School of Medicine, St. Louis, MO 63110.
restores phage growth to a normal level and causes the production of a clear plaque.
By in vitro directed mutagenesis of the cloned initiation determinant, it will be possible to define more precisely the n' recognition sequence and to identify possible contiguous sequencesthatmight be
required
for primosomeassembly or movement but notfor n' recognition per se. The chimeric phages constructed here represent a first step in the genetic analysisof the4X174 initiationdeterminant.MATERIALS ANDMETHODS
Bacterialandphagestrains. Thebacterial strainsusedare
derived from E. coli K-12. The strains used were K37 (Hfr supD), W1485 (F+ supE), RL108(F+ Tetrleu met rk
mk+
recA56). The construction ofbacteriophages M13bla6l andM13z\E101
hasbeendescribed previously(10, 13). Bacterio-phages M13 andkX174am3
werelaboratory stocks.Enzymes and chemicals. T4 DNA ligase and restriction enzymes HaeIII,
Hinfl,
Pstl, and HindII, were purchased from Bethesda Research Laboratories (Gaithersburg, Md.). EcoRIandAluIrestriction enzymesweregenerously provid-edbyM. Komaromy and J. Kaguni,respectively. Bacterial alkalinephosphatasewaspurchasedfrom Worthington Diag-nostics (Freehold, N.J.), DNA polymerase I (E. coli) from New England Biolabs (Beverly,Mass.), [a-32P]dATPat 800 Ci/mmol and[ot-32P]dCTP
at 670 Ci/mmol from NewEn-gland Nuclear Corp. (Boston, Mass.), and deoxyribonucleo-side triphosphates and the
2',3'-dideoxynucleoside
triphos-phates from P-L Biochemicals (Milwaukee, Wis.). Avian myeloblastosis virus reversetranscriptase,
EcoRI linker (CATGAATTCATG), and theoligonucleotide primer
(CGCAACGTTGTT) were generouslyprovided by
J. W. Beard, Life Science Inc. (St. Petersburg, Fla.) and K.Itakura, City of Hope Medical Center (Duarte,
Calif.),
respectively. Sea Plaque agarose was purchased fromMa-rine Colloids Division, FMC Corp.
(Rockville,
Md.), PstI linker (GCTGCAGC) from Collaborative Research, Inc. (Waltham, Mass.), and rifampin from Sigma Chemical Co. (St. Louis,Mo.).178
on November 10, 2019 by guest
http://jvi.asm.org/
Media and buffers. M9 and TYE media, TYE plates, agarose gels, electrophoresis buffer, TE, top agar, and Triton lyticmix have been described previously (10, 20).
Analysis of phage and phageDNAs.Preparationandplaque assay of M13 phages, rapid gel analysis of chimeric phage DNAs, minilysate analysis of intracellularreplicative forms (RF), preparation ofphage RF andplasmid DNAs(10, 12), and conditions for restriction enzyme cleavage ofDNA (8, 24)have been previouslydescribed as cited.
Purification ofDNA fragments. MixturesofDNA restric-tion fragments were separated by electrophoresis through SeaPlaque agarose gels. The desired fragment was recov-ered by excisingtheappropriate section ofthe gel, melting the agarose at 65°C, bringing the NaCl concentration to 0.3 M,andextracting twice withphenol to remove agarose. The agarose-free aqueous layer was extracted twice with ether, andthe DNAwasrecoveredby precipitation with2volumes of ethanol at -20°C.
Phosphatase treatment.Bacterial alkaline phosphatasewas used to removethe5'-terminal phosphate from the ends of DNA fragments. The DNA wasdialyzed intoa buffer of 10 mMTris,pH 8.0, and treated with0.25 Uofenzyme per ,ug offragments for30min at 37°C. The enzyme was removed subsequently by phenol extraction.
Ligation, transformation, sequence analysis, and prepara-tion of probe. Preparation of
32P-labeled
phage (9), linker ligation (15), cohesive end ligation (5), transformation of RL108 (10), and DNA sequence analysis by the dideoxynu-cleotide substitution method by a modification (15) of the procedure ofSangeret al. (21) have been described previ-ously.RESULTS
Selective cloning of 4X174 initiation determinants. To identify SS initiation determinants (ssi) that
might
becon-tained within either the viral or
complementary
strand of ,X174 duplex DNA, we cloned restrictionfragments
of4X174
RFinto theuniqueEcoRI site of M13AE101RF. The M13AE101 phage gives a turbid plaque, whereas chimeric derivativescontainingan ssideterminantgive
clearplaques
(19).
4X174 RFwascleaved withAluI,HaeIII,orHindll, and the resultingfragments were mixed to form a
pool.
EcoRI linkers were ligated to thefragment ends,
followedby
cleavage with EcoRI andsubsequent
ligation
to EcoRI-cleaved, alkalinephosphatase-treated
M13AE101
RF. The ligated DNA was used to transfectCaCl2-treated
E. coli RL108forplaque formation.Fifty-eight
clearplaques
wereobtainedonthe
plates
inabackground
of greater than 1,000 turbid plaques. Fifteen of theclear-plaque
isolates wereselectedfor further
study.
Gelelectrophoresis
ofviral DNA contained in culture supernatants revealed four sizeclasses,
threeof whichwereclearly larger
thanthevectorM13AE101 (datanot shown).Restrictionenzyme
analysis
andpartial
sequencing
of the inserts in the RFs of thesephages
revealed that three differentinsertshadbeen obtainedbythis selection(Fig. 1): the HaeIII-1 fragment (seven isolates), the AluI-2fragment (four isolates), and the Hindllfragments
1 plus 9 (one isolate, fragmentorderasfound in the genome), with three isolates containing no inserts (pseudorevertants of M13AE101 to clearplaque morphology). In all isolates the XX174viral strand had beenjoinedto the M13 viral strand. Thesechimericphageswereusedassize markersforscreen-ing 43 additional clear-plaque isolates from the
original
transformation plates, and no other size classes wereob-0/5386
FIG. 1. Location of the cloned fragments on the 4X174 physical and genetic map. The shaded area(s) of each ring shows the fragment(s)that confer aclear plaque morphology when cloned into the vector M13AE101. The cloned fragments are AluIlfragment 2 (853 bp),HaeIIIfragment 1 (1,353 bp), andHindIl fragments 1 and 9 (1,057 plus 162 bp). All fragments giving rise to clear plaques contain 4X174viral strand sequences cloned into the M13 viral strand. The genesof4X174areindicatedby capital letters;intergenic spaces are indicatedby cross-hatches. The 181-bp fragment subcloned in Fig. 5 isindicatedby a bracket and the symbol rri.
served. These threefragmentsallcontainedthe 55-basepair (bp) sequence previously identified as the effector for the protein n' dATPase (22;Fig. 1).
Restoration of wild-type phage growth by cloned
4X174
sequences.Figure2showsphagegrowth curvesobtained for M13AE101 and thechimericphagesM13+X102,M134X103,
and M13+X106. The cloning vector M13AE101 shows aninitial lag ofmorethan 30 min in theproduction of phageand
ayield of only2 x
101
1 PFU/ml after20 hofgrowth (13). In contrast, the chimeric phages rapidly produced phage and showed final yields ranging from 5- to 40-fold greater than thatof the vector.Subcloning ofthe
4X174
initiation determinant. We have shown previously thata 1,565-bp HaeII fragmentof4X174 RFcloned intoan HaeII site withinthe intergenic space of M13 conferred a rifampin-resistant mechanism of initiation of SS to RF on the chimericviral
DNA (11, 18). This fragment spans the region identified here as giving clear plaques when cloned into the M13AE101 vector. We have used this HaeIIfragment for subcloningtodefinefurther the initiation determinant located in this region of the4XX174
genome. Because this determinant confersarifampin-resis-tantinitiation mechanism, we term it an rri determinant (a subclass ofssideterminants) (19).
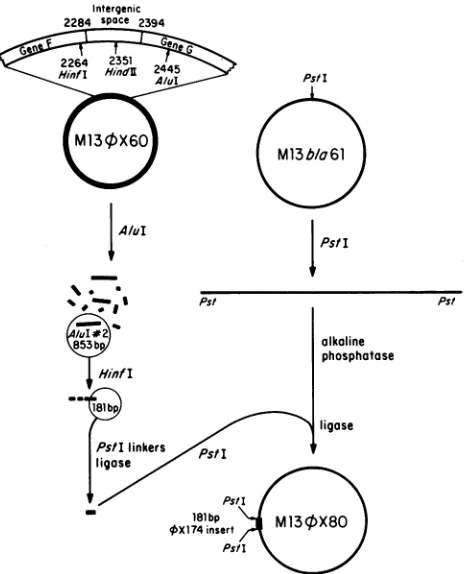
The 4X174rrideterminant has been subclonedasoutlined in Fig. 3. An 853-bp AluI fragment corresponding to the
XX174 AluI-2 fragment was isolated from M134X60RF by electrophoresis of an AluI digest through Sea Plaque
ag-arose. The appropriate bandwas excised from thegel, and the DNA was isolated as described above. This AluI
frag-mentwascleaved withHinfl, and the3-bp overhangingend wasfilledin with DNA polymerase Iplus all four deoxynu-cleotides. PstI linkers [d(G-C-T-G-C-A-G-C)] were ligated
onto theblunt ends, and the resulting fragments were then treated withPstI andligated toPstI-cleaved, alkaline
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.314.551.77.271.2]10
D 1010
L.L
109
108
1071
0 1 2 3 20
TIME
(hours)
FIG. 2. Restoration of normal phage growth by the cloned OX
sequences. E. coli K37 cells were grown in M9 medium containing 0.2% glucoseand0.5% CasaminoAcids. At a cell density of 2x 108 cells perml, samples were infected with M13+X102, M134X103, M134X106, orM13AE101 phage at amultiplicity of infection of 1. Sampleswereremoved from each culture at intervals and assayed forPFU.
phatase-treated M13bla6l RF. M13bla6l isan M13
cloning
vector containing the Tn31-lactamase
gene and a unique PstIsite (10). The ligated DNAwasusedtotransfect E. coliRL108. Forty random plaques were selected and screened forchimeric phages containing thecorrect size insert. Five isolates were used forpreparingRFforfurtherstudy. Upon
treatmentwithPstI,all RFisolateswerefoundtocontain the
same 181-bp insert. Representative isolates in which either the viral or complementary strand of this 181-bp fragment
was inserted into the viral strand ofM13bla6l have been termed
M134X81
and M13+X80,respectively.
The
4X174
sequences contained in both M13+X80 and M13+X81weredeterminedbytheprocedure ofSangeretal. (21) with the synthetic oligodeoxynucleotide primer CGCAACGTTGTT(23). Bothphageswerefoundtocontain only the 181-bp AluI-to-Hinflfragment that spansthe inter-genic regionbetween the4X174
genesF andG andasingle PstIlinkeron each endof the fragment.Rifampin-resistantRFformation in vivo.Theabilityof32p_ labeled
M134X80
andM13+X81 todirect initiation ofcom-plementary strand synthesis by arifampin-resistant mecha-nismwascarriedout aspreviouslydescribed (11, 17).E.coli K37cellswereinfected with eachphageseparately in either
the presence or absence of 200
[Lg
of rifampin per ml.Infected cellswereharvestedat15 minafterinfection, lysed,
and sedimented through high-salt sucrose gradients. The resulting sedimentation profiles are shown in Fig. 4. Like wild-type M13, the
M134X80
phageforms parental RF in the absence of rifampin but not in its presence. In contrast, M13+X81 is converted to the duplex form in either the presence or the absence ofrifampin. These results indicate the presence of an rri determinant in the viral strand of the 181-bp fragment, but not in the complementary strand. This observation is in agreement with the finding that XX174 sequences capable of giving rise to clear plaques in M13AE101 areexclusively of viral strand origin.Strand specificity of the
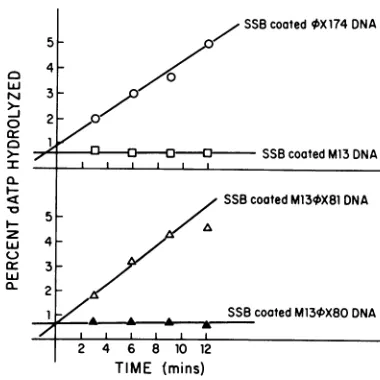
4X174
primosome loading site.M13+X80 and M13fX81 viral DNAs were assayed for their ability both to function as effectors of the dATPase activity of the purified n' protein and to mediate the assembly of a functional primosome. The results (Fig. 5) indicate that M134X81 and XX174 viral DNAs coated with SSB are
equallygood effectors ofthen' dATPase, whereas M13 and M13+X80 DNAs are not.Similarly, M13+X81 DNA, but not M134X80 DNA, wasfound to direct SS to RFsynthesis in
ntergenic 2284 space 2394
Hi,7f I
Ai44
M
3
|
/I
PstIPS,
alkaline phosphatase
FIG. 3. Outline of the construction of M13+X80. M134X60
carriesa 4X174HaeII fragment(1,565bp) that contains the inter-genicspacebetween genes F and G.M134X60RF wascleaved with Alul, and the resulting fragments were separated by agarose gel electrophoresis. The853-bpAlulfragment was extracted from the agarose and treated with Hinfl, giving rise to four restriction fragments. The cohesive ends produced by Hinfl were filled by usingDNApolymerase I (as outlined in the text), and thefragments were ligated toPstIlinkers which were subsequently cleaved with PstItoproduce cohesive ends. The vector DNA,M13bIa6l RF(10), was cut at its single PstI site and treated with bacterial alkaline phosphatasetopreventself-closure. Afterligationof theAluI-Hinfl fragments into the PstI-cut vector, the ligated DNA was used to
transfectCaCl2-treatedE.coli K37 cells.Phage DNAs from individ-ual plaqueswere screenedby agarose gelanalysis (10)to identify chimericphages containingasinglecopyof the cloned 181-nucleo-tide sequence.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.119.264.77.394.2] [image:3.612.326.558.287.574.2]oSl
I,1
vf,4
I 10
lo
20 30 40 1 10 20 30 40E 10
(b)
SS
M13PX80
- 20-(d)
sS Ml3kX8-_ \ +rifampicin _ +rifampicin
5 -10
RFI
1 10 20 30 40 1 10 20 30 40
Fraction Number
FIG. 4. Effectofrifampin (rifampicin)onparentalRFformation in vivoby M134¢X80 and M134X81.E. coliK37cellswere grown
withaerationat37°Cin glucose minimal mediumtoanabsorbanceat
595 nm of 0.4 and were infected with 32P-labeled phage in the presenceorabsence of 400,ugofrifampin perml. Fifteen minutes
afterinfection, the cells (5 ml) werepoured over5 ml ofice-cold TENCNbuffer, centrifuged,and washed three times with TENCN bufferat0°C. Thewashed pelletsweresuspended in 0.5 ml TENCN
buffer, and lysozyme was added to 400 ,ug/ml. After a 10-min
incubation at 37°C, sarkosylwasgentlymixed into the lysatetoa
final concentration of 1% (wt/vol). After an additional 10-min
incubation at37°C, Sarkosyl wasgently mixed into the lysate toa at -20°Corlayereddirectlyonto5to20%sucrosegradients inTE
buffercontaining 1.0 MNaCl. Sedimentation wasfor 4 hat5°C at 40,000 rpmina SW40rotor. Samples of fractions were spottedon
Whatmanno. 1 paperand counted.
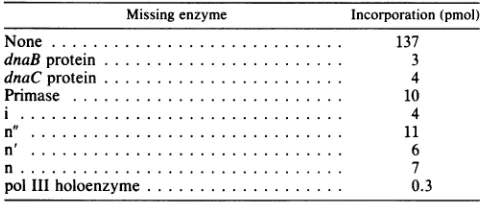
vitro by using purified primosome proteins and pol III
holoenzyme. By omitting one component at a time, it was found that each of the primosome proteins wasrequiredfor M134X81 RF synthesis (Table 1).
DISCUSSION
An exonuclease VII-resistant, 55-nucleotide fragment of
XX174 viral DNA located in an untranslated region of the PX174genome between genes Fand G has been shown to contain the single recognition sitefor the E. coli n' protein within the ~X174 viral strand (22). Recognition of this
specific sequence in 4X174 DNA is accompanied by n'
ATPase (dATPase) activity and the destabilization of a fraction of the SSB molecules bound to the viral DNA.
Bindingof n' to itsrecognition sequenceis followed bythe
assembly ofamultiprotein primosomewhichmoves
proces-sively around the circular SS viral DNA to prime repeated initiations of thecomplementary strand(1, 3).
Synthesis ofthe 4X174 viral strand occurs by a rolling-circle mechanism (7) inwhich the viral strand of theduplex RF is continuously synthesized with the circular
comple-mentarystrandasthetemplate.The initiationofviral strand synthesis occurs at a unique nick introduced into the viral strand of the RFbytheaction of the kX174 gene Aprotein. In contrast,invivopulse-labeling experimentsareconsistent with a discontinuous mechanism of viral strand synthesis. Basedon ananalysisof the sequenceof the (X174 comple-mentarystrand, Matthesetal.(14) have identified three sites in the complementary strand that have some homology to
the single n' site in the viral strand. These sites could possiblybeinvolved inadiscontinuoussynthesisof the viral strand in vivo by a mechanism involving the primosome. Alternatively, such initiations might occur by still other mechanisms, such as adnaB-primase reaction (2).
The phage M13AE101 isa viabledeletion mutant of M13 from which the complementary strand origin has been de-leted (13). The mutant is extremely defective in the conver-sionof the viral DNAto aduplex RF. Phage DNAsynthesis is reduced20-fold, andconsequently, the phage forms very turbid plaques. We have found that DNA sequencescapable ofdirecting initiation on an SS template will rescue this defect whencloned into the mutant genome (14). We have, therefore, made use of this property ofM13AE101 to search for initiation determinants that could possibly be located within the 4X174 complementary strand and which would restore normal phagegrowth and lead to the formation of a clearplaque(15).
By cloningHaeIII,Aliil,andHindlldigests of 4X174RF, we were able to construct chimeric phage that give a clear plaque and a high phage yield. However, in all cases, the cloned fragment overlapped the known n' recognition site within the untranslated region between genes F and G. Furthermore, in all cases, the XX174 strandcontainedwithin the M13 viral strand was the viral strand and not its complement. These results imply, but do not prove, an
absence ofprimosome loading sites in the 4X174 comple-mentary strand. The successfulcloning of other primosome loading sites from each of the two strands ofduplex ColEl DNA(15,16) support thisconclusion.Wethereforepropose that if (X174 viral strand synthesis is indeeddiscontinuous invivo, the mechanism differs significantly from that of the
SSB coated 0X174DNA
5 -O
0 2
-13
c072L_SSB coatedM13DNA
I l
a.
I-< / SSB coatedM13OX81DNA
z wi 4 'r 3
0. 2
17A SSB coatedM13tX80 DNA
2 4 6 8 10 12
TIME (mins)
FIG. 5. dATPaseactivity ofthe n' proteinwith differentDNAs aseffectors.Assaymixtures(25 ,ul)contained 240pmol (as nucleo-tide) of the indicated SSB-coated SS DNAs. Production of
[3H]dADPwas measuredbypolyethyleneimine-cellulose thin-layer chromatography.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.54.291.76.365.2] [image:4.612.331.521.497.687.2]TABLE 1. Requirement forM13+X81 chimericphage SS to RF synthesis in vitro'
Missing enzyme Incorporation (pmol)
None ... 137
dnaBprotein ... ... 3
dnaC protein ... ... 4
Primase ... ... 10
i ... 4
n" ... 11
n'... 6
n... 7
pol IIIholoenzyme ... 0.3
aInvitroDNAreplication assay wascarriedout asdescribedby
Nomura et al. (16), using M13+X81 as a template for SS-*RF
synthesis. Trichloroacetic acid-precipitable counts weremeasured after incubation for 10minat30°C.
complementary strand and probably does not involve the primosome.
The cloned viral strand primosome assembly site func-tioned asefficientlyas(X174viral DNAbothinvivoandin vitro. Recombinants containing the complement of the ac-tiven'sitewereobtainedby plaquehybridizationtechniques and were shown to give a turbid plaque and to have no significant n' dATPase effector activity or primosome as-sembly capability. Similarly,asynthetic55-mer correspond-ingto thecomplementarycopy ofthe 55-mer n' site is also inert as an dATPase effector (R. L. Low, R. Crea, and A. Kornberg, unpublished data). The lack of activity by the sequencecomplementarytothatof theprimosome assembly site on the viral strand suggests that specific sequence information as well as nucleic acid secondary structure is required for a functional site. This is also true of the primosome assembly sites in ColEl DNA (15, 16).
ACKNOWLEDGMENTS
Thisworkwassupportedby PublicHealthServicegrantAl10752 fromtheNationalInstitutes ofHealth.M.S. was a trainee on a cell and molecular biology training grant GM 07185 from the National InstitutesofHealth.
WethankArthurKornberg, inwhoselab the primosomalproteins werepurified,for encouragement.
LITERATURE CITED
1. Arai, K., N. Arai,J. Shlomai, J.Kobori,L.Polder, R. Low,U. Hubscher, L. Bertsch,and A. Kornberg. 1981. Enzyme studies of
4X174
DNAreplication. Prog. Nucleic AcidsRes. 26:9-32. 2. Arai, K., and A. Kornberg. 1979. A general priming systememploying only dnaBproteinandprimase forDNAreplication.
Proc. Natl.Acad. Sci. U.S.A. 76:4308-4312.
3. Arai, K.,R.Low,J. Kobori, J.Shlomai,and A.Kornberg.1981. Mechanism of dnaB protein action. V. Association of dnaB protein, protein n',and otherprepriming proteinsin the primo-someof DNAreplication. J.Biol. Chem. 256:5273-5280. 4. Arai, N.,L.Polder,K.-I.Arai, A. Kornberg. 1981. Replication
of
4OX174
DNAwithpurifiedenzymes. II.Multiplication ofthe duplex form by coupling of continuous and discontinuous synthetic pathways.J. Biol. Chem. 256:5239-5246.5. DeVries, F. A. J., C.J. Collins,and D. A.Jackson.1976.Joining of simian virus 40 DNA molecules at endonuclease R Eco RI sites by polynucleotide ligase and analysis of the products by agarose gel electrophoresis. Biochim. Biophys. Acta 435:213-227.
6. Eisenberg,S.,J.Griffith,andA.Kornberg.1977.4X174cistron
A proteinisa multifunctional enzyme in DNA replication. Proc. Natl. Acad. Sci. U.S.A. 74:3198-3202
7. Eisenberg, S., J. F. Scott, and A. Kornberg. 1978. An enzyme system for replicating the duplex replicative form of4X174 DNA, p. 287-302. In D. T. Denhart, D.Dressler, and D. S. Ray (ed.), The single-stranded DNA phages. Cold Spring Harbor Laboratory, Cold SpringHarbor, N.Y.
8. Endlich, B., and S. Linn. 1981. Type 1 restriction enzymes, p. 137-157. InP. B. Boyer (ed.), The enzymes, 3rd ed. Academic Press,Inc., NewYork.
9. Fidanian, H. M.,and D. S. Ray. 1972.Replication of bacterio-phage M13. VII. Requirement of the gene 2 protein for the accumulation of aspecificRFIIspecies. J. Mol.Biol.72:51-63. 10. Hines, J. C., and D. S. Ray. 1980.Construction and character-ization of new coliphage M13 cloning vectors.Gene 11:207-218. 11. Kaguni, J., L. S. LaVerne, M. Strathearn, and D. S. Ray. 1980. Cloning and expression offoreign replication origins in the single stranded DNA phage M13, p. 35-44. In S. Zadrajil and J. Sponar(ed.), DNA recombination interactions and repair. Per-gamon Press, Inc., Elmsford, N.Y.
12. Katz, L., D. T. Kingsbury, and D. R. Helinski.1973.Stimulation by cyclicadenosine monophosphate ofplasmid deoxyribonucle-ic acid replication and catabolite repression of the plasmid deoxyribonucleic acid-proteinrelaxationcomplex.J. Bacteriol. 114:577-591.
13. Kim, M. H., J. C. Hines, and D. S. Ray. 1981.Viabledeletions oftheM13complementarystrandorigin.Proc. Natl. Acad. Sci. U.S.A.78:6784-6788.
14. Matthes,M., P. J. Weisbeek, and D. T. Denhardt. 1982. Mecha-nism ofreplication ofbacteriophage (X174. XIX.Initiation of
4X174 viral strand DNA synthesis at internal sites on the genome. J. Virol. 42:301-305.
15. Nomura, N., R. L. Low, andD. S. Ray. 1982.Selectivecloning of ColEl DNA initiation sequences using the cloning vector M13AE101. Gene18:239-246.
16. Nomura, N., R. L. Low, and D. S. Ray. 1982. Identification of ColElDNA sequence thatdirectsingle strand-to-doublestrand conversionbya4X174 typemechanism. Proc.Natl.Acad. Sci. U.S.A.79:3153-3157.
17. Nomura, N., and D. S. Ray.1980. Expression ofaDNAstrand initiation sequenceof ColEl plasmidin asingle-stranded DNA phage. Proc.Natl. Acad. Sci. U.S.A. 77:6566-6570.
18. Ray, D. S., J. M. Cleary, J. C.Hines,M. H.Kim, M.Strathearn, L. S.Kaguni, and M. Roark. 1981. DNA initiationdeterminants of bacteriophage M13 and of chimeric derivatives carrying foreign replicationdeterminants,p.169-172.In D.S.Ray(ed.), Theinitiation ofDNAreplication. Academic Press, Inc., New York.
19. Ray, D.S., J.C.Hines,K. M.Kim,R.Imber,and N. Nomura. 1982. M13 vectors for selectivecloningof sequencesspecifying initiationofDNAsynthesisonsingle-strandedtemplates. Gene 18:231-238.
20. Ray,D.S.,and K. Kook. 1978. Insertionof theTn3transposon intothe genome ofthe single-stranded DNAphageM13. Gene 4:109-119.
21. Sanger,F., S.Nicklen,and A.R.Coulson. 1977.DNA sequenc-ing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U.S.A. 74:5463-5467.
22. Shlomai, J., and A. Kornberg. 1980. An Escherichiacoli replica-tionprotein thatrecognizesauniquesequence withinahairpin regionof4)X174 DNA. Proc. Natl. Acad. Sci. U.S.A. 77:799-803.
23. Wallace, R. B., M. J. Johnson, S. V.Suggs, K.-I.Miyoshi, R. Bhatt, and K. Itakura. 1981. A setofsynthetic oligodeoxyribo-nucleotide primers forDNAsequencing in theplasmid vector pBR322. Gene16:21-26.
24. Wells, R. D., R. D. Klein, and C.K. Singleton. 1981. Type II restriction enzymes, p. 157-192. In P. B. Boyer (ed.), The enzymes, 3rd ed. AcademicPress, Inc.,NewYork.