0022-538X/07/$08.00⫹0 doi:10.1128/JVI.00053-07
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
The ATM/ATR Signaling Effector Chk2 Is Targeted by Epstein-Barr
Virus Nuclear Antigen 3C To Release the G
2
/M Cell Cycle Block
䌤
Tathagata Choudhuri, Subhash C. Verma, Ke Lan, Masanao Murakami, and Erle S. Robertson*
Department of Microbiology and The Tumor Virology Program of the Abramson Comprehensive Cancer Center, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania
Received 9 January 2007/Accepted 27 March 2007
Epstein-Barr virus (EBV) infects most of the human population and persists in B lymphocytes for the lifetime of the host. The establishment of latent infection by EBV requires the expression of a unique repertoire of genes. The product of one of these viral genes, the EBV nuclear antigen 3C (EBNA3C), is essential for the growth transformation of primary B lymphocytes in vitro and can regulate the transcription of a number of viral and cellular genes important for the immortalization process. This study demonstrates an associated function of EBNA3C which involves the disruption of the G2/M cell cycle checkpoint. We show that
EBNA3C-expressing lymphoblastoid cell lines treated with the drug nocodazole, which is known to block cells at the G2/M transition, did not show a G2/M-specific checkpoint arrest. Analyses of the cell cycles of cells expressing
EBNA3C demonstrated that the expression of this essential EBV nuclear antigen is capable of releasing the G2/M checkpoint arrest induced by nocodazole. This G2/M arrest in response to nocodazole was also abolished
by caffeine, suggesting an involvement of the ATM/ATR signaling pathway in the regulation of this cell cycle checkpoint. Importantly, we show that the direct interaction of EBNA3C with Chk2, the ATM/ATR signaling effector, is responsible for the release of this nocodazole-induced G2/M arrest and that this interaction leads
to the serine 216 phosphorylation of Cdc25c, which is sequestered in the cytoplasm by 14-3-3. Overall, our data suggest that EBNA3C can directly regulate the G2/M component of the host cell cycle machinery, allowing for
the release of the checkpoint block.
Epstein-Barr virus (EBV) is a human gammaherpesvirus which targets predominantly B cells and epithelial cells and is associated with a number of human cancers, including Burkitt’s lymphoma, nasopharyngeal carcinoma, Hodgkin’s disease, AIDS-associated and transplant-associated immunoblastic lymphoma, and somewhat controversially, invasive breast car-cinoma (15, 29). EBV is also known to be the causative agent of infectious mononucleosis (15, 29). The in vitro infection of primary B cells with EBV gives rise to lymphoblastoid cell lines (LCLs) which express a subset of 12 latent viral transcripts (15, 29). These 12 transcripts encode six EBV nuclear antigens (EBNAs), three latent membrane proteins (LMPs), two EBV RNAs, and the BARF transcripts (15, 29). Among these, six proteins, EBNA1, EBNA-LP, EBNA2, EBNA3A, EBNA3C, and LMP1, have been shown to be important or critical for the growth transformation and immortalization of human primary B cells in vitro (7, 14, 15, 29, 35).
The EBNA3 gene family comprises a set of three genes arranged in tandem on the EBV genome (15). The EBNA3 family of proteins (EBNA3A, EBNA3B, and EBNA3C) are nuclear phosphoproteins with molecular masses ranging from 130 to 160 kDa and are thought to act as transcriptional reg-ulators (15, 30). These EBNA3 proteins function primarily as transcriptional regulators and contain similar structural motifs as well as a region of limited homology in the amino terminus
(15, 40). This domain contains the binding site for the cellular repressor RBP-J, also referred to as CSL (30). EBNA3C can prevent the binding of RBP-Jto EBNA2 and can downregu-late the EBNA2-modifiedtrans-activation of the LMP1 pro-moter through the association of EBNA3C with RBP-J(30). Furthermore, EBNA3C can regulate Cp, one of the major latent promoters controlling EBNA expression, through RBP-Jand other corepressors (21, 22). EBNA3C also ap-pears to have repressor functions potentially mediated via its interaction with histone deacetylases (28). In addition, studies have shown that EBNA3C is the product of an immortalizing oncogene and is capable of cooperating with H-ras in cotrans-formation assays and of overriding Rb-regulated cellular events (25). EBNA3C has also been shown to play a role in regulating the acetylation and coactivation activity of the p300-ProT␣complex and can function as a transcriptional repressor (34, 41).
Following infection with EBV, B cells become activated as a result of the expression of the EBV latent antigens, con-tributing to the indefinite proliferation of the latently in-fected lymphoblasts (15). These immortalized LCLs express a characteristic repertoire of latent antigens (EBNAs and LMPs), which coordinately act to reprogram the cell cycle controls, leading to the proliferation of the infected cells. EBNA2 and EBNA-LP are expressed within the first 24 to 32 h following EBV infection, coinciding with the transition from G0 to G1 (19). Between 32 and 48 h postinfection, LMP1 and LMP2 are expressed and the levels of EBNA3A, EBNA3B, and EBNA3C begin to rise, corresponding to the movement of cells into S phase and progression through the cell cycle (19).
The cell division cycle is driven by the sequential activation
* Corresponding author. Mailing address: Department of Microbi-ology and Abramson Comprehensive Cancer Center, University of Pennsylvania School of Medicine, 201E Johnson Pavilion, 3610 Hamilton Walk, Philadelphia, PA 19104. Phone: (215) 746-0116. Fax: (215) 898-9557. E-mail: erle@mail.med.upenn.edu.
䌤Published ahead of print on 4 April 2007.
6718
on November 8, 2019 by guest
http://jvi.asm.org/
of a series of cyclin-dependent kinases (CDKs) (5). The timing of the activation of the different CDK isoforms determines the order of occurrence of the major cell cycle transitions: entry into G1phase, the onset of DNA replication, and entry into mitosis (23). An intricate network of redundant control mech-anisms exists to establish the precise timing and sequence of the activation of the various CDK isoforms (5). These regula-tory mechanisms, known as checkpoints, ensure that progres-sion through key cell cycle phase transitions occurs only after the successful completion of the preceding phase by regulating the CDK and cyclin activities responsible for normal cell cycle progression (9). The disruption of host cell checkpoint mech-anisms is common in cancer cells and cells infected with trans-forming DNA viruses, which include simian virus 40, adenovi-rus, and papillomavirus (3).
EBV recombinant viruses and in vitro assays have been used to determine the latent genes responsible for interfering with the host cell cycle machinery (3), and to date most of this work has focused on the G0/G1 transition and the G1 restriction point (31). The expression of EBNA2 and EBNA-LP leads to the upregulation of cyclin D2, important in driving resting B cells from G0to G1(32). The G1restriction point is thought to be facilitated by EBNA3C via the disruption of the cyclin/cdk-pRB-E2F pathway (25). While the mechanism for G2/M pro-gression in cells transformed with EBV has not been charac-terized, studies have suggested that EBNA3C may also facilitate this stage of the cell cycle. In NIH 3T3 fibroblasts arrested by serum depletion, the expression of EBNA3C main-tains cell proliferation and induces nuclear division in the ab-sence of cytokinesis (26). In addition, the NIH 3T3 fibroblast study showed that EBNA3C can abolish the mitotic spindle checkpoint activated by a microtubule-destabilizing drug (26). Recently, Wade and Allday demonstrated that EBV is capable of suppressing the G2/M checkpoint activated by genotoxic drugs (38), and the expression of LMP1 in B-cell lines has been shown to produce a G2/M block in cells, although a clear mechanism for such activities has not been elucidated (11).
Resistance to the cytotoxic action of nocodazole is the con-sequence of an intact G2/M checkpoint, but a high proportion of immortalized and tumor cell lines are defective for this checkpoint arrest and are consequently sensitive to killing by nocodazole (39). The sensitivity of the EBV-immortalized LCLs tested suggests that EBV is capable of bypassing or releasing the G2/M checkpoint response to nocodazole, possi-bly in a manner similar to the reported disruption of the G2/M checkpoint response to mitomycin C, cisplatin, and azelaic bishydroxamine (19, 38). In order to further define the mech-anism by which EBV compromises cell cycle checkpoints, we have examined in more detail the ability of the EBV latent antigen EBNA3C to disrupt the nocodazole-induced G2/M checkpoint response to identify specific factors that may be involved. This study identifies a new function of the EBNA3C protein, which shows an ability to release the G2/M checkpoint arrest. We also show that this release from the G2/M block involves the interaction of EBNA3C with Chk2, the specific effector of the ataxia telangiectasia mutated (ATM)/ATM-Rad3-related (ATR) signaling pathway involved in cell cycle regulation.
MATERIALS AND METHODS
Cell lines, antibodies, and constructs.BJAB cells are EBV-negative B cells isolated from Burkitt’s lymphoma patients and were provided by Elliott Kieff (Brigham and Women’s Hospital, Boston, MA). BJAB cell lines and EBV-positive LCL1 and LCL2 and were grown in RPMI 1640 medium (HyClone, Logan, UT) supplemented with 10% bovine growth serum, 2 mM glutamine, and 25 U/ml penicillin/streptomycin. 293 cells were maintained in Dulbecco’s mod-ified Eagle’s medium (HyClone, Logan, UT) supplemented with 10% bovine growth serum, 2 mM glutamine, and 25 U/ml penicillin/streptomycin.
Polyclonal rabbit anti-phospho-Tyr15 Cdc2 (catalog no. 9111), rabbit poly-clonal anti--actin, mouse monoclonal Cdc2 (catalog no. 9116), mouse mono-clonal cyclin B1, and rabbit polymono-clonal Cdc25c antibodies were all purchased from Cell Signaling Inc. (Danvers, MA). Mouse monoclonal 14-3-3 antibody (sc-1657) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The affinity-purified antihemagglutinin (anti-HA) rabbit polyclonal antibody (A190-108A) was obtained from Bethyl Laboratories Inc. (Montgomery, TX). A10 monoclonal antibody reactive with EBNA3C and anti-myc ascites fluid reactive with myc have been described previously (17).
The pTP557Chk2 plasmid provided by Tanya Paull (University of Texas, Austin) was used to construct pCDNA3.1HAChk2 by PCR and subcloning into pCDNA3.1HA. The specific truncation constructs of Chk2 were generated by PCR amplification and were subcloned into the pGEX2T plasmid (GE Health-care, Piscataway, NJ). The ATR-wt and ATR-kd inducible cell lines were a gift from Paul Nghiem (Massachusetts General Hospital, Cambridge).
Transfection.BJAB and 293 cells were transfected by electroporation using a Bio-Rad Gene Pulser II electroporator. Ten million cells were collected and washed once in phosphate-buffered saline (PBS). The cells were then resus-pended in 400l of either Dulbecco’s modified Eagle’s medium or RPMI 1640 medium containing DNA normalized to balance the total DNA, and transfection efficiency was determined by evaluating green fluorescent protein expression as an internal control. Once resuspended, the cells were transferred to 0.4-cm electroporation cuvettes and electroporated at 975F and 220 V for BJAB cells and 210 V for 293 cells. Following electroporation, the cells were plated in 10 ml of supplemented medium and grown at 37°C and 5% CO2for 20 h before being
harvested.
Western blotting.Western blotting assays were performed as previously re-ported (6, 36). Western blot analysis was performed by using antibodies specific to EBNA3C and Nm23-H1 and infrared dye secondary antibodies (Rockland, Inc., Gilbertsville, PA), followed by detection with an Odyssey imager (LI-COR, Inc., Lincoln, NE).
siRNA transfection.RNA interference in Chk2 was performed by using 21-bp (including a two-deoxynucleotide overhang) small interfering RNA (siRNA) duplexes purchased from Dharmacon Inc. (Lafayette, CO). The coding strand for Chk2 siRNA was GAACCUGAGGACCAAGAAC-deoxynucleotide-deoxyn ucleotide. For transfection, BJAB and LCL2 cells were seeded into 6-well plates and transfected with 200 nM siRNA duplexes by using Lipofectamine 2000 according to the recommendations of the manufacturer (Invitrogen, Inc., Carls-bad, CA). Cells treated with Lipofectamine 2000 (mock treated) or transfected with the control, nonspecific siRNA duplex VIII (Dharmacon Inc., Lafayette, CO; ACUCUAUCUGCACGCUGACUU) were used as controls for direct comparison. Twenty-four hours after transfection, cells were treated with nocodazole (200 ng/ml). Cells were collected, washed with PBS, and pro-cessed for the analysis of cell cycle distribution or immunoblotting.
We also cloned the short hairpin RNA (shRNA) into pSIREN (Invitrogen, Inc., Carlsbad, CA). Firefly luciferase gene shRNA was used as a control shRNA (BD Clontech, Mountain View, CA). shRNA for Chk2 (5⬘-GATCCGGAACC TGAGGACCAAGAACTTCAAGAGAGTTCTTGGTCCTCAGGTTCCT TTTTTGATATCG-3⬘) was cloned into pSIRENRetroP (BD Clontech, Moun-tain View, CA) after annealing with its complementary sequence (indicated by underlining). Ten million BING cells were transfected with 15g of either pSIRENRetroP Chk2 shRNA or pSIRENRetroP Luc shRNA. At 24 h post-transfection, cells were selected with complete medium containing puromycin (3
g/ml). Medium containing virion particles was centrifuged to remove cell debris and then filtered through 0.45-m-pore-size syringe filters. Virions were pelleted at 20,000 rpm for 2 h and resuspended in 1⫻PBS; LCL2 cells were transduced with the virus, treated with nocodazole, harvested, fixed, and processed for the analysis of cell cycle distribution or immunoblotting.
Real-time quantitative PCR.Total RNA from BJAB cells at 24 h after trans-fection with the control vector and EBNA3C was isolated by using TRIzol reagent according to the instructions of the manufacturer (Invitrogen, Inc., Carlsbad, CA). cDNA was made by using a Superscript II reverse transcriptase kit according to the instructions of the manufacturer (Invitrogen, Inc., Carlsbad,
VOL. 81, 2007 EBNA3C RELEASES G2/M ARREST 6719
on November 8, 2019 by guest
http://jvi.asm.org/
CA). The specific primers for Chk2 and Chk1, respectively, were as follows: sense, 5⬘-CGGATGTTGAGGCTCACGA-3⬘, and antisense, 5⬘-TATGCCCTG GGACTGTGAGG-3⬘, and sense, 5⬘-CCCGCACAGGTCTTTCCTT-3⬘, and an-tisense, 5⬘-GGCTGGGAAAAGCTGATCC-3⬘, yielding 252- and 292-bp PCR products, respectively.-actin was amplified by using the following primers: sense, 5⬘-GCTCGTCGTCGACAACGGCTC-3⬘, and antisense, 5⬘-CAAACAT GATCTGGGTCATCTTCTC-3⬘, yielding a 352-bp PCR product. The target gene was amplified from cDNA by using SYBR green real-time master mix (MJ Research Inc., Waltham, MA), 1 mM (each) primer, and 1l of the cDNA product in a total volume of 20l. Thirty-five cycles of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C, followed by 10 min at 72°C, were performed in an Opticon II thermocycler (MJ Research Inc., Waltham, MA). Each cycle was followed by two plate readings, with the first at 72°C and the second at 85°C. A melting curve analysis was performed to verify the specificity of the products, and the values for the relative quantitation were calculated by the⌬⌬Ctmethod (6).
The experiment was performed in triplicate.
GlutathioneS-transferase (GST) fusion protein preparation, in vitro binding assays, and cellular lysate binding.Escherichia colistrain BL21 cells were trans-formed with pGex2T-Chk2 plasmids expressing four different truncations of Chk2 and selected on an ampicillin plate. A culture grown overnight from a single colony was inoculated into 500 ml of Luria-Bertani medium and grown to mid-exponential phase with shaking. The cells were then induced with 1 mM
isopropyl--D-thiogalactopyranoside (IPTG) overnight at 30°C with shaking. The cells were subsequently harvested and sonicated, and the protein was solubilized. The lysate was then incubated with glutathione-Sepharose beads overnight at 4°C with rotation. The beads were collected by centrifugation and then washed four times with NETN (20 mM Tris䡠HCl [pH 8.0]–100 mM NaCl–1 mM EDTA– 0.5% Nonidet P-40) containing protease inhibitors. The protein-bound beads were then stored at 4°C in NETN containing protease inhibitors.
The full-length pA3M clone of the EBNA3C gene was transcribed and trans-lated in vitro with [35
S]methionine-cysteine in the T7 TNT system (Promega, Inc., Madison, WI). The in vitro-translated proteins were first precleared with glutathione-Sepharose beads in binding buffer (1⫻PBS, 0.1% NP-40, 0.5 mM dithiothreitol, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 2g of apro-tinin per ml, 1g of pepstatin A per ml, 2g of leupeptin per ml) for 30 min at 4°C with rotation, and the beads were removed by centrifugation. A second preclearing with GST-bound glutathione-Sepharose beads followed for 1 h at 4°C with rotation, with the beads removed by centrifugation. The preclear pro-tein was then incubated with truncation mutant constructs of GST-Chk2 for 16 h at 4°C with rotation. The beads were then pelleted by centrifugation and washed four times with the binding buffer. The beads and bound protein were then denatured with sodium dodecyl sulfate (SDS)--mercaptoethanol lysis buffer with boiling, followed by SDS-polyacrylamide gel electrophoresis (PAGE). The
FIG. 1. DNA cell cycle profiles of LCLs treated with nocodazole show that cells transformed with EBV can bypass the G2/M block
induced by nocodazole. LCL1 and LCL2 along with BJAB cells were treated with nocodazole for 24 h; the cells were then harvested and stained with PI, and their cell cycle profiles were determined by FACSCaliber analysis. Untreated and treated cells of the two cell lines transformed with EBV, LCL1 and LCL2, showed similar cell cycle profiles, with increases in the proportions of sub-G0cells seen at high
concentrations of nocodazole. BJAB cells, an EBV-negative B-cell line, were used as a control and showed a distinctive increase in the proportion of cells at G2/M, indicating a block by nocodazole. Results
[image:3.585.83.243.66.365.2]for the untreated and treated cells are shown, together with the per-centages of cells in each stage of the cell cycle. Means and standard deviations were derived from three independent experiments. Conc., concentration.
FIG. 2. DNA cell cycle profiles of BJAB and 293 cells transfected with EBNA3C and treated with nocodazole show that EBNA3C is capable of releasing cells from the G2/M cell cycle block. (A and B)
BJAB cells (stably transfected with the pA3M vector and pA3MEBNA3C) (A) and 293 cells (transfected with the pA3M vector and pA3MEBNA3C) (B) were treated with nocodazole (200 ng/ml) for 24 h and then harvested. The DNA cell cycle distribution patterns were determined by FACSCalibur analysis. Results for the untreated con-trol cells and treated cells are shown, together with the percentages of cells in each stage of the cell cycle. Means and standard deviations were derived from three independent experiments.⫹, present; ⫺, absent. (C) Western blot analysis showed the expression of EBNA3C in the BJAB and 293 stably transfected cell lines.␣-EBNA3C, anti-EBNA3C.
on November 8, 2019 by guest
http://jvi.asm.org/
gel was then dried and exposed to a storage phosphor screen (Amersham Bio-sciences Inc., Piscataway, NJ).
The LCL nuclear extract used in the binding experiments was prepared as described previously (16). The nuclear extract was precleared with glutathione-Sepharose beads for 30 min at 4°C with rotation. The lysates were additionally precleared with GST-bound glutathione-Sepharose beads for 1 h at 4°C with rotation. The lysates were then incubated with GST-Chk2 truncation construct-bound beads, equivalent to the GST-construct-bound beads used for preclearing, and rotated overnight at 4°C. SDS lysis buffer with heating was used to elute the bound protein from the beads, followed by 10% SDS-PAGE. Western blotting using the specific anti-EBNA3C antibody A10 was performed to detect EBNA3C.
Flow cytometric analysis of cell cycle phase distribution of nuclear DNA.For the determination of the cell cycle phase distribution of nuclear DNA, BJAB and 293 cells (106
cells each) were harvested. Cells were fixed with 3% paraformal-dehyde and permeabilized with 0.5% Triton X-100, and nuclear DNA was labeled with propidium iodide (PI; 125 mg/ml) after RNase treatment. The cell cycle phase distribution of nuclear DNA was determined with a FACSCalibur (Becton Dickinson Inc., San Diego, CA) by using CellQuest software (Becton Dickinson Inc., San Diego, CA) and a fluorescence (FL2-A) detector equipped with a 488-nm-wavelength argon laser light source and a 623-nm-wavelength band-pass filter (linear scale). A total of 10,000 events were acquired for analysis. Histograms displaying DNA content (as indicated by PI fluorescence;xaxis) versus counts (yaxis) were created. CellQuest statistics were employed to quan-titate the data at different phases of the cell cycle. The quantification data were approximated to the nearest number.
Immunofluorescence.Immunofluorescence assays were performed essentially as described previously (6, 33). Briefly, fixed cells were blocked in the appropri-ate serum and then incubappropri-ated with the specific primary mouse monoclonal EBNA3C antibody (A10) and rabbit polyclonal anti-HA for Chk2 (the Chk2
gene was originally cloned upstream of three HA epitopes in the pCDNA3.1 vector) for 1 h. Cells were washed and then further incubated with the appro-priate secondary antibody conjugated to Alexa Fluor 488 (green) and Alexa Fluor 596 (red) at 1:1,000 dilutions in PBS for 1 h. Slides were washed, visualized with an Olympus FluoView 300 IX81 inverted confocal microscope, and photographed with a digital camera and FluoView software (Olympus Inc., Melville, NY).
In-cell Western analysis.In-cell Western assays were performed as described previously, with some modifications (6). In-cell Western analysis was used for the evaluation of the Tyr15 phosphorylation of Cdc2. BJAB cells were stably trans-fected with EBNA3C or with the empty control vector and grown to exponential phase. The cells were collected and diluted to a concentration of 105cells/ml in
RPMI 1640 medium. Two hundred microliters of the cell suspension was dis-pensed into round-bottom 96-well plates (20,000 cells/well), and the cells were kept in culture overnight. Cells were fixed with 4% formaldehyde for 20 min after a thorough washing with 1⫻Tris-buffered saline. This step was followed by permeabilization and washing with 0.1% Triton X-100 in 1⫻Tris-buffered saline. Cells were then incubated with both Cdc2 (rabbit polyclonal; 1:1,000) and phos-pho-Tyr15 Cdc2 (mouse monoclonal; 1:1,000) antibodies overnight. Secondary antibodies goat anti-rabbit infrared dye 800 and goat anti-mouse infrared dye 680 (Molecular Probes, Inc., Eugene, OR) were used (1:800), and the mixtures were further incubated for 1 h. Plates were then dried in the dark and scanned for detection in both the 700 and 800 channels by using the Odyssey infrared scanning system (LI-COR, Inc., Lincoln, NE).
RESULTS
EBV is capable of disrupting the G2/M checkpoint arrest
[image:4.585.58.527.69.345.2]induced by nocodazole. Two EBV-immortalized LCLs were
FIG. 3. Nocodazole suppresses the phosphorylation of Cdc2 at Tyr15. (A) Western blot analysis of G2/M cell cycle regulators cyclin B1 and
Cdc2 in BJAB control cells and EBNA3C (E3C)-transfected BJAB cells following exposure to nocodazole. Stably transfected cells were treated with nocodazole (200 ng/ml) for 24 h and then harvested, and total cell lysates were prepared. Following electrophoresis and transfer onto membranes for Western blot analysis, the membranes were probed with antibodies (␣) for cyclin B1, total Cdc2, Tyr15-phosphorylated Cdc2 [Cdc2(Tyr15)], and-actin as a protein control.⫹, present;⫺, absent. (B) Results of in-cell Western analysis showing the phosphorylation at Tyr15 of Cdc2 in the presence of EBNA3C. BJAB cells carrying the vector control or stably transfected with EBNA3C were grown in round-bottom 96-well plates. The cells were then treated with nocodazole. The results of in-cell Western analysis for Tyr15 phosphorylation are presented with respect to the basal level of Cdc2 and are expressed as relative intensities. Means and standard deviations were derived from three independent experiments.
VOL. 81, 2007 EBNA3C RELEASES G2/M ARREST 6721
on November 8, 2019 by guest
http://jvi.asm.org/
treated with nocodazole in a dose-dependent manner for 24 h, and their cell cycle distribution patterns were determined by flow cytometry (Fig. 1, upper and middle panels). Untreated control cells showed a normal, log-phase-growth cell cycle dis-tribution pattern. Following nocodazole treatment, the two LCLs showed drastic decreases in the percentages of cells in the S and G2/M phases and increases in the populations of cells with less than a diploid set of DNA, representing dead and dying cells (Fig. 1). This distribution pattern was consistently observed in both independently generated LCLs. Among BJAB cells, the same concentrations of nocodazole led to G2/M-arrested populations (Fig. 1, bottom panel). These re-sults suggest that the EBV immortalization of B cells leads to the release of the nocodazole-induced blockage at the G2/M checkpoint of the cell cycle. Additionally, as the concentration of the drug increased, the sensitivity to the cytotoxic effects of nocodazole also increased.
The essential EBV nuclear antigen EBNA3C is capable of disrupting the G2 checkpoint block induced by nocodazole.
Given that EBV was capable of disrupting the G2/M check-point response to nocodazole, it was important to establish which of the EBV latent nuclear proteins was responsible for this effect. As the G2/M checkpoint response is intact in BJAB and 293 cells, they were ideal cell lines for the evaluation of the effects of critical EBV latent proteins. A previous study had suggested the involvement of an EBNA3 family member in regulating G2(19). We therefore decided to focus our atten-tion initially on critical proteins in this family. The distant open reading frame for EBNA3C on the EBV genome is known to be important for B-cell immortalization, and therefore, we chose it as the first candidate for our study. A panel of BJAB cells stably transfected with the empty vector alone or the vector expressing EBNA3C were selected and then treated with nocodazole for 24 h (Fig. 2A). Notably, the expression of EBNA3C alone did not affect the normal cell cycle distribu-tion, and the treatment of empty vector-transfected BJAB con-trol cells with nocodazole for 24 h produced the expected G2/M arrest and little cell death. In contrast, the nocodazole treatment of BJAB cells expressing the EBNA3C protein failed to arrest cells in the G2/M phase and corresponded to an increase in the percentage of cells with less than diploid DNA content (Fig. 2A). This response was similar to that observed in LCLs expressing the full set of EBV latent genes. The same trend was observed in 293 cells expressing the EBNA3C pro-tein (Fig. 2B). These results indicated that EBNA3C is capable of releasing the G2/M checkpoint block induced by nocodazole, thereby allowing cell cycle progression. Additionally, a small percentage of the cells were noted to have increased sensitivity to the cytotoxic effects of nocodazole in the presence of EBNA3C (Fig. 2B).
EBNA3C inhibits the downregulation of Cdc2 Tyr15 phos-phorylation triggered by nocodazole. To further assess these checkpoint responses, biochemical analyses of markers of the G2/M cell cycle checkpoint were conducted. Nocodazole in-duced G2/M arrest, indicated by the observed accumulation of BJAB cells in G2/M phase, and the 293 cells described above also demonstrated a blockage in the cell cycle machinery that controls progression into mitosis (10, 20). The ultimate target of the G2/M checkpoint is a block in the activation of cyclin B-Cdc2, the critical regulator of mitosis, detected as an
accu-mulation of the inactive Tyr15-phosphorylated form of the cyclin B-Cdc2 complex (1). Therefore, the inhibition of the checkpoint response to each of these agents by EBNA3C should result in the disruption of the mechanism blocking cyclin B-Cdc2 activation and a lack of accumulation of this inactive complex. To investigate this effect, the levels of cyclin B1 and the inactive Tyr15-phosphorylated form of Cdc2 were analyzed by immunoblotting of lysates of equal numbers of untreated and nocodazole-treated EBNA3C-expressing BJAB cells (Fig. 3A). In EBNA3C-expressing cells, the levels of cy-clin B and Cdc2 remained the same in both treated and un-treated cells. However, a dramatic change in the level of Cdc2 phosphorylated at Tyr15 was observed (Fig. 3A). Small de-creases in cyclin B1 and Cdc2 levels in nocodazole-treated cells relative to those in untreated control cells in the absence of EBNA3C were also detected (Fig. 3A). Interestingly, the acti-vation of mitosis starts with the phosphorylation of Cdc2 at Tyr15 (1). We observed that Tyr15 phosphorylation in Cdc2 was drastically downregulated in B cells in the presence of
FIG. 4. (A) The bypassing of nocodazole-induced G2 arrest by
EBNA3C is inhibited by caffeine. BJAB cells (stably transfected with the pA3M vector and pA3MEBNA3C) and 293 cells (transfected with the pA3M vector and pA3MEBNA3C) were treated with nocodazole (200 ng/ml) with and without caffeine (5 mM) for 24 h and then harvested, and their DNA cell cycle distributions were determined by FACSCalibur analysis. Results for the untreated control cells and treated cells are shown, together with the percentages of cells in each stage of the cell cycle. These data represent the means of results from three separate experiments.⫹, present;⫺, absent. (B) ATR-wt U20S and ATR-kd U20S cell lines were treated with nocodazole in the presence and absence of EBNA3C and then harvested, and their DNA cell cycle distribution patterns were determined by FACSCalibur anal-ysis. The cell cycle distributions of untreated control cells and treated cells are shown, together with the percentages of cells in each stage of the cell cycle. Means and standard deviations were derived from three independent experiments and quantifications were rounded to the nearest number.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.315.526.65.323.2]nocodazole, suggesting G2arrest (Fig. 3A). In EBNA3C-ex-pressing BJAB cells, no change in Cdc2 Tyr15 phosphorylation upon treatment with nocodazole was observed (Fig. 3A). To further corroborate the results of the immunoblot analysis, which showed an inhibition of Cdc2 Tyr15 phosphorylation repression, an “in-cell” Western assay was performed. Again, we observed a dramatic reduction in Cdc2 Tyr15 phosphory-lation in the presence of nocodazole. Furthermore, when EBNA3C was expressed under those conditions in nocodazole-treated cells, this reduction was eliminated, indicating a role for EBNA3C in relieving the cell cycle block at G2through the regulation of Cdc2 Tyr15 phosphorylation (Fig. 3).
EBNA3C is capable of disrupting the G2 checkpoint block
induced by nocodazole through the ATM/ATR signaling path-way.We noted that BJAB cells expressing EBNA3C failed to stably arrest at G2phase in the presence of nocodazole. Pre-viously it was reported that nocodazole and other cytotoxic drugs used for synchronizing cell cycle phases may affect the DNA damage response pathway, specifically the ATM/ATR
pathway (26). The ATM/ATR DNA damage response pathway is a network of interacting pathways that act at the G2phase of the cell cycle to block G2/M progression by ultimately blocking the activation of cyclin B-Cdc2 (19). The ability of EBNA3C to inhibit the G2/M checkpoint blockage response to a range of different agents that produce very different cellular effects sug-gested that EBNA3C might be targeting a common pathway used in response to all these agents. This possibility was exam-ined by using the known sensitivity of both ATM and ATR to inhibition by caffeine (19). The ATM/ATR signaling pathway was utilized in response to nocodazole treatment, suggesting that this signaling pathway may be a target for regulation by EBNA3C. Caffeine released the nocodazole-induced G2phase arrest in both BJAB and 293 cells (Fig. 4A). Interestingly, the effect of caffeine on nocodazole-induced cell cycle arrest was similar to that of EBNA3C (Fig. 4A). To address this effect more specifically, we used doxycycline-inducible ATR-wt and ATR-kd U20S cell lines (24). As expected, the doxycycline induction of the ATR-kd cell line failed to release the
nocoda-FIG. 4—Continued.
VOL. 81, 2007 EBNA3C RELEASES G2/M ARREST 6723
on November 8, 2019 by guest
http://jvi.asm.org/
[image:6.585.47.542.69.491.2]zole-induced cell cycle arrest (Fig. 4B). Also as expected, the expression of EBNA3C in the ATR-wt cell line abolished the nocodazole-induced G2/M phase arrest, whereas no major ef-fect in the ATR-kd cell line was seen (Fig. 4B).
EBNA3C interacts directly with Chk2.The data given above suggest that EBNA3C regulates the function of a cellular pro-tein that is an important component of the ATM/ATR path-way. A microarray analysis of the EBNA3C-expressing BJAB cells demonstrated the downregulation of Chk1 and Chk2 rel-ative to the levels of these kinases in BJAB cells transfected with the empty vector alone (data not shown), and this finding was validated by semiquantitative real-time PCR. The results of the PCR analysis suggested that Chk2 was dramatically downregulated in the presence of EBNA3C, but any effect on Chk1 was not evident by this real-time PCR assay (Fig. 5A). This kinase is activated in response to DNA damage and rep-licative arrest in both Saccharomyces cerevisiae and humans and is capable of phosphorylating Cdc25c, which inhibits the ability of Cdc25c to activate the cyclin B-Cdc2 complexes nec-essary for G2/M transition (19). To determine if there was an
association between EBNA3C and Chk2, coimmunoprecipita-tion was performed with BJAB and 293 cells expressing EBNA3C and Chk2 from a heterologous system. Chk2 tagged with the HA epitope was found to be coimmunoprecipitated with myc-tagged EBNA3C from both cell types by using anti-myc antibodies for immunoprecipitation (Fig. 5B). This finding was corroborated by the results of immunofluorescence anal-ysis, which showed the colocalization of Chk2 and EBNA3C at specific sites within the nucleus (Fig. 5C).
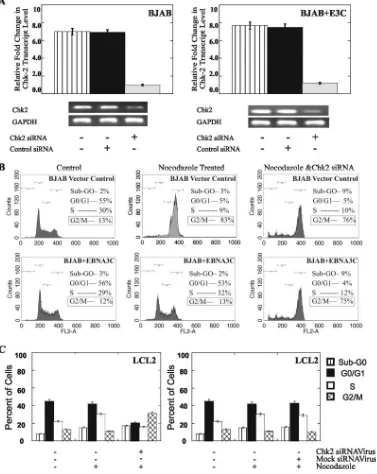
siRNA-mediated downregulation of Chk2 expression inhib-its EBNA3C-mediated release of nocodazole-induced G2/M
[image:7.585.113.472.71.349.2]cell cycle arrest.To experimentally verify the role of Chk2 in the EBNA3C-mediated release of the G2/M phase block, we used siRNA technology to suppress Chk2 expression. Trans-fection with an siRNA targeted to Chk2 suppressed Chk2 expression by 90% in both BJAB and BJAB EBNA3C-express-ing cell lines compared with the expression in the mock-treated controls (Fig. 6A). The expression of Chk2 was not affected in cells transfected with a nonspecific control siRNA (Fig. 6A). Chk2 siRNA-transfected cells and control cells (BJAB cells
FIG. 5. EBNA3C interacts directly with Chk2. (A) Total RNA was extracted from BJAB cells 24 h after transfection with the control vector and EBNA3C (E3C) and was used to make cDNA. Specific primers for Chk1 and Chk2 were used (see Materials and Methods), and the cDNA was amplified by using SYBR green real-time master mix. The data show that in EBNA3C-expressing cells, the levels of Chk2 transcripts were significantly reduced compared to those in cells transfected with the vector alone. In EBNA3C-expressing cells, Chk1 transcript levels were close to those in empty vector-transfected cells. Means and standard deviations were derived from three independent experiments.⫹, present;⫺, absent; EtBr, ethidium bromide; GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) Coimmunoprecipitation of Chk2 and EBNA3C. BJAB and 293 cells were cotransfected with the expression constructs pCDNA3.1HAChk2 and pA3MEBNA3C, and coimmunoprecipitation from the cell lysates was performed by using anti-myc (␣-myc) antibodies. The coimmunoprecipitates were separated by electrophoresis, transferred onto nitrocellulose membranes, and then probed with HA antibodies (␣-HA) for Chk2. Chk2 was found to immunoprecipitate with EBNA3C from both cell types. (C) Chk2 and EBNA3C colocalize in U20S cells. U20S cells were cotransfected with the expression constructs pCDNA3.1HAChk2 and pA3MEBNA3C. Following transfection, the cells were grown overnight on coverslips and fixed. EBNA3C and Chk2 were detected by using specific primary mouse monoclonal antibody against EBNA3C (A10) and rabbit polyclonal anti-HA antibody for Chk2, followed by appropriate secondary antibodies conjugated to Alexa Fluor 488 (green) and Alexa Fluor 596 (red), respectively. The merge panel shows that Chk2 and EBNA3C colocalize in nuclear foci. The DAPI (4⬘,6⬘-diamidino-2-phenylindole) panel shows that both proteins are nuclear, as expected.
on November 8, 2019 by guest
http://jvi.asm.org/
and BJAB cells stably transfected with EBNA3C) were then treated with nocodazole, and their cell cycle distribution pat-terns were assessed after 24 h (Fig. 6B). The treatment of siRNA-transfected BJAB cells with nocodazole resulted in
[image:8.585.100.477.72.546.2]slightly less accumulation of the cells in G2phase and a small increase in the percentage of dead cells (Fig. 6B, upper pan-els). Interestingly, in EBNA3C-expressing BJAB cells, Chk2 downregulation facilitated the nocodazole-induced G2 phase
FIG. 6. Suppression of Chk2 levels inhibits the ability of EBNA3C to bypass the G2/M checkpoint block imposed by nocodazole. (A) Real-time
semiquantitative PCR for Chk2 showed the specific effect of Chk2 siRNA on Chk2 transcripts levels. Total RNA from the cells was isolated to make cDNA. A specific primer set for Chk2 was used, and the cDNA was amplified by using SYBR green real-time master mix. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ⫹, present; ⫺, absent. (B) BJAB cells stably transfected with both the pA3M vector and pA3MEBNA3C were treated with nocodazole with and without Chk2 siRNA for 24 h and then harvested, and their DNA cell cycle distribution patterns were determined by FACSCalibur analysis. Results for the untreated control cells and treated cells are shown, together with the percentages of cells in each stage of the cell cycle. These data are representative of results from three separate experiments and quantifications were rounded to the nearest number. (C) LCL2 cells were treated with nocodazole with and without virus containing Chk2 siRNA along with the mock virus for 24 h and then harvested. The chromatin DNA was stained with PI prior to cell cycle distribution analysis by fluorescence-activated cell sorting. Results for the untreated control cells and treated cells are shown, together with the percentages of cells in each stage of the cell cycle. Means and standard deviations were derived from three independent experiments.
VOL. 81, 2007 EBNA3C RELEASES G2/M ARREST 6725
on November 8, 2019 by guest
http://jvi.asm.org/
arrest, suggesting a requirement for Chk2 in the EBNA3C-mediated bypass of the G2/M block induced by nocodazole (Fig. 6B). We also used the Chk2 siRNA expression vector systems in BING cells to produce viruses which were used to transduce LCL2 cells and knock out Chk2 expression. Virus-containing, Chk2 siRNA-transfected cells were transduced and selected by using puromycin, and the selected cells were treated with nocodazole. The data showed that the G2 /M-phase cell population increased by more than 50% and the G1-phase population decreased by approximately 50% in LCL2 when Chk2 levels were knocked down (Fig. 6C, left panel). As expected, the use of a control mock siRNA virus produced little or no change in the G0/G1, S, and G2/M
pop-ulations (Fig. 6C, right panel). Thus, the suppression of Chk2 levels in the system allows nocodazole to induce the G2/M block in LCLs transformed with EBV, and the essential EBV latent nuclear antigen EBNA3C may utilize Chk2 to release the G2/M block.
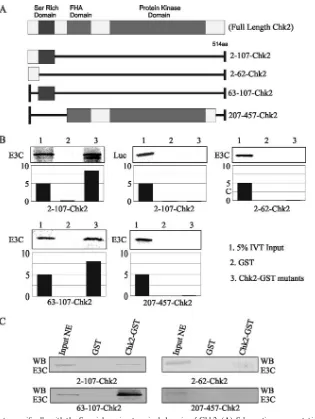
[image:9.585.135.450.64.483.2]EBNA3C binds to the Ser-rich amino-terminal domain of Chk2. In vitro-translated EBNA3C protein labeled with 35S was incubated with a number of truncated mutant forms of the GST-Chk2 polypeptide to map the binding region. The results of the binding assays indicated that the region of Chk2 that interacts with EBNA3C lies between amino acids 63 and 107, a fragment that encompasses the Ser-rich domain of Chk2 (Fig. 7A and B). This interaction region also overlaps with the
FIG. 7. EBNA3C interacts specifically with the Ser-rich amino-terminal domain of Chk2. (A) Schematic representation of full-length domains along with the different truncation constructs. FHA is the forkhead association domain. aa, amino acids. (B) In vitro-translated EBNA3C (E3C) protein was35S labeled and tested for binding with a number of truncated mutant GST-Chk2 constructs spanning the binding region (amino acids
2 to 107, 2 to 62, 63 to 107, and 621 to 1351). The results show that the region of EBNA3C responsible for interacting with Chk2 lies between amino acids 65 and 107, a stretch that encompasses the Ser-rich region of Chk2. Relative binding levels are shown in the bar diagrams. IVT, in vitro translation. (C) Lysates from EBV-positive LCL1 cells expressing EBNA3C were incubated with the GST-Chk2 constructs described above. The pull-down assay again showed a preference for the region located between amino acids 63 and 107, which includes the serine-rich domain. WB, Western blot; NE, nuclear extract.
on November 8, 2019 by guest
http://jvi.asm.org/
forkhead association domain (amino acids 93 to 201). Addi-tionally, the data indicated that the carboxy-terminal region comprising amino acids 207 to 457 of Chk2 is not involved in binding, as there was little or no detectable interaction be-tween this region and EBNA3C (Fig. 7). The results of a separate assay using lysates from EBV-positive LCLs express-ing EBNA3C support the in vitro data showexpress-ing an association of EBNA3C with the serine-rich region of Chk2 (Fig. 7C).
The cellular phosphatase Cdc25c is localized predominantly to the cytoplasm in the presence of EBNA3C.Chk2 is activated in response to replicative arrest and is capable of phosphory-lating Cdc25c, which inhibits the Cdc25c-mediated activation of the cyclin B-Cdc2 complexes necessary for the G2/M tran-sition (2). Negative regulation of the Cdc25c protein phos-phatase by phosphorylation at Ser216, the 14-3-3 binding site also bound by Chk2 (2), is an important regulatory mechanism used by cells to block mitotic entry under normal conditions and after DNA damage (2). During mitosis, Cdc25c becomes hyperphosphorylated at multiple sites apart from Ser216, which results in a predominantly nuclear localization pattern (2). The phosphorylation of Cdc25c at Ser216 by Chk2 allows Cdc25c to bind 14-3-3, which is known to function as a scaffold
[image:10.585.56.525.69.337.2]protein involved in regulating the cytoplasmic-to-nuclear shut-tling of proteins with which Cdc25c and 14-3-3 interact (2). In asynchronous cells, treatment with nocodazole triggered the nuclear translocation of Cdc25c (Fig. 8A, lane 3). Although Cdc25c is not phosphorylated at Ser216 during mitosis, it is phosphorylated at multiple sites within its amino terminus. Hyperphosphorylation allows for Cdc25c to have retarded mobility on SDS-PAGE gels (Fig. 8A, lanes 3 and 5). In EBNA3C-expressing cells, most of the Cdc25c is in the cyto-plasm, a pattern which represents a reduction in the phos-phorylation level and shows a lack of synchronization of the cells after treatment with nocodazole (Fig. 8A, lanes 4 and 5). Immuno-fluorescence analysis to further corroborate the above-described results showed the nuclear exclusion of Cdc25c and the colocal-ization of Cdc25c and 14-3-3 (Fig. 8C). The coimmunoprecipita-tion of Cdc25c and 14-3-3 from EBNA3C-expressing nocodazole-treated cells also supports the above-mentioned finding (Fig. 8B). Anti-14-3-3 antibodies immunoprecipitated Cdc25c from EBNA3C-expressing cells treated with nocodazole but showed no association when cells were treated with nocodazole in the absence of EBNA3C (Fig. 8B, lanes 3 and 6). In addi-tion, a close look at the immunofluorescence results supported
FIG. 8. Cdc25c is translocated predominantly to the cytoplasm in the presence of EBNA3C. (A) Western blots for the detection of Cdc25c in BJAB control cells and EBNA3C (E3C)-transfected BJAB cells following exposure to nocodazole and hydroxyurea (HU) are shown. Stably transfected cells were treated with nocodazole for 24 h and harvested, and total cell lysates were prepared, fractionated on SDS-PAGE gels, and then transferred onto membranes. The membranes were probed to detect Cdc25c and phospho-Ser216 Cdc25c, EBNA3C, and-actin as a loading control. Numbers at the left of the blots are molecular size markers. Relative density levels are shown in the bar diagram below.⫹, present;⫺, absent; hyperphospho Cdc25c, hyperphosphorylated Cdc25c; Cdc25(Ser216), Ser216-phosphorylated Cdc25c. (B) Coimmunoprecipitation of 14-3-3 and Cdc25c from EBNA3C-expressing cells. BJAB cells were transfected with the pA3MEBNA3C construct and treated with nocodazole for 24 h. Coimmunoprecipitation from cell lysates was performed by using anti-14-3-3 antibodies. The complexes were separated by electrophore-sis, transferred onto membranes, and then probed with specific antibodies to detect Cdc25c (␣-Cdc25c) and 14-3-3 (␣-14 3 3). Cdc25c was found to immunoprecipitate with 14-3-3 from nocodazole-treated EBNA3C-expressing cells but not from the control cells. IP, immunoprecipitate. (C) Immunofluorescence analysis of Cdc25c and 14-3-3 from U20S cells with or without nocodazole (Noco) treatment and also from EBNA3C-expressing nocodazole-treated U20S cells shows the predominant localization of Cdc25c in the presence of EBNA3C when U20S cells were treated with nocodazole. DAPI, 4⬘,6⬘-diamidino-2-phenylindole.
VOL. 81, 2007 EBNA3C RELEASES G2/M ARREST 6727
on November 8, 2019 by guest
http://jvi.asm.org/
the exclusion of Cdc25c from the nucleus in the presence of EBNA3C (Fig. 8C, lower panel). These results strongly suggest that EBNA3C can release the nocodazole-induced G2/M phase arrest of cells. This release occurs through the interac-tion of EBNA3C with Chk2, which results in the inhibiinterac-tion of the phosphorylation of the cellular phosphatase at Ser216 and the progression of the cell cycle.
DISCUSSION
Transformation with EBV requires the coordinate and timely expression of a number of latent genes that function in the establishment and maintenance of the immortalized state. The function of these individual viral genes, including the EBNA3C gene, required during these events has been the focus of numerous years of research. However, the func-tional role of EBNA3C in EBV-induced B-cell transforma-tion has yet to be fully understood. Previous studies have demonstrated that EBNA3C functions as a transcriptional regulator (6, 40) and can bind and modulate the function of the cellular DNA binding protein RBP-J, a known repres-sor of transcription (22). EBNA3C also activates the tran-scription of a number of cellular genes and is essential for the transformation of primary B lymphocytes with EBV in vitro (35).
To enable EBV to drive resting B cells to progress into proliferating lymphoblasts, it would be necessary to specif-ically express EBV proteins capable of manipulating the host cell cycle machinery. The initial G0/G1 transition is thought to be controlled by the expression of EBNA2 and EBNA-LP through their ability to upregulate cyclin D2 (32). Progression into S phase is preceded by the expression of LMP1 and LMP2 and EBNA3A, ENBA3B, and EBNA3C. Earlier studies have suggested that the expression of LMP1 can lead to cytostatic effects, impose a block on cells in the G2/M phase of the cell cycle, and lead to reduced cell growth (11, 26). EBV is reported to be able to release the G2/M checkpoint block and induce cell cycle division which is divorced from cytokinesis (26). The expression by EBV of specific proteins capable of overcoming the effects of LMP1 would be necessary to produce the immortalized state of the cells. The results presented here suggest that overcoming the effects of LMP1 is likely to be one of the functions of EBNA3C in its ability to bind and inactivate Chk2, thereby inactivating the checkpoint response and so preventing the accumulation of cells in G2/M phase.
An analysis of the effect of nocodazole on LCLs demon-strated that these cells do not arrest at the G2/M cell cycle checkpoint but instead can induce cell death, a response similar to that previously seen in tumor and transformed cell lines that have a dysfunctional checkpoint (27). Nocodazole induces G2/M arrest in cells that have a functional point response, whereas in cells that have a defective check-point, no arrest but some cell death is seen. This finding suggests that EBV-positive LCLs may have a dysfunctional G2/M checkpoint, which was previously shown by Wade and Allday (38). It was also suggested that one of the EBV-encoded proteins is likely to be responsible for this effect. The present work described in this report revealed that EBNA3C can contribute to and may be a major player in the
release from the G2/M checkpoint arrest imposed in re-sponse to treatment with nocodazole. This suggestion is strongly supported by the results of the biochemical analysis of the kinase Cdc2, which showed the recovery of the levels of active Tyr15-phosphorylated cyclin B1-Cdc2 in the pres-ence of EBNA3C.
Nocodazole disrupts the chromatin structure and can ar-rest cells in G2/M. This checkpoint response pathway is targeted by EBNA3C, as shown by this study. We have shown that the G2/M checkpoint response to nocodazole is sensitive to inhibition by caffeine, implicating the role of ATM/ATR, and more specifically, ATR. One of the check-point kinases downstream of ATM/ATR is Chk2, and our real-time PCR data suggested that EBNA3C can downregu-late Chk2 expression. It is also known that Chk2 is activated in response to other agents that initiate G2/M phase arrests, such as the plant isoflavone genistein (8), and the G2 /M-phase DNA damage checkpoint is defective in Chk2⫺/⫺ embryonic stem cells (13), supporting a role for Chk2 in the G2 checkpoint response. Our studies demonstrated the in-volvement of the ATR-dependent pathway by using an ATR-kd inducible cell line. Thus, EBNA3C is capable of bypassing the nocodazole-induced G2/M block by a mecha-nism which is likely not to be directly linked to the effect of nocodazole on the microtubule disruption.
The observed physical interaction between EBNA3C and Chk2 in EBNA3C-expressing BJAB cells suggests that EBNA3C can disrupt the G2checkpoint response by directly blocking normal Chk2 function. This idea is supported by the results of the studies which showed that the downregu-lation of Chk2 by siRNA diminished the ability of EBNA3C to mediate the release of the nocodazole-induced G2/M arrest and that this disruption of the G2/M checkpoint re-sponse was an immediate effect of EBNA3C expression rather than an adaptive response to longer-term expression. EBNA3C and Chk2 are both located in the nucleus (4). We have demonstrated that EBNA3C binds directly to the Ser-rich domain within the amino-terminal domain of Chk2. The functional relevance of this specific domain is yet to be elucidated, but it is likely that this domain may be altered or regulated by EBNA3C in EBV-positive cells. EBNA3C binding to Chk2 may result in destabilization, increasing the turnover of Chk2, similar to the effect of the human papil-lomavirus 18 E6 protein that directly binds and destabilizes p53, thereby mimicking the role of MDM2 in regulating p53 levels and function (37). Some recent studies in our lab have shown a role for EBNA3C in regulating cyclins, including cyclin A, as well as the inhibitor of cyclin-Cdk2, p27, and the retinoblastoma protein (18). It would be interesting to note if Chk2 is another potential target for cell cycle regulation by EBNA3C utilizing the proteosome degradation pathway. Alternatively, the mechanism of action may be similar to that reported for the human papillomavirus 18 E7 protein that binds to the pocket region of the Rb-related proteins, thereby blocking their ability to sequester the E2F family of proteins, although E7 binding has also been reported to destabilize Rb-related proteins (12). Either of these two mechanisms may also account for the lower level of Chk2 protein observed in the BJAB cell lines expressing the EBNA3C protein. While EBNA3C has some level of
on November 8, 2019 by guest
http://jvi.asm.org/
scriptional effects, the specific contribution of these effects of EBNA3C to Chk2 activity and expression remains to be elucidated.
This study has identified a new role for EBNA3C in its ability to disrupt the DNA damage and replication G2/M checkpoint. These findings are in agreement with previous observations which showed that EBNA3C can disrupt mul-tiple cell cycle checkpoints (26) and that EBV can suppress a G2/M checkpoint activated by genotoxic agents (38). How-ever, there were no direct mechanisms associated with the role of EBNA3C in this process. This study has now identified some initial players involved in the release of the G2/M block medi-ated by EBNA3C, and we put forth a basic mechanism for this activity (Fig. 9). EBNA3C interacts directly with Chk2, an effector of the ATM/ATR signaling pathway. This interaction leads to phosphorylation predominantly at Ser216 on the cel-lular phosphatase Cdc25c, which is then sequestered in the cytoplasm by 14-3-3 and so allows for cylin B-Cdc2 activation and the bypassing of the G2/M checkpoint.
ACKNOWLEDGMENTS
We thank Tanya Paull (University of Texas, Austin) and Paul Nghiem (Massachusetts General Hospital, Cambridge) for providing the plasmid for the GST-Chk2 constructs and the ATR-wt and ATR-kd cell lines, respectively.
This work was supported by National Institutes of Health grants NCI CA72150-07, NCI CA91792-01, NIDCR DE14136-01, and NCI CA108461 to E.S.R. E.S.R. is a scholar of the Leukemia and Lym-phoma Society of America.
REFERENCES
1.Borgne, A., and L. Meijer.1996. Sequential dephosphorylation of p34(cdc2) on Thr-14 and Tyr-15 at the prophase/metaphase transition. J. Biol. Chem.
271:27847–27854.
2.Bulavin, D. V., Y. Higashimoto, Z. N. Demidenko, S. Meek, P. Graves, C. Phillips, H. Zhao, S. A. Moody, E. Appella, H. Piwnica-Worms, and A. J. Fornace, Jr.2003. Dual phosphorylation controls Cdc25 phosphatases and mitotic entry. Nat. Cell Biol.5:545–551.
3.Cannell, E. J., P. J. Farrell, and A. J. Sinclair.1996. Epstein-Barr virus exploits the normal cell pathway to regulate Rb activity during the im-mortalisation of primary B-cells. Oncogene13:1413–1421.
4.Chehab, N. H., A. Malikzay, M. Appel, and T. D. Halazonetis.2000. Chk2/ hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev.14:278–288.
5.Choudhuri, T., S. Pal, T. Das, and G. Sa.2005. Curcumin selectively induces apoptosis in deregulated cyclin D1-expressed cells at G2 phase of cell cycle in a p53-dependent manner. J. Biol. Chem.280:20059–20068.
6.Choudhuri, T., S. C. Verma, K. Lan, and E. S. Robertson.2006. Expression of alpha V integrin is modulated by Epstein-Barr virus nuclear antigen 3C and the metastasis suppressor Nm23-H1 through interaction with the GATA-1 and Sp1 transcription factors. Virology351:58–72.
7.Cohen, J. I., F. Wang, J. Mannick, and E. Kieff.1989. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA86:9558–9562.
[image:12.585.130.457.70.368.2]8.Darbon, J. M., M. Penary, N. Escalas, F. Casagrande, F. Goubin-Gramatica, C. Baudouin, and B. Ducommun.2000. Distinct Chk2 activation pathways are triggered by genistein and DNA-damaging agents in human melanoma cells. J. Biol. Chem.275:15363–15369.
FIG. 9. A hypothetical model shows the putative mechanisms for the bypassing of the nocodazole-induced G2arrest by EBNA3C (E3C).
Nocodazole treatment reduces the level of phosphorylated Cdc2. The viral nuclear antigen EBNA3C binds directly to Chk2, which results in the phosphorylation of Cdc25c at Ser216. Cdc25c, which is phosphorylated predominantly at Ser216, is sequestered in the cytoplasm and is now unable to regulate the phosphorylation of nuclear Cdc2. The resulting effect leads to the activation of cyclin B-Cdc2 and progression through the G2/M
stage, releasing the block imposed by nocodazole. The overall effect is the release of the nocodazole-induced G2arrest.
VOL. 81, 2007 EBNA3C RELEASES G2/M ARREST 6729
on November 8, 2019 by guest
http://jvi.asm.org/
9.Elledge, S. J.1996. Cell cycle checkpoints: preventing an identity crisis. Science274:1664–1672.
10.Enoch, T., and P. Nurse.1990. Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell60:665–673. 11.Floettmann, J. E., K. Ward, A. B. Rickinson, and M. Rowe.1996. Cytostatic effect of Epstein-Barr virus latent membrane protein-1 analyzed using tet-racycline-regulated expression in B cell lines. Virology223:29–40. 12.Goodwin, E. C., and D. DiMaio.2000. Repression of human papillomavirus
oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc. Natl. Acad. Sci. USA97:
12513–12518.
13.Hirao, A., Y. Y. Kong, S. Matsuoka, A. Wakeham, J. Ruland, H. Yoshida, D. Liu, S. J. Elledge, and T. W. Mak.2000. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science287:1824–1827.
14.Kaye, K. M., K. M. Izumi, and E. Kieff.1993. Epstein-Barr virus latent membrane protein 1 is essential for B-lymphocyte growth transformation. Proc. Natl. Acad. Sci. USA90:9150–9154.
15.Kieff, E.1996. Epstein-Barr virus and its replication, p. 2343–2395.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, 3rd ed., vol. 2. Lippincott-Raven, Philadelphia, PA.
16.Knight, J. S., M. A. Cotter II, and E. S. Robertson.2001. The latency-associated nuclear antigen of Kaposi’s sarcoma-latency-associated herpesvirus trans-activates the telomerase reverse transcriptase promoter. J. Biol. Chem.276:
22971–22978.
17.Knight, J. S., K. Lan, C. Subramanian, and E. S. Robertson.2003. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and asso-ciates with the corepressors mSin3A and NCoR in human B-cell lines. J. Virol.77:4261–4272.
18.Knight, J. S., and E. S. Robertson.2004. Epstein-Barr virus nuclear antigen 3C regulates cyclin A/p27 complexes and enhances cyclin A-dependent ki-nase activity. J. Virol.78:1981–1991.
19.Krauer, K. G., A. Burgess, M. Buck, J. Flanagan, T. B. Sculley, and B. Gabrielli.2004. The EBNA-3 gene family proteins disrupt the G2/M check-point. Oncogene23:1342–1353.
20.Kumagai, A., and W. G. Dunphy.1991. The cdc25 protein controls tyrosine dephosphorylation of the cdc2 protein in a cell-free system. Cell64:903–914. 21.Lin, J., E. Johannsen, E. Robertson, and E. Kieff.2002. Epstein-Barr virus nuclear antigen 3C putative repression domain mediates coactivation of the LMP1 promoter with EBNA-2. J. Virol.76:232–242.
22.Marshall, D., and C. Sample.1995. Epstein-Barr virus nuclear antigen 3C is a transcriptional regulator. J. Virol.69:3624–3630.
23.Morgan, D. O.1995. Principles of CDK regulation. Nature374:131–134. 24.Nghiem, P., P. K. Park, Y. Kim, C. Vaziri, and S. L. Schreiber.2001. ATR
inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal pre-mature chromatin condensation. Proc. Natl. Acad. Sci. USA98:9092–9097. 25.Parker, G. A., T. Crook, M. Bain, E. A. Sara, P. J. Farrell, and M. J. Allday.
1996. Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7. Oncogene13:2541–2549.
26.Parker, G. A., R. Touitou, and M. J. Allday. 2000. Epstein-Barr virus
EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis. Oncogene19:700–709.
27.Qiu, L., A. Burgess, D. P. Fairlie, H. Leonard, P. G. Parsons, and B. G. Gabrielli.2000. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell11:2069–2083. 28.Radkov, S. A., R. Touitou, A. Brehm, M. Rowe, M. West, T. Kouzarides, and
M. J. Allday.1999. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol.73:5688–5697. 29.Rickinson, A. B., and E. Kieff.1996. Epstein-Barr virus. Lippincott-Raven,
Philadelphia, PA.
30.Robertson, E. S., J. Lin, and E. Kieff.1996. The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ. J. Virol.70:3068–3074.
31.Sinclair, A. J., M. Fenton, and S. Delikat. 1998. Interactions between Epstein-Barr virus and the cell cycle control machinery. Histol. His-topathol.13:461–467.
32.Sinclair, A. J., I. Palmero, G. Peters, and P. J. Farrell.1994. EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus. EMBO J.13:3321–3328. 33.Subramanian, C., M. A. Cotter II, and E. S. Robertson.2001. Epstein-Barr virus nuclear protein EBNA-3C interacts with the human metastatic sup-pressor Nm23-H1: a molecular link to cancer metastasis. Nat. Med.7:350– 355.
34.Subramanian, C., S. Hasan, M. Rowe, M. Hottiger, R. Orre, and E. S. Robertson.2002. Epstein-Barr virus nuclear antigen 3C and prothymosin alpha interact with the p300 transcriptional coactivator at the CH1 and CH3/HAT domains and cooperate in regulation of transcription and histone acetylation. J. Virol.76:4699–4708.
35.Tomkinson, B., E. Robertson, and E. Kieff.1993. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol.67:2014–2025.
36.Verma, S. C., K. Lan, T. Choudhuri, and E. S. Robertson.2006. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen modulates K1 expression through itscis-acting elements within the terminal repeats. J. Virol.80:3445–3458.
37.Villa, L. L.1997. Human papillomaviruses and cervical cancer. Adv. Cancer Res.71:321–341.
38.Wade, M., and M. J. Allday.2000. Epstein-Barr virus suppresses a G(2)/M checkpoint activated by genotoxins. Mol. Cell. Biol.20:1344–1360. 39.Warrener, R., H. Beamish, A. Burgess, N. J. Waterhouse, N. Giles, D.
Fairlie, and B. Gabrielli.2003. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB J.17:1550–1552.
40.Zhao, B., D. R. Marshall, and C. E. Sample.1996. A conserved domain of the Epstein-Barr virus nuclear antigens 3A and 3C binds to a discrete domain of J. J. Virol.70:4228–4236.
41.Zhao, B., and C. E. Sample.2000. Epstein-Barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an Spi-1/Spi-B binding site. J. Virol.74:5151–5160.