Binding Specificity in Genogroup I Noroviruses
Sreejesh Shanker,aRita Czako,bBanumathi Sankaran,dRobert L. Atmar,b,cMary K. Estes,b,cB. V. Venkataram Prasada,b
Verna Marrs McLean Department of Biochemistry and Molecular Biology,aDepartment of Molecular Virology and Microbiology,band Department of Medicine,cBaylor College of Medicine, Houston, Texas, USA; Berkeley Center for Structural Biology, Lawrence Berkeley National Laboratory, Berkeley, California, USAd
ABSTRACT
Human noroviruses (NoVs) cause acute epidemic gastroenteritis. Susceptibility to the majority of NoV infections is determined by genetically controlled secretor-dependent expression of histo-blood group antigens (HBGAs), which are also critical for NoV attachment to host cells. Human NoVs are classified into two major genogroups (genogroup I [GI] and GII), with each geno-group further divided into several genotypes. GII NoVs are more prevalent and exhibit periodic emergence of new variants, sug-gested to be driven by altered HBGA binding specificities and antigenic drift. Recent epidemiological studies show increased ac-tivity among GI NoVs, with some members showing the ability to bind nonsecretor HBGAs. NoVs bind HBGAs through the protruding (P) domain of the major capsid protein VP1. GI NoVs, similar to GII, exhibit significant sequence variations in the P domain; it is unclear how these variations affect HBGA binding specificities. To understand the determinants of possible strain-specific HBGA binding among GI NoVs, we determined the structure of the P domain of a GI.7 clinical isolate and compared it to the previously determined P domain structures of GI.1 and GI.2 strains. Our crystallographic studies revealed significant struc-tural differences, particularly in the loop regions of the GI.7 P domain, altering its surface topography and electrostatic land-scape and potentially indicating antigenic variation. The GI.7 strain bound to H- and A-type, Lewis secretor, and Lewis nonse-cretor families of HBGAs, allowing us to further elucidate the structural determinants of nonsenonse-cretor HBGA binding among GI NoVs and to infer several contrasting and generalizable features of HBGA binding in the GI NoVs.
IMPORTANCE
Human noroviruses (NoVs) cause acute epidemic gastroenteritis. Recent epidemiological studies have shown increased preva-lence of genogroup I (GI) NoVs. Although secretor-positive status is strongly correlated with NoV infection, cases of NoV infec-tion associated with secretor-negative individuals are reported. Biochemical studies have shown that GI NoVs exhibit genotype-dependent binding to nonsecretor histo-blood group antigens (HBGAs). From our crystallographic studies of a GI.7 NoV, in comparison with previous studies on GI.1 and GI.2 NoVs, we show that genotypic differences translate to extensive structural changes in the loop regions that significantly alter the surface topography and electrostatic landscape of the P domain; these fea-tures may be indicative of antigenic variations contributing to serotypic differentiation in GI NoVs and also differential modula-tion of the HBGA binding characteristics. A significant finding is that the threshold length and the structure of one of the loops are critical determinants in the binding of GI NoVs to nonsecretor HBGAs.
H
uman noroviruses (NoVs) are the leading cause of nonbacte-rial acute gastroenteritis worldwide (1–3). They belong to the familyCaliciviridae and are divided into 6 genogroups (geno-group I [GI] to GVI) (4). Each genogroup is phylogenetically fur-ther divided into several genotypes. GI and GII contain the epide-miologically predominant human NoVs (5–7). Norwalk virus (NV), the prototype human NoV, belongs to GI, genotype 1 (GI.1) (8). GI NoVs, apart from NV, are not as well studied and understood as their GII counterparts due to their lower preva-lence. Recent epidemiological studies, however, show an increase in the prevalence of GI outbreaks worldwide, with different geno-types such as GI.4, GI.6, GI.3, and GI.7 predominating in different geographical regions (9–13). A study from Alberta, Canada, showed that GI NoVs were responsible for⬃37% of all NoV in-fections from late 2012 to early 2013, with the majority being caused by GI.6 and GI.7. This was an⬃15-fold-higher prevalence than in 2010 (12). Studies from Brazil have also reported up to ⬃48% of NoV infections as being due to GI NoVs (13,14). In the United States, the Centers for Disease Control and Prevention (CDC) also reported increased prevalence of GI viruses, with a 6-fold increase in GI.6 strains compared to the prevalence in 2010(10), whereas the GI.7 genotype ranked 4th within all GI out-breaks (15). In addition, GI NoVs have recently been reported to be a common cause of traveler’s diarrhea among U.S. adults trav-eling to Mexico (16).
Several studies have shown that susceptibility to human NoVs is determined by genetically controlled expression of histo-blood group antigens (HBGAs), which are also critical for NoV attach-ment to host cells (17,18). HBGAs are oligosaccharides that are classified into different types (types 1 to 4) based on their carbo-hydrate compositions and the glycosidic linkages of their
precur-Received21 January 2014 Accepted11 March 2014
Published ahead of print19 March 2014
Editor:D. S. Lyles
Address correspondence to B. V. Venkataram Prasad, vprasad@bcm.edu.
Supplemental material for this article may be found athttp://dx.doi.org/10.1128 /JVI.00201-14.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.00201-14
on November 7, 2019 by guest
http://jvi.asm.org/
sor disaccharides (19,20). Both the terminal carbohydrate moi-eties and the internal precursor glycosidic linkage are thought to contribute as recognition sites for NoVs (21,22). HBGAs localize to the surface of epithelial cells and in mucosal secretions of secre-tor-positive individuals. Secresecre-tor-positive status is a susceptibility factor for the prototype NV (23,24). Nonsecretors were initially thought to be resistant to human NoVs, but recent epidemiolog-ical and structural studies with other GI genotypes have chal-lenged this paradigm (11,13,25). The secretor status of an indi-vidual is determined by expression of a functional fucosyl transferase 2 (FUT2) enzyme that adds an␣-fucose (SeFuc) to the -galactose (-Gal) of the disaccharide precursor to form the se-cretor epitope, or H-type HBGA. The H-type HBGA can be fur-ther modified by enzyme A or B by addingN-acetylgalactosamine (GalNAc) or Gal to the precursor-Gal to form A- or B-type HBGA, respectively (20). Similarly, the Lewis (Le) status is deter-mined by the activity of the fucosyl transferase 3 (FUT3) enzyme, which adds an ␣-fucose (LeFuc) to the N-acetylglucosamine (GlcNAc) of the disaccharide precursor to form the Lewis epitope. The sequential addition of carbohydrate moieties by FUT2 and FUT3 along with enzymes A and B gives rise to the secretor and nonsecretor Lewis and ABH families of HBGAs (26).
The HBGA binding site in the NoVs is located at the distal end of the protruding (P) domain of the major capsid protein VP1. Crystallographic studies of the P domain in complex with HBGAs show that the HBGA binding sites in GI and GII are structurally distinct (27,28). In GII NoVs, accumulation of sequence varia-tions and associated structural changes on the surface of the P domain result in altered HBGA binding specificities and antigenic drift. In the GII.4 genotype, this has been associated with the pe-riodic emergence of new variants (18,29–31). GI NoVs also ex-hibit extensive sequence changes in the vicinity of the HBGA bind-ing site, which could potentially alter HBGA bindbind-ing specificities and antigenicity and thus contribute to GI NoV evolution. How-ever, unlike the case with GII NoVs, the effect of sequence varia-tions in GI has not been well characterized. Recent studies have shown that in contrast to GI.1, a GI.2 strain has the ability to bind nonsecretor Lewis HBGAs (25). A longer variable loop region to-gether with a glutamine residue (Q389) is proposed to be critical for such differential HBGA binding, as the Q389N mutation abol-ished nonsecretor HBGA binding. This raised questions on whether other GI members with distinct sequence variations at these regions can bind nonsecretor HBGAs and, if so, what deter-minants modulate glycan-binding preferences among GI NoVs and how genotypic differences in GI alter the P domain structure and influence HBGA binding specificities. To answer these ques-tions, we studied a GI.7 NoV clinical isolate, as it showed signifi-cant sequence changes in the P domain compared to GI.1 and GI.2, including a Pro (P388) at the position equivalent to Q389 in GI.2, and carried outin vitrobinding studies with GI.7 virus-like particles (VLPs) together with systematic crystallographic analysis of the P domain by itself and in complex with various secretor and nonsecretor HBGAs.
Our enzyme-linked immunosorbent assay (ELISA) binding studies clearly showed that GI.7 VLPs bind to nonsecretor HBGAs in addition to secretor HBGAs, despite the Pro residue substitu-tion. Our crystallographic studies showed that sequence varia-tions result in extensive structural differences in the loop regions and that these differences contribute not only to modulating HBGA binding specificities but also possibly to antigenic
varia-tions among the GI NoVs. Our studies strongly suggest that it is not the residue composition corresponding to position 389 but the threshold length and structure of the P-loop that are the crit-ical determinants for nonsecretor HBGA binding in GI NoVs.
MATERIALS AND METHODS
Preparation of recombinant VLPs. The cDNA segments ORF2 and ORF3, coding for major and minor capsid proteins VP1 and VP2 of the respective NoV strains, were cloned into the pVL1392 baculovirus trans-fer vector (Invitrogen). The proteins were overexpressed by infection of Hi-5 cells with recombinant baculovirus, and self-assembled virus-like particles (VLPs) were purified by sequential ultracentrifugation first on a sucrose cushion and then in a CsCl gradient. Following dialysis into phos-phate buffer, protein expression and the integrity of resultant VLPs were assessed by Western blotting and electron microscopy. VLPs representa-tive of both GI.1 (GenBank accession numberNC_001959) and GI.7 (GenBank accession numberJN005886) genotypes were produced. VLPs with the point mutation W375A in the glycan binding site were produced using a previously described recombinant baculovirus construct (28).
ELISA.All reagents were diluted in 0.1 M sodium phosphate buffer, pH 6.1, with 0.25% fatty acid-free bovine serum albumin (Sigma-Al-drich), and all assay volumes were 100 l. Neutravidin-coated, pre-blocked, 96-well plates (Pierce Thermo Fisher Scientific) were coated with the following synthetic biotinylated multivalent glycans for 2 h at 25°C: H type 1-polyacrylamide (PAA)-biotin, A antigen-PAA-biotin, or Lea
-PAA-biotin (Glycotech). Negative-control wells were coated with -PAA-biotin hydra-zide reagent (Pierce Thermo Fisher Scientific). VLPs representing GI.1, W375A mutant GI.1, and GI.7 (10g/ml) were added to the plate and allowed to bind for 2 h at 4°C. The plates were then washed with assay buffer 4 times between each incubation. Bound VLPs were detected using NV-specific polyclonal rabbit serum for 1 h at 4°C, followed by horserad-ish peroxidase-conjugated goat anti-rabbit antibody (Sigma-Aldrich) for 1 h at 4°C. The color was developed by adding tetramethylbenzidine per-oxidase liquid substrate (Pierce Thermo Fisher Scientific). The reaction was stopped after 10 min at 25°C by adding 1 M phosphoric acid. Absor-bance at 450 nm was measured using a SpectraMax M5 plate reader (Mo-lecular Devices).
Purification and crystallization of GI.7 P domain.The P domain (amino acids [aa] 226 to 526) construct of a GI.7 strain (TCH-060) was cloned into expression vector pMal-C2E (New England BioLabs) and overexpressed inEscherichia coli, followed by protein purification using chromatography techniques as described previously (30). The purified P domain was then concentrated to⬃10 mg/ml and stored in 20 mM Tris-HCl buffer (pH 8.0) containing 250 mM NaCl, 1 mM dithiothreitol (DTT), and 5 mM MgCl2for crystallization trials. Crystallization
condi-tions were screened with commercially available screens using the nano-liter handling system Mosquito (TTP LabTech) at 20°C by the hanging-drop vapor diffusion method. The crystallization hanging-drops were examined using a Rock Imager (Formulatrix). Unliganded protein (⬃10 mg/ml) crystallized under the condition that had 0.2 M sodium formate, 0.1 M bis-tris propane (pH 6.5), and 20% (wt/vol) polyethylene glycol 3350 (PEG 3350). Crystals measuring 0.1 to 0.2 mm were obtained in 2 to 3 weeks. Crystals of P domain that bound to each of the ligands, H-type 2 trisaccharide, A-type 2 tetrasaccharide, A-type 3 tetrasaccharide, Ley
tet-rasaccharide, Lextetrasaccharide, and Leatetrasaccharide (Dextra
Labo-ratories) were obtained by soaking the unliganded P domain crystals for 30 min in a reconstituted crystallization reservoir solution containing 80 mM ligand and 20% glycerol as a cryoprotectant, followed by flash freez-ing in liquid nitrogen.
Diffraction, data collection, and structure determination.X-ray dif-fraction data for the unliganded (native) and liganded P domain crystal data were collected on the 5.0.1 beamline at Advanced Light Source (ALS), Lawrence Berkeley National Laboratory. Diffraction data were processed using either HKL2000 (32), D*TREK (33), or IMOSFLM (34). The space group in each case was confirmed using POINTLESS (35). An initial elec-Determinants of HBGA Binding among GI Noroviruses
on November 7, 2019 by guest
http://jvi.asm.org/
tron density map was obtained by molecular replacement (MR) using the previously published GI.1 P domain structure (PDB identifier [ID] 2ZL5) as the phasing model using PHASER (36) as implemented in the CCP4 suite (35). This was followed byab initioautomated model building and solvent addition using ARP/Warp (37) to reduce model bias. Further model building was carried out using COOT (38) software, followed by iterative cycles of refinement and model building using Refmac (39) or PHENIX (40). During the refinement, both translation, liberation, and screw (TLS) parameters and noncrystallographic symmetry (NCS) con-straints were included. The liganded P domain structures were deter-mined similarly except that the native GI.7 P domain structure was used as the initial phasing model. The oligosaccharide moieties of the HBGAs were generated using the SWEET2 package of the Glycosciences.de server (http://www.glycosciences.de), modeled into the electron density using COOT and validated by computing simulated annealing omit maps using PHENIX. Following each cycle of refinement, the model was corrected based on theFobs-Fcalcdifference maps. Stereochemistry of the structures
was confirmed using COOT modules and PROCHECK (41). The stereo-chemistry of the oligosaccharides, including the allowed conformational angles, was checked using CARP package in the Glycosciences.de server (http://www.glycosciences.de). Data collection and refinement statistics are provided inTable 1. Electrostatic potential surface was calculated us-ing the PyMOL (http://www.pymol.org) software package, which was also used for generating the final figures. Superposition of the various struc-tures was carried out using either COOT or PyMoL. Ligand interactions were analyzed using COOT and LIGPLOT (42) with donor-to-acceptor distances of between 2.6 Å and 3.3 Å for hydrogen bonding interactions and carbon-carbon distances of between 3.4 Å and 4.5 Å for hydrophobic interactions.
PDB accession codes.The coordinates and structure factors for the protein structures solved in this study have been deposited in the Protein Data Bank (PDB) under the accession codes 4P12 for the unliganded GI.7 P domain structure and P1V (H-type 2), 4P26 (A-type 2), 4P25 (Ley),
4P2N (Lex), and 4P3I (Lea) for the P domain-ligand complexes.
RESULTS
Sequence variations in VP1 of GI NoVs.The TCH-060 strain of
NoV used in our studies was isolated from an 11-year-old boy in 2003 at the Texas Children’s Hospital, Houston, TX (43). Phylo-genetic analysis of the VP1 amino acid sequence (GenBank acces-sion numberAEQ77282.1) confirmed that it belonged to the GI.7 genotype and showed that it is distant from the structurally char-acterized GI.1 and GI.2 strains (data not shown). To examine the extent and nature of sequence changes in VP1 among GI NoVs particularly in the vicinity of HBGA binding sites, as determined from the crystallographic analysis of HBGA-bound GI.1 and GI.2 P domain structures, we carried out sequence alignment of repre-sentative GI VP1 sequences, including that from our GI.7 strain
(Fig. 1). This analysis showed that the majority of the significant
[image:3.585.42.548.76.401.2]sequence alterations occur in the regions that correspond to the outermost loop regions of the P domain. These loops are labeled A-, B-, P-, and S-loops, following the same convention as in the previous structural study on a GI.2 P domain (25). In addition to the changes in these loops, our sequence analysis indicated con-siderable variation in the region neighboring Q389, which we have labeled as the T-loop. This Q389 residue, which is shown to be
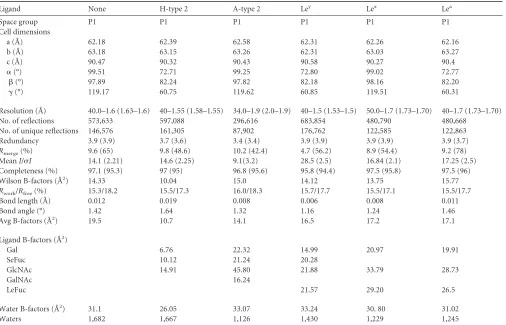
TABLE 1Data processing and refinement statistics of the GI.7 P domaina
Ligand None H-type 2 A-type 2 Ley Lex Lea
Space group P1 P1 P1 P1 P1 P1
Cell dimensions
a (Å) 62.18 62.39 62.58 62.31 62.26 62.16
b (Å) 63.18 63.15 63.26 62.31 63.03 63.27
c (Å) 90.47 90.32 90.43 90.58 90.27 90.4
␣(°) 99.51 72.71 99.25 72.80 99.02 72.77
(°) 97.89 82.24 97.82 82.18 98.16 82.20
␥(°) 119.17 60.75 119.62 60.85 119.51 60.31
Resolution (Å) 40.0–1.6 (1.63–1.6) 40–1.55 (1.58–1.55) 34.0–1.9 (2.0–1.9) 40–1.5 (1.53–1.5) 50.0–1.7 (1.73–1.70) 40–1.7 (1.73–1.70) No. of reflections 573,633 597,088 296,616 683,854 480,790 480,668
No. of unique reflections 146,576 161,305 87,902 176,762 122,585 122,863 Redundancy 3.9 (3.9) 3.7 (3.6) 3.4 (3.4) 3.9 (3.9) 3.9 (3.9) 3.9 (3.7) Rmerge(%) 9.6 (65) 9.8 (48.6) 10.2 (42.4) 4.7 (56.2) 8.9 (54.4) 9.2 (78) MeanI/I 14.1 (2.21) 14.6 (2.25) 9.1(3.2) 28.5 (2.5) 16.84 (2.1) 17.25 (2.5) Completeness (%) 97.1 (95.3) 97 (95) 96.8 (95.6) 95.8 (94.4) 97.5 (95.8) 97.5 (96) Wilson B-factors (Å2) 14.33 10.04 15.0 14.12 13.75 15.77 Rwork/Rfree(%) 15.3/18.2 15.5/17.3 16.0/18.3 15.7/17.7 15.5/17.1 15.5/17.7 Bond length (Å) 0.012 0.019 0.008 0.006 0.008 0.011
Bond angle (°) 1.42 1.64 1.32 1.16 1.24 1.46
Avg B-factors (Å2) 19.5 10.7 14.1 16.5 17.2 17.1
Ligand B-factors (Å2)
Gal 6.76 22.32 14.99 20.97 19.91
SeFuc 10.12 21.24 20.28
GlcNAc 14.91 45.80 21.88 33.79 28.73
GalNAc 16.24
LeFuc 21.57 29.20 26.5
Water B-factors (Å2) 31.1 26.05 33.07 33.24 30. 80 31.02
Waters 1,682 1,667 1,126 1,430 1,229 1,245
a
Values in parentheses are for the highest-resolution shell. The wavelength was 0.97935 ˚ for all. Waters, number of water molecules.
on November 7, 2019 by guest
http://jvi.asm.org/
critical for nonsecretor HBGA binding in the GI.2 strain, is not conserved in all the GI strains. The GI.1 and GI.7 strains have a Pro residue at this position (Fig. 1), as does a GI.3 strain (JKPG) asso-ciated with infection in nonsecretors (11), whereas some others have Asn (Fig. 1). Further, the P-loop region, whose longer length has been identified as critical for nonsecretor HBGA binding in GI.2, exhibits considerable variation. Compared to GI.2, this re-gion has a 4-residue deletion in GI.1 and a 2-residue deletion in GI.7.
In vitroanalysis of HBGA binding in GI.7 NoV.To investi-gate how these sequence changes in the P domain may influence HBGA binding in GI.7 NoV, we carried outin vitroELISA binding studies of GI.7 VLPs with various secretor and nonsecretor HBGAs (Fig. 2). Our results show (Fig. 2) that both GI.1 and GI.7 VLPs bound secretor HBGAs, whereas in the W375A mutant VLPs, all HBGA binding interactions were ablated. In contrast to GI.I, the GI.7 VLPs also bind Lea, a nonsecretor HBGA, despite a
Pro residue at the position corresponding to Q389 in GI.2. Known
HBGA binding profiles of various GI NoVs, including GI.7 (pres-ent study), are summarized inTable 2. To understand how the sequence variations in the GI.7 P domain translate to structural changes that result in the observed HBGA binding, we carried out systematic crystallographic analysis of the unliganded GI.7 P do-main, followed by its complex with various HBGAs, including nonsecretor HBGAs.
Unliganded GI.7 P domain structure.The recombinant GI.7
P domain (residues 225 to 526) was cloned and overexpressed inE. coliand purified to homogeneity for crystallization trials. Crystals of the P domain belonging to the P1 space group with four molecules in the asymmetric unit diffracted to⬃1.6 Å. The structure was determined using molecular replacement tech-niques and refined to a finalRfac/Rfreeof 0.15/0.18 (Table 1).
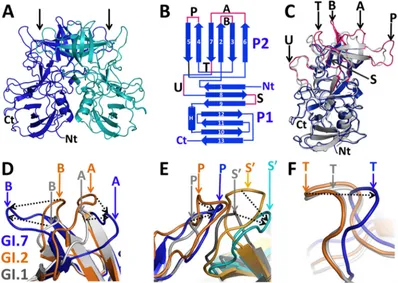
Struc-tural comparison of the four molecules in the asymmetric unit showed that they are very similar to one another, with an average root mean square deviation (RMSD) of⬃0.5 Å. These four mol-ecules in the asymmetric unit form two very similar dimers
FIG 1Amino acid sequence alignment of representative GI variants. Only residues from aa 281 to 446 (GI.7 numbering) in the P domain of VP1 are shown, and they represent primarily the P2 subdomain (aa 281 to 410) and a part of the PI subdomain (aa 411 to 446) labeled and shown as a dashed line. The GI.7 strain (GI.7_TCH_2003) used in this study is shown as the first sequence, and the GI.1 (GI.1_NV) and GI.2 (GI.2_FUV258) strains used to compare and contrast in this study are shown as second and third sequences, respectively. Regions of high sequence variability, shown inside the boxes, are named as A-, B-, P-, S-, T-, and U-loops based on previous GI P domain structures. The residues corresponding to Q389 (circled in black) in the GI.2 FUV 258 strain that was shown to be critical for nonsecretor binding is shown with an arrow. The amino acids participating in HBGA interactions in two or more strains are shown with asterisks, and those unique to GI.7 are shown with pound signs. Every residue is shown by a dot above the sequences, every 5th residue is indicated by a dash, and every 10th residue is numbered according to the GI.7 numbering. The GenBank accession numbers for the various sequences represented here are as follows:AEQ77282.1for GI.7_TCH 2003,NP_056821.2for GI.1_NV,
BAC05516.1for GI.2_FUV258,ACX33982.1for GI.3_JKPG,AAA16285.1for GI.3_DSV,BAB18267.1for GI.4_Chiba,AFH88383.1for GI.6_2010,AFC89659.1for GI.7_2010,ADB54834.1for GI.8_2008,AAN15140.1for GI.8_2008 Boxer, andAEY77023.1for GI.9_2008.
Determinants of HBGA Binding among GI Noroviruses
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.45.543.64.409.2](Fig. 3A). The overall structure of the GI.7 P domain is similar to the P domain structures of other GI NoVs. As in other P domain structures (25,27,44), the GI.7 P domain consists of a well-de-fined P2 subdomain (residues 280 to 412) composed of a six-stranded antiparallel-barrel with elaborate loops connecting the -strands that is inserted between the N and C termini of the P1 subdomain (residues 225 to 279 and 413 to 526) consisting of a twisted antiparallel-sheet and an␣-helix (Fig. 3B).
Structural comparison between the GI P domain structures.
To examine whether the sequence alterations in the GI.7 P domain translate into structural changes, we superimposed one of the sub-units from the GI.7 P dimer with the corresponding subunit of the P dimer of GI.1 (Fig. 3C) and GI.2 strains (data not shown). These structures superimposed with an average RMSD of⬃1.5 Å. Al-though the overall P domain structure is conserved, significant structural differences (⬎2 Å) are observed in the outermost loop regions in the P2 subdomain that include the A-loop (aa 368 to 378), B-loop (aa 295 to 303), P-loop (aa 338 to 350), and T-loop (aa 387 to 395) (indicated in magenta inFig. 3BandC). In addi-tion, noticeable changes are also observed in the S-loop (aa 432 to 442), which is in the P1 subdomain, and in another loop (aa 403 to 412) at the junction of the P1 and P2 subdomains that is named the U-loop.
The most prominent difference is observed in the A- and B-loops. The distal end of the A-loop in the GI.7 P domain deviates by an average of⬃8 Å from the A-loop in the GI.1 and GI.2 P domains and by about⬃7.5 Å from the B-loop (Fig. 3D). This structural comparison also revealed that these two loops, which lie in close proximity with each other at the top of the P2 subdomain, can exist in at least two conformations: a closed conformation as observed in the GI.1 and GI.2 P domain structures and a novel open conformation as observed in the GI.7 P domain structure
(Fig. 3D). While these two loops are separated by⬃6 Å in the GI.1
and GI.2 structures, they are separated by as much as⬃17.5 Å in the GI.7 P domain structure. Such a large distance between these two loops in the GI.7 P domain exposes a considerably larger surface area underneath that can potentially contribute to
differ-ential antigenic presentations. The open conformation in the GI.7 P domain is the result of sequence variations in these loops that lead to longer antiparallel -sheets than in the GI.1 and GI.2 strains.
The P- and S-loops also show considerable differences. In the context of the dimer, the P- and S-loops from the opposing sub-units lie in close proximity (Fig. 3E). In the GI.2 P domain dimer, these two loops, which are longer than in the GI.1 P domain dimer, are involved in the formation of the nonsecretor HBGA binding site. In the GI.7 P domain dimer, these two loops are of intermediate length; while the P-loop lies at an intermediate po-sition compared to GI.1 and GI.2 dimers, the S-loop deviates by ⬃8 Å. Structural alterations in the T-loop (Fig. 3F) are unique to GI.7. The T-loop is shorter in the GI.7 P domain and deviates by ⬃4.5 Å compared to the case with GI.1 and GI.2 strains, bringing this loop closer to the HBGA binding site.
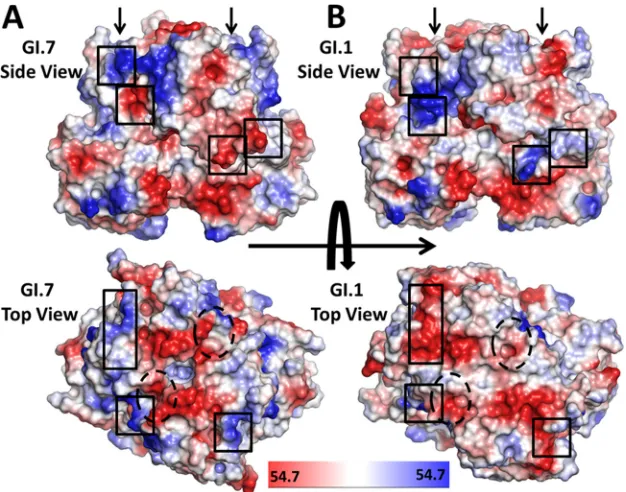
These sequence and structural variations between these GI strains also translate to significant changes in surface topography and differential electrostatic charge distribution. A comparison of the electrostatic potential surfaces computed from the GI.7 and GI.1 P domain structures shows dramatic charge reversals in the surface-exposed regions of the P2 subdomain, and some of these changes are in close proximity to the HBGA binding site (Fig. 4). These electrostatic potential surface differences along with surface topography changes likely indicate antigenic variations among GI NoVs.
HBGA binding in GI.7.To examine how the observed
[image:5.585.43.284.65.209.2]struc-tural changes in the GI.7 P domain affect HBGA binding specific-ities, we crystallized the GI.7 P domain with glycans of ABH secre-tor, Lewis secresecre-tor, and Lewis nonsecretor HBGA families. The HBGAs used were mainly of type 2 precursor backbones, in con-trast to the type 1 precursor HBGAs used in the previous struc-tural studies, so as to understand the strucstruc-tural basis of recogni-tion of different HBGA types (1–3) among GI NoVs. Crystals of the P domain-HBGA complexes were obtained by soaking native crystals of the GI.7 P domain (P1 space group with four molecules in the asymmetric unit) with an excess of each carbohydrate li-gand. Each of these crystals diffracted to a resolution similar to that of the unliganded P domain crystals. Statistics of data collec-tion and structure refinement for each of the complex structures are given inTable 1. In each case, the ligand density was clearly visualized (see Fig. S1 in the supplemental material), and unless mentioned otherwise, all the carbohydrate residues in the ligand could be modeled unambiguously into the density map. The bound ligand was validated further using simulated annealing omit maps.
TABLE 2HBGA binding profiles of GI NoVsa
Strain genotype or mutation
Binding to secretor HBGA Binding to nonsecretor
HBGA Lex/a Reference(s) A-type H-type Ley/b
GI.7 ⫹ ⫹ ⫹ ⫹ Current study GI.1 ⫹ ⫹ ⫹ ⫺ 18,28,52
GI.1W375A ⫺ ⫺ ⫺ ⫺ 28
GI.2 ⫹ ⫹ ⫹ ⫹ 25
GI.2 Q389N ⫺ ⫺ ⫹ ⫺ 25
GI.3 ⫹ ⫹ ⫹ ⫹ 11
GI.4 ⫹ ⫹ ⫹ ⫹ 22
GI.8 ⫹ ⫹ ⫹ ⫹ 22
a⫹, binding activity reported;⫺, no binding activity.
FIG 2GI.7 VLPs bind secretor and nonsecretor HBGAs. Shown is binding of GI.7 VLPs to multivalent synthetic carbohydrate reagents conjugated with biotin. Binding of GI.1 VLPs (NV) and NV VLPs with a point mutation in the glycan binding site (W375A) is shown for comparison. Each condition was tested with six replicates, and the entire experiment was reproduced in tripli-cate. Each bar represents the mean absorbance value. The error bars represent 1 standard deviation of the mean. The carbohydrate reagents used were trisac-charide containing A antigen, trisactrisac-charide containing H-type 1 antigen, tri-saccharide containing Leaantigen, and biotin hydrazide reagent.
on November 7, 2019 by guest
http://jvi.asm.org/
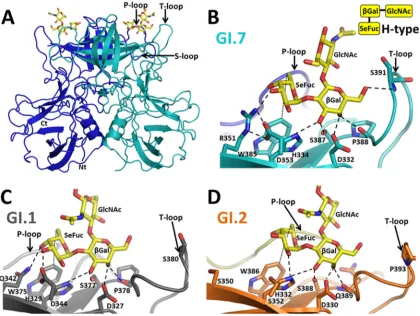
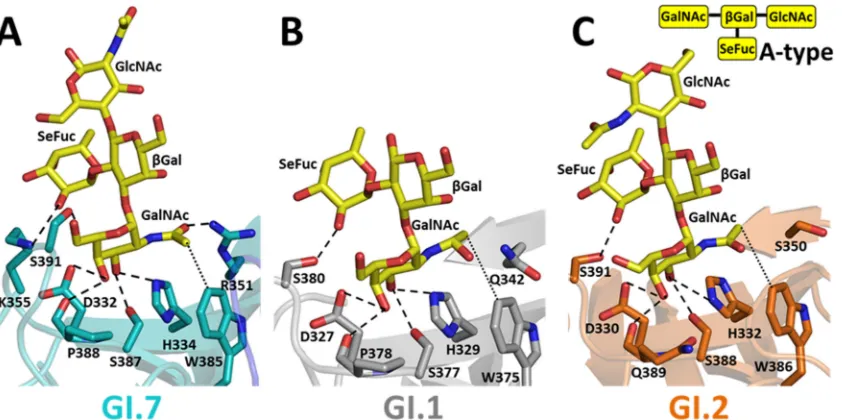
[image:5.585.298.546.599.714.2]H- and A-type HBGA binding to GI.7.We determined the structure of GI.7 in complex with H-type 2 and A-type 2 HBGAs. The HBGA binding site in GI.7, similar to those of other GI strains, is located on the outermost surface-exposed region of the P2 subdomain in close proximity to the variable P-, S-, and T-loops (Fig. 5A). The H- and A-type HBGA binding predominantly involves direct interactions with residues contributed from indi-vidual subunits of the P domain dimer. However, residues from the opposing subunit in the dimer participate in ligand binding through water-mediated interactions (data not shown). Although the H- and A-type binding in GI.7 is generally similar to that observed in GI.1 and GI.2, there are noticeable differences because of the sequence variations that can potentially confer differential affinities for these HBGAs.
The H-type HBGA interacts with the P domain primarily through-Gal and SeFuc; the GlcNAc moiety of the trisaccharide does not participate in direct interactions. -Gal is firmly an-chored in the binding site by an extensive network of hydrogen bond interactions involving its exocyclic hydroxyl groups and res-idues D332, H334, S387, P388, and S391 (Fig. 5B). A comparison with the H-type HBGA-bound P domain structures of GI.1 (Fig. 5C) and GI.2 (Fig. 5D) shows that residues D332, H334, and S387
are structurally conserved. These residues are also totally con-served across the GI NoVs as determined by sequence comparison
(Fig. 1). The P388 residue, which structurally corresponds to P378
in GI.1 and Q389 in GI.2, shows significant variation across GI NoVs (Fig. 1). The S391 residue interaction is unique to GI.7. This residue in the T-loop is positioned in close proximity to-Gal because of the conformational change, allowing it to participate in the hydrogen bond interaction. Analogous interactions in GI.1 and GI.2 are not possible, as the T-loop lies at a distance of 7 Å from-Gal. The GI.7 P domain also exhibits variations from GI.1 and GI.2 with respect to its interactions with the SeFuc of the H-type. In GI.7, the side chain of R351 makes a hydrogen bond with SeFuc. This residue is also involved in hydrogen bond inter-actions with the D353 residue. Both these residues show signifi-cant variations across GI NoVs. In GI.1, each member of the cor-responding pair of residues (Q342 and D344) contributes directly to HBGA binding, whereas in GI.2, the corresponding residues (S350 and S352) do not participate in hydrogen bonding to SeFuc due to their shorter side chains. A conserved feature, however, is the hydrophobic interaction involving SeFuc. The hexose ring of the SeFuc moiety makes van der Waals contact with the indole ring of W385, which, in turn, is involved in a cation-pi interaction
FIG 3The GI.7 P domain shows significant structural changes. (A) Overall structure of the GI.7 P domain dimer, with one subunit shown in blue and the other in cyan. The N and C termini of one of the subunits in the dimer are labeled Nt and Ct, respectively. The HBGA binding site is located on the P2 subdomain and is indicated by black arrows. (B) Topology diagram of the GI.7 P domain highlighting the locations of the variable loops (magenta) connecting various-strands (blue arrows) numbered starting from the N terminus. The lone helix is shown as a cylinder and labeled H. The-strands in the P2 subdomain are shown as vertical arrows and those in the P1 subdomain as horizontal arrows. (C) Superposition of the P domain structures from one subunit of the GI.7 dimer (blue) onto one subunit from the GI.1 dimer (gray) (PDB ID 2ZL5). The outermost loop regions in the GI.7 P2 subdomain that show significant differences are highlighted in magenta, marked with black arrows, and labeled as loops A, B, P, S, T, and U. (D) Close-up view of variable loops A and B of the GI.7 strain (blue) superimposed on the corresponding loop regions in GI.1 (gray) and GI.2 (orange). (E) Close-up view of the superposition of P-loop (blue) and S-loop (cyan) from the opposing subunit (indicated as S=) in GI.7 with those in GI.1 (gray) and GI.2 (orange). (F) Close-up view of the superposition of GI.7 T-loop (blue) with the corresponding loop regions in GI.1 (gray) and GI.2 (orange). In panels D, E, and F, the positions of individual loops in the GI.7 (blue), GI.2 (orange), and GI.1 (gray) P domain structures are indicated by downward-pointing arrows, and their relative movements are indicated by dashed black arrows.
Determinants of HBGA Binding among GI Noroviruses
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.93.491.68.351.2]with H334. Both these interactions are observable in the GI.1 and GI.2 structures, and the two residues are also exceptionally well conserved across GI NoVs.
With respect to A-type tetrasaccharide binding, while three of the four saccharide moieties that showed interactions with the P domain were clearly visible at a contour level of 3, the density for the terminal GlcNAc was weak and required lowering of the con-tour level to 2.5and showed no interactions with the P domain. As observed in the case of H-type binding, sequence variations in the P domain result in several differential interactions along with some conserved features. In GI.7 (Fig. 6A), as in GI.1 (Fig. 6B) and GI.2 (Fig. 6C), binding of the A-type to the P domain is primarily mediated by theN-acetylgalactosamine (GalNAc) moiety. A-type binding shares several similarities with H-type binding. In GI.7, the hexose ring (Gal) of GalNAc shows hydrogen bond interac-tions with residues D332, H334, S387, P388, and S391 similar to those shown by-Gal in H-type binding, and theN-acetyl arm participates in interactions similar to those of SeFuc of the H-type, including hydrophobic interactions with W385 and hydrogen bonding interactions with R351 (Fig. 6A). This hydrogen bond interaction involving R351 is not observed in GI.1 and GI.2, as the structurally corresponding residues Q342 in GI.1 and S350 in GI.2, with their shorter side chains, cannot be in close proximity to theN-acetyl arm. However, some of the other GI NoVs have a lysine at this position that could make hydrogen bonds similar to those made by R351 (Fig. 1). In addition to interactions involving GalNAc, in GI.1, GI.2, and GI.7, SeFuc of the A trisaccharide par-ticipates in binding through hydrogen bonding interactions in-volving a P domain residue. While serine residues in GI.1 (S380) and GI.2 (S391) participate in the hydrogen bonding, in GI.7, due to structural changes, a lysine residue (K355) participates in the
hydrogen bonding. These results clearly show that although the GI strains can bind A- and H-type HBGAs, the affinity to these li-gands might be modulated in a strain-dependent manner based on the sequence variations and structural changes in the P do-main.
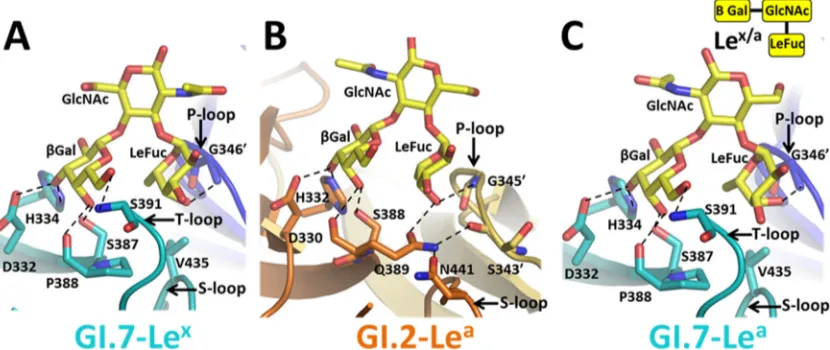
P domain-secretor Lewis HBGA binding.To examine how
sequence and structural variations in GI.7 affect the binding of difucosyl HBGA, we determined the structure of GI.7 in com-plex with the Ley(type 2) tetrasaccharide. The structural anal-ysis showed that SeFuc and-Gal of Leyoccupy the same
po-sitions as described for the H-type above, making nearly identical interactions with the P domain residues (Fig. 7A). In addition to these interactions involving SeFuc and-Gal, Le-Fuc of difucosyl Leyparticipates in a direct hydrogen bonding
interaction with the G346=(the prime indicates that the residue is from the opposing subunit) residue located on the P-loop of the opposing subunit in the dimer (Fig. 7A). Thus, in contrast to A- and H-type HBGA binding, Leybinding involves residues
from both subunits of the dimer. The only other structure for comparison with GI.7 in regard to difucosyl binding is that of GI.2 with Leb(Fig. 7B). With respect to the SeFuc and-Gal moiety interactions, the GI.7 and GI.2 structures exhibit the same differential interactions as described above for H-type binding. With regard to LeFuc, a similar hydrogen bond inter-action is observed with G346=in GI.7 as was described for GI.2 involving G345=, despite the shorter P-loop in GI.7. The main difference, however, is that the hydrogen bonding interaction between LeFuc and Q389 observed in GI.2 is absent in GI.7 with a Pro in the corresponding position. Although biochem-ical studies show that GI.1 can bind to difucosyl HBGAs such as Ley(45), the structural basis of this interaction has not been
FIG 4Comparison of the electrostatic potential surfaces of the GI.7 and GI.1 P domain dimers. Shown are side views and top views displaying the plotted electrostatic potential surfaces of the P domain from GI.7 (A) and GI.1 (B) strains (PDB ID 2ZL5). The electrostatic potential variation from negative (red) to positive (blue) is indicated by the scale bar at the bottom. Black boxes indicate regions within the two variants that show significant charge reversals. The HBGA binding sites are indicated by black arrows on the side views and by black dashed ovals on the top views.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.136.449.66.312.2]determined. In the absence of the structure, we modeled the GI.7-bound Leyonto the GI.1 P domain structure (PDB ID 2ZL6) (Fig. 7C). The model shows that GI.1 can interact with SeFuc and-Gal similarly to GI.7 or GI.2, with the same dif-ferential interactions as noted above for H-type binding. How-ever, the modeling suggests that the analogous hydrogen bond interaction between LeFuc and a Gly residue as observed in GI.2 and GI.7 structures is unlikely because the the P-loop in GI.1 is significantly shorter than the P-loop in either GI.7 or GI.2.
GI.7 P domain binding to nonsecretor Lewis HBGAs.To
ex-amine if monofucosyl nonsecretor Lewis HBGAs can bind to the GI.7 P domain in the absence of the Q389 residue and a longer P-loop, which are shown to be critical for nonsecretor HBGA binding in the case of GI.2 (25), we determined the structure of the GI.7 P domain in complex with both Lex(type 2) and Lea(type 1). These nonsecretor HBGAs are monofucosyl with only LeFuc linked to GlcNAc of the precursor disaccharide. Despite the ab-sence of the Q389 and a longer P-loop, our results show that the GI.7 P domain can bind to both Lexand Leanonsecretor HBGAs. In the Lex-bound GI.7 P domain structure (Fig. 8A), both the
-Gal and LeFuc moieties are involved in hydrogen bonding
in-teractions with the P domain residues. The inin-teractions involving the hexose ring of the-Gal are essentially similar to those ob-served with the other HBGA interactions described above, and the hydrogen bond interactions involving LeFuc and G346=are also the same as those observed with LeFuc of the difucosyl Lewis HBGA. The differential interactions because of the sequence and structure variations in GI.7 in comparison with GI.2 also remain the same (Fig. 8B). Modeling of Lexonto GI.1 (data not shown) suggests that while GI.1 can bind the-Gal moiety, the interac-tions with LeFuc are unlikely to be due to the shorter P-loop in GI.1.
In the P domain structure bound to nonsecretor Lea(type 1)
(Fig. 8C), Leabinds in a manner similar to that for Lex. The-Gal
and LeFuc moieties of Leaoccupy identical positions and partici-pate in hydrogen bonding interactions similar to those of Lex. The
difference between the two structures is with respect to the orien-tation of the GlcNAc moiety that flips 180° between the two struc-tures. The flip is due to the different glycosidic linkage in the precursor disaccharide. It is noteworthy, however, that in the GI.7-Leacomplex structure, although the density for GlcNAc, Le-Fuc, and-Gal are clearly represented at a contour level of⬃3in one of the molecules, in the other 3 molecules of the asymmetric
FIG 5Differential hydrogen bonding patterns in H-type HBGA binding among GI strains. (A) Cartoon representation of the GI.7 P domain dimer with bound H-type 2 HBGA (shown here and in subsequent panels in yellow stick representation with oxygen atoms in red and nitrogen atoms in blue). The two subunits in the dimer are shown in blue and cyan. The HBGA binding site is indicated with black arrows. (B to D) Close-up views of hydrogen bonding interactions between the GI.7 P domain and H-type 2 (cyan) (B), GI.1 P domain (gray) and H-type 1 (PDB ID 2ZL6) (C), and GI.2 P domain (orange) and H-type 1 (PDB ID 3ASQ) (D). The interacting P domain residues are individually labeled and shown as sticks with oxygen and nitrogen atoms in red and blue, respectively. The hydrogen bond (satisfying the donor-to-acceptor distance criterion of 2.6 to 3.2 Å) and hydrophobic (satisfying C-C distance criterion of 3.4 to 4.5 Å) interactions are shown in black dashed and dotted lines, respectively (here and in subsequent figures). A cartoon representation of the H-type trisaccharide is shown at the top right corner.
Determinants of HBGA Binding among GI Noroviruses
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.83.503.67.383.2]unit, the densities are weaker and required lowering of the contour levels to 2.5to model the HBGAs.
DISCUSSION
Human NoVs are suggested to evolve based on changing glycan binding specificities and antigenic drift, resulting from the accu-mulation of sequence variations on the outermost surface-ex-posed P domain of VP1. This has been particularly well studied in the GII NoVs, with the most prevalent genotype, GII.4, shown to undergo epochal evolution (30,46,47). Such studies are limited for the GI NoVs, in which it remains poorly understood how sequence variations that affect the P domain structure can lead to altered glycan binding specificities and possible antigenic
varia-tion. In this study, we determined the P domain structure from a member of the GI.7 genotype that bound to secretor and nonse-cretor HBGAs. By comparing the GI.7 structure to the available GI.1 and GI.2 P domain structures, we were able to characterize how sequence variations affect the P domain structure and differ-entially alter the glycan binding and to identify strain-dependent structural determinants of HBGA binding among the GI NoVs.
Sequence variations results in significant structural changes
in the P domain of GI NoVs.A striking observation from our
structural studies is that although the overall P domain structure is conserved among the GI NoVs, sequence variations profoundly alter the outermost loop regions (Fig. 3). These changes also
sig-FIG 6Comparison of A-type HBGA binding to the P domain among GI strains. Shown are close-up views of hydrogen bond interactions between the GI.7 P domain and A-type 2 HBGA (cyan) (A), GI.1 P domain and A-type 1 HBGA (gray) (PDB ID 2ZL7) (B), and GI.2 P domain (orange) and A-type 1 HBGA (PDB ID 3ASP) (C). A cartoon representation of the A-type tetrasaccharide is shown at the top right corner.
FIG 7Comparison of Le secretor HBGA binding in GI strains. Shown are close-up views of hydrogen bonding interactions with secretor Lewis family HBGAs: GI.7 P domain (cyan and blue) and Ley(A), GI.2 P domain (orange and olive) and Leb(PDB ID 3ASS) (B), and GI.1 P domain (light gray and dark gray) and modeled Ley(C). In the interactions with secretor Lewis HBGA, SeFuc and-Gal bind in a way similar to that of the H-type, and similar to the case with H-type binding, all hydrogen bond interactions with these two moieties are maintained in both GI.7 and GI.2. Additionally, LeFuc is involved in hydrogen bonding to a Gly residue (G346 for GI.7 and G345 for GI.2) from the other monomer. The modeling of Leyonto the GI.1 P domain shows that Leycan interact with SeFuc and-Gal in a way similar to that of the H-type, and similar to the case with the H-type, all hydrogen bonding interactions are possible (data not shown). A cartoon representation of the secretor Lewis tetrasaccharide is shown at the top right corner.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.85.506.66.276.2] [image:9.585.83.509.498.658.2]nificantly affect the surface topography of the P2 subdomain, ac-companied by several electrostatic charge reversals (Fig. 4). One major difference is in the A- and B-loops, which lie in close prox-imity to each other. In the GI.7 P domain structure, these two loops are well separated in a distinctly open conformation, expos-ing a large surface area underneath, in contrast to the closed con-formation found in the GI.1 and GI.2 strains. Sequence alignment suggests that the A- and B-loops are likely even longer in the GI.8 and GI.9 strains (Fig. 1). Interestingly, in GI.1 NV, the B loop contains a residue critical for binding of HBGA blocking antibod-ies (48), and the corresponding loops in the P domains of murine NoV (GV) (49,50) and rabbit hemorrhagic disease virus (animal calicivirus) (51) contain the neutralization antigenic sites. Thus, this region can be a major site for potential differential antigenic presentations contributing to serotypic differentiation among the GI variants, a factor that may have to be considered in designing antiviral strategies for GI NoVs.
The P- and S-loops also show significant structural changes among the structurally characterized GI P domains and are seen to deviate in a concerted manner (Fig. 3D). Although these loops are distant within each subunit in the dimer, they come closer to each other in the context of the dimer and lie in the close vicinity of the HBGA binding site. Sequence comparison among the representa-tive GI variants shows that these loops vary in both length and composition among the representative GI strains. Our studies also revealed another loop region (T-loop) that is susceptible to signif-icant structural changes (Fig. 3F). In contrast to the GI.1 and GI.2 P domains, the sequence variations in the GI.7 P domain bring the T-loop closer to the HBGA binding site. A sequence comparison of the T-loops among different GI strains shows that the T-loop is shorter in most GI strains, including GI.6, GI.8, and GI.9 strains
(Fig. 1). These structural changes as a result of sequence variations
among the GI variants thus could allow GI NoVs to fine-tune and modulate both antigenicity and glycan binding affinities and con-tribute to the evolution of GI NoVs. Similar alterations in surface topology and electrostatic charge reversals along with variations in HBGA binding specificities are also observed among the more prevalent GII.4 genotype and have been linked to epochal evolu-tion in these variants (30).
Sequence variations in the GI P domain can potentially
mod-ulate HBGA binding affinities.Our studies on GI.7 with
mono-fucosyl and dimono-fucosyl secretor HBGAs substantiate the previous observation that HBGA binding in GI NoV involves primarily a Gal moiety, in contrast to the Fuc moiety in GII NoVs, and a highly conserved hydrophobic interaction. Despite the significant sequence variations and consequent structural changes, the resi-dues involved in Gal binding (D332, H334, and S387) and hydro-phobic interactions (W385) are positioned in nearly identical po-sitions in the structurally conserved-strands in all GI P domain structures. These residues are also exceptionally well conserved in all the GI NoVs, suggesting that they all should exhibit binding to similar sets of HBGAs. However, comparative analysis of GI P domain structures with HBGAs indicate that the relative affinities of HBGA binding are likely to be differentially modulated by the other residues in the loop regions that show significant structural changes because of associated sequence variations. This is clearly evident in GI.7, in which, for example, the structural changes in the T-loop allows residue S391 to hydrogen bond to the Gal moi-ety, whereas an analogous interaction is absent in the case of GI.1 and GI.2. Another example is in the GI.1 structure, in which Q342 and D344 contribute to hydrogen bonding to the SeFuc moiety. In the GI.7 P domain structure, Q342 is replaced by R351, which contributes to hydrogen bonding to SeFuc, and D353, which is in the position of GI.1 D344, hydrogen bonds with R351 instead of with SeFuc. In GI.2, these residues are replaced by S350 and S352 and do not interact with SeFuc (Fig. 5). In the binding of the difucosyl Lewis HBGAs also, in addition to the above-mentioned differences in Gal binding, the GI.7 and GI.2 structures show dif-ferential interactions with LeFuc. Although the contributions of such differential interactions to relative HBGA binding affinities may be subtle in the context of the P domain, in the context of the virion during infection, the differences in binding affinities may be more pronounced and discriminatory considering the added avidity effects.
How and whether such differential affinities in HBGA binding contribute to relative differences in productive infection and/or prevalence between GI strains (also true for GII strains) is an in-teresting question. While the interplay between antigenic
varia-FIG 8Comparison of nonsecretor Lewis HBGA binding in GI strains. Shown is a close-up view of hydrogen bond interactions with monofucosyl Lewis family HBGAs: GI.7 P domain (cyan/blue) and Lex(A), GI.2 P domain (orange and olive) and Lea(PDB ID 3ASR) (B), and GI.7 P domain (cyan and blue) and Lea(C). A cartoon representation of the nonsecretor trisaccharide is shown at the top right corner.
Determinants of HBGA Binding among GI Noroviruses
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.87.502.69.244.2]tion and HBGA binding specificity is one major factor, productive infection and/or prevalence may be dependent upon a certain threshold affinity for a particular HBGA or set of HBGAs and their abundance in the gut during infection. Experimental verification of such a hypothesis requires an infectivity model for human NoVs, which is not currently available.
The P-loop structure is the determinant in strain-dependent
binding of nonsecretor Lewis HBGAs.While many studies have
clearly indicated that the majority of the GI NoVs bind to secretor Lewis HBGAs, only a subset of GI NoVs bind nonsecretor Lewis HBGAs. As a structural basis for why only a subset of GI NoVs bind to nonsecretor Lewis HBGAs, based on the studies with GI.2 NoV, it was suggested that Q389 and a longer P-loop are critical determinants for binding (25). Ourin vitrobinding studies using GI.7 VLPs and our crystallographic studies clearly showed that despite a Pro residue (P388) in the structurally equivalent position of Q389, the GI.7 P domain can indeed bind nonsecretor Lewis HBGA (Fig. 8). Despite a shorter P-loop in GI.7 than that in the GI.2, the main-chain amide group of G346=is in place to hydrogen bond with LeFuc as in GI.2. Thus, it is very likely that the threshold length and the structure of the P-loop, which allow main-chain interaction with LeFuc of this HBGA, are the sole critical determi-nants in nonsecretor HBGA binding. In G1.1 NV, the P loop is significantly shorter and cannot make analogous interactions with LeFuc of the nonsecretor HBGAs. In the GI.3 NoV, recent epide-miological studies have shown a secretor-independent infection pattern (11). Although this strain has a Pro residue corresponding to Q389 in GI.2 residues, the P-loop length is predicted to be the same as in GI.7 on the basis of the sequence, further supporting the idea that the threshold length of P-loop is critical. From these observations, it can be predicted that genotypes such as GI.4, GI.8, and GI.9 would have the appropriate P-loop length to bind non-secretor HBGAs, and indeed, Shirato et al. (22) have demon-strated binding of GI.4 and GI.8 VLPs to Lea.
HBGA precursor types have minimal structural effects on
binding.Our studies also provide valuable structural insight into
whether the type of glycoside linkage in the precursors has any effect on HBGA binding to GI NoVs. Most of our studies on GI.7 were done with HBGAs with a type 2 disaccharide precursor ( -Gal1-4GlcNac) and, in the case of Lea, with a type 1 precursor (-Gal1-3GlcNAc), whereas previous studies on GI.1 and GI.2 have used HBGAs with a type 1 disaccharide precursor. Our stud-ies in comparison with GI.1 and GI.2 show that all the hydrogen bond and hydrophobic interactions remain invariant and are not affected by the glycosidic linkage of the precursor. GlcNAc does not make any direct contact with the P domain in any of the structures. The linkage affects the orientation of the terminal GlcNAc moiety and flips this moiety 180° to reverse the direction of itsN-acetyl arm. Our structural studies of GI.7 with A-type 3 HBGA (-Gal1-3GalNac) also showed (data not shown) the same interactions as with the A-type 2 HBGA, further confirming that the type of precursor has minimal effects on the P domain-HBGA interactions. However, previous studies have indicated qualitative differences between HBGAs with different precursor types (22). Although no changes have been observed structurally, the reversal of theN-acetyl arm could lead to an additional solvent-mediated hydrogen bond(s) with one type and not the other to bring about small affinity changes, which may be accentuated in the context of VLPs due to avidity effects.
Conclusion.Recent epidemiological studies have shown
in-creased prevalence of GI NoVs. Although secretor-positive status is strongly correlated with NoV infection, cases of NoV infection associated with secretor-negative individuals are reported. Bio-chemical studies have shown that GI NoVs exhibit genotype-de-pendent binding to nonsecretor HBGAs. Our main focus was to understand how genotypic differences in GI NoVs affect the P domain structure and HBGA binding specificities. Our studies of GI.7 in comparison with similar studies on GI.2 and GI.1 demon-strate how genotypic differences translate to extensive structural changes in the loop regions that significantly alter the surface to-pography, perhaps reflective of antigenic variations, and also dif-ferentially alter the HBGA binding characteristics. We have iden-tified two loops in the P2 subdomain that exhibit large conformational changes and suggested that they represent major sites for differential antigenic presentations contributing to sero-type differentiation. A significant finding from our studies is that the threshold length and the structure of the P loop are critical determinants in the binding of GI NoVs to nonsecretor HBGAs. Further epidemiological studies are required to assess the contri-bution of such genotype-dependent binding to nonsecretor HBGAs in GI NoVs during NoV infection.
ACKNOWLEDGMENTS
This work was supported by the following grants from the NIH: PO1 AI057788 to M.K.E., R.L.A., and B.V.V.P., P30DK56638 to M.K.E., and P30 CA125123, which funds the Recombinant Protein and Monoclonal Antibody Production Shared Resource at Baylor College of Medicine. This work was also supported by the Robert Welch Foundation (grant Q1292 to B.V.V.P.) and the John S. Dunn Research Foundation (funds to R.L.A.). R.C. was supported in part by Agriculture and Food Research Initiative Competitive Grant no. 2011-68003-30395 from the USDA Na-tional Institute of Food and Agriculture.
We acknowledge the use of the synchrotron beamlines (5.0.1) at the Advanced Light Source, Lawrence Berkeley National Laboratory, for dif-fraction data collection and thank their staff for excellent help. The Berke-ley Center for Structural Biology is supported in part by the NIH, NIGMS, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sci-ences, of the U.S. Department of Energy under contract number DE-AC02-05CH11231.
REFERENCES
1.Hall AJ, Lopman BA, Payne DC, Patel MM, Gastanaduy PA, Vinje J, Parashar UD.2013. Norovirus disease in the United States. Emerg. Infect. Dis.19:1198 –1205.http://dx.doi.org/10.3201/eid1908.130465. 2.Patel MM, Hall AJ, Vinje J, Parashar UD.2009. Noroviruses: a
compre-hensive review. J. Clin. Virol.44:1– 8.http://dx.doi.org/10.1016/j.jcv.2008 .10.009.
3.Ramani S, Atmar RL, Estes MK.2014. Epidemiology of human norovi-ruses and updates on vaccine development. Curr. Opin. Gastroenterol.
30:25–33.http://dx.doi.org/10.1097/MOG.0000000000000022. 4.Kroneman A, Vega E, Vennema H, Vinje J, White PA, Hansman G,
Green K, Martella V, Katayama K, Koopmans M.2013. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol.158:2059 – 2068.http://dx.doi.org/10.1007/s00705-013-1708-5.
5.Clarke IN, Estes MK, Green KY, Hansman G, Knowles NJ, Koopmans M, Matson DO, Meyers G, Neill JD, Radford A, Smith AW, Studdert MJ, Thiel HJ, Vinje J.2012. Caliciviridae, p 977–986. InKing AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: classification and nomenclature of viruses: ninth report of the International Committee on Taxonomy of Viruses. Elsevier, San Diego, CA.
6.Glass RI, Parashar UD, Estes MK.2009. Norovirus gastroenteritis. N. Engl. J. Med.361:1776–1785.http://dx.doi.org/10.1056/NEJMra0804575.
7.Hoa Tran TN, Trainor E, Nakagomi T, Cunliffe NA, Nakagomi O.
2013. Molecular epidemiology of noroviruses associated with acute
on November 7, 2019 by guest
http://jvi.asm.org/
radic gastroenteritis in children: global distribution of genogroups, geno-types and GII.4 variants. J. Clin. Virol.56:185–193.http://dx.doi.org/10 .1016/j.jcv.2012.11.011.
8.Zheng DP, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS.
2006. Norovirus classification and proposed strain nomenclature. Virol-ogy346:312–323.http://dx.doi.org/10.1016/j.virol.2005.11.015. 9.Bruggink LD, Oluwatoyin O, Sameer R, Witlox KJ, Marshall JA.2012.
Molecular and epidemiological features of gastroenteritis outbreaks in-volving genogroup I norovirus in Victoria, Australia, 2002–2010. J. Med. Virol.84:1437–1448.http://dx.doi.org/10.1002/jmv.23342.
10. Leshem E, Barclay L, Wikswo M, Vega E, Gregoricus N, Parashar UD, Vinje J, Hall AJ.2013. Genotype GI.6 norovirus, United States, 2010–2012. Emerg. Infect. Dis.19:1317–1320.http://dx.doi.org/10.3201/eid1908.130445. 11. Nordgren J, Kindberg E, Lindgren PE, Matussek A, Svensson L.2010.
Norovirus gastroenteritis outbreak with a secretor-independent suscepti-bility pattern, Sweden. Emerg. Infect. Dis.16:81– 87.http://dx.doi.org/10 .3201/eid1601.090633.
12. Hasing ME, Lee BE, Preiksaitis JK, Tellier R, Honish L, Senthilselvan A, Pang XL.2013. Emergence of a new norovirus GII.4 variant and changes in the historical biennial pattern of norovirus outbreak activity in Alberta, Canada, from 2008 to 2013. J. Clin. Microbiol.51:2204 –2211.http://dx .doi.org/10.1128/JCM.00663-13.
13. Vicentini F, Denadai W, Gomes YM, Rose TL, Ferreira MS, Le Moullac-Vaidye B, Le Pendu J, Leite JP, Miagostovich MP, Spano LC.2013. Molecular characterization of noroviruses and HBGA from infected Qui-lombola children in Espirito Santo State, Brazil. PLoS One8:e69348.http: //dx.doi.org/10.1371/journal.pone.0069348.
14. Soares CC, Santos N, Beard RS, Albuquerque MC, Maranhao AG, Rocha LN, Ramirez ML, Monroe SS, Glass RI, Gentsch J.2007. Norovirus detection and genotyping for children with gastroenteritis, Brazil. Emerg. Infect. Dis.13:1244– 1246.http://dx.doi.org/10.3201/eid1308.070300.
15. Vega E, Barclay L, Gregoricus N, Shirley SH, Lee D, Vinje J. 2014. Genotypic and epidemiologic trends of norovirus outbreaks in the United States, 2009 to 2013. J. Clin. Microbiol.52:147–155.http://dx.doi.org/10 .1128/JCM.02680-13.
16. Ajami NJ, Kavanagh OV, Ramani S, Crawford SE, Atmar RL, Jiang ZD, Okhuysen PC, Estes MK, Dupont HL.2014. Seroepidemiology of noro-virus-associated travelers’ diarrhea. J. Travel Med.21:6 –11.http://dx.doi .org/10.1111/jtm.12092.
17. Hutson AM, Atmar RL, Graham DY, Estes MK.2002. Norwalk virus infection and disease is associated with ABO histo-blood group type. J. Infect. Dis.185:1335–1337.http://dx.doi.org/10.1086/339883.
18. Huang P, Farkas T, Zhong W, Tan M, Thornton S, Morrow AL, Jiang X.2005. Norovirus and histo-blood group antigens: demonstration of a wide spectrum of strain specificities and classification of two major bind-ing groups among multiple bindbind-ing patterns. J. Virol.79:6714 – 6722.http: //dx.doi.org/10.1128/JVI.79.11.6714-6722.2005.
19. Marionneau S, Ruvoen N, Le Moullac-Vaidye B, Clement M, Cailleau-Thomas A, Ruiz-Palacois G, Huang P, Jiang X, Le Pendu J. 2002. Norwalk virus binds to histo-blood group antigens present on gastrodu-odenal epithelial cells of secretor individuals. Gastroenterology122:1967– 1977.http://dx.doi.org/10.1053/gast.2002.33661.
20. Ruvoën-Clouet N, Belliot G, Le Pendu J.2013. Noroviruses and histo-blood groups: the impact of common host genetic polymorphisms on virus transmission and evolution. Rev. Med. Virol.23:355–366.http://dx .doi.org/10.1002/rmv.1757.
21. Shirato H.2012. Norovirus recognition sites on histo-blood group antigens. Front. Microbiol.3:177.http://dx.doi.org/10.3389/fmicb.2012.00177. 22. Shirato H, Ogawa S, Ito H, Sato T, Kameyama A, Narimatsu H, Xiaofan
Z, Miyamura T, Wakita T, Ishii K, Takeda N.2008. Noroviruses distin-guish between type 1 and type 2 histo-blood group antigens for binding. J. Virol.82:10756 –10767.http://dx.doi.org/10.1128/JVI.00802-08. 23. Hutson AM, Airaud F, LePendu J, Estes MK, Atmar RL.2005. Norwalk
virus infection associates with secretor status genotyped from sera. J. Med. Virol.77:116 –120.http://dx.doi.org/10.1002/jmv.20423.
24. Lindesmith L, Moe C, Marionneau S, Ruvoen N, Jiang X, Lindblad L, Stewart P, LePendu J, Baric R.2003. Human susceptibility and resistance to Norwalk virus infection. Nat. Med.9:548 –553.http://dx.doi.org/10 .1038/nm860.
25. Kubota T, Kumagai A, Ito H, Furukawa S, Someya Y, Takeda N, Ishii K, Wakita T, Narimatsu H, Shirato H.2012. Structural basis for the recognition of Lewis antigens by genogroup I norovirus. J. Virol.86:
11138 –11150.http://dx.doi.org/10.1128/JVI.00278-12.
26. Marionneau S, Airaud F, Bovin NV, Le Pendu J, Ruvoen-Clouet N.
2005. Influence of the combined ABO, FUT2, and FUT3 polymorphism on susceptibility to Norwalk virus attachment. J. Infect. Dis.192:1071– 1077.http://dx.doi.org/10.1086/432546.
27. Cao S, Lou Z, Tan M, Chen Y, Liu Y, Zhang Z, Zhang XC, Jiang X, Li X, Rao Z.2007. Structural basis for the recognition of blood group tri-saccharides by norovirus. J. Virol.81:5949 –5957.http://dx.doi.org/10 .1128/JVI.00219-07.
28. Choi JM, Hutson AM, Estes MK, Prasad BV.2008. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. U. S. A.105:9175–9180.http://dx .doi.org/10.1073/pnas.0803275105.
29. Donaldson EF, Lindesmith LC, Lobue AD, Baric RS.2010. Viral shape-shifting: norovirus evasion of the human immune system. Nat. Rev. Mi-crobiol.8:231–241.http://dx.doi.org/10.1038/nrmicro2296.
30. Shanker S, Choi JM, Sankaran B, Atmar RL, Estes MK, Prasad BV.
2011. Structural analysis of histo-blood group antigen binding specificity in a norovirus GII.4 epidemic variant: implications for epochal evolution. J. Virol.85:8635– 8645.http://dx.doi.org/10.1128/JVI.00848-11. 31. Lindesmith LC, Donaldson EF, Baric RS.2011. Norovirus GII.4 strain
antigenic variation. J. Virol. 85:231–242.http://dx.doi.org/10.1128/JVI .01364-10.
32. Otwinowski Z, Minor W.1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol.276:307–326.http://dx .doi.org/10.1016/S0076-6879(97)76066-X.
33. Pflugrath J.1999. The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr.55:1718 –1725.http://dx.doi.org/10 .1107/S090744499900935X.
34. Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG.2011. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr.67:271–281.http: //dx.doi.org/10.1107/S0907444910048675.
35. Dodson EJ, Winn M, Ralph A.1997. Collaborative Computational Project, number 4: providing programs for protein crystallography. Methods Enzymol.
277:620–633.http://dx.doi.org/10.1016/S0076-6879(97)77034-4.
36. McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ.2007. Phaser crystallographic software. J. Appl. Crystallogr.40:
658 – 674.http://dx.doi.org/10.1107/S0021889807021206.
37. Morris RJ, Perrakis A, Lamzin VS. 2003. ARP/wARP and automatic interpretation of protein electron density maps. Methods Enzymol.374:
229 –244.http://dx.doi.org/10.1016/S0076-6879(03)74011-7.
38. Emsley P, Cowtan K.2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr.60:2126 –2132.http://dx .doi.org/10.1107/S0907444904019158.
39. Murshudov GN, Vagin AA, Dodson EJ.1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystal-logr.53:240–255.http://dx.doi.org/10.1107/S0907444996012255.
40. Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH.
2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr.66:213–221.http://dx.doi .org/10.1107/S0907444909052925.
41. Laskowski RA, Macarthur MW, Moss DS, Thornton JM.1993. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crys-tallogr.26:283–291.http://dx.doi.org/10.1107/S0021889892009944.
42. Wallace AC, Laskowski RA, Thornton JM.1995. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng.
8:127–134.http://dx.doi.org/10.1093/protein/8.2.127.
43. Koo HL, Neill FH, Estes MK, Munoz FM, Cameron A, Dupont HL, Atmar RL.2013. Noroviruses: the most common pediatric viral enteric pathogen at a large university hospital after introduction of rotavirus vac-cination. J. Pediatric Infect. Dis. Soc.2:57– 60.http://dx.doi.org/10.1093 /jpids/pis070.
44. Prasad BV, Hardy ME, Dokland T, Bella J, Rossmann MG, Estes MK.
1999. X-ray crystallographic structure of the Norwalk virus capsid. Science
286:287–290.http://dx.doi.org/10.1126/science.286.5438.287.
45. Hutson AM, Atmar RL, Estes MK.2004. Norovirus disease: changing epidemiology and host susceptibility factors. Trends Microbiol.12:279 – 287.http://dx.doi.org/10.1016/j.tim.2004.04.005.
46. Siebenga JJ, Vennema H, Renckens B, de Bruin E, van der Veer B, Siezen RJ, Koopmans M.2007. Epochal evolution of GGII.4 norovirus Determinants of HBGA Binding among GI Noroviruses
on November 7, 2019 by guest
http://jvi.asm.org/
capsid proteins from 1995 to 2006. J. Virol.81:9932–9941.http://dx.doi .org/10.1128/JVI.00674-07.
47. Lindesmith LC, Donaldson EF, Lobue AD, Cannon JL, Zheng DP, Vinje J, Baric RS.2008. Mechanisms of GII.4 norovirus persistence in human popula-tions. PLoS Med.5:e31.http://dx.doi.org/10.1371/journal.pmed.0050031. 48. Chen Z, Sosnovtsev SV, Bok K, Parra GI, Makiya M, Agulto L, Green KY,
Purcell RH.2013. Development of Norwalk virus-specific monoclonal anti-bodies with therapeutic potential for the treatment of Norwalk virus gastro-enteritis. J. Virol.87:9547–9557.http://dx.doi.org/10.1128/JVI.01376-13. 49. Katpally U, Wobus CE, Dryden K, Virgin HW, IV, Smith TJ.2008.
Structure of antibody-neutralized murine norovirus and unexpected dif-ferences from viruslike particles. J. Virol.82:2079 –2088.http://dx.doi.org /10.1128/JVI.02200-07.
50. Taube S, Rubin JR, Katpally U, Smith TJ, Kendall A, Stuckey JA, Wobus CE.2010. High-resolution x-ray structure and functional analysis of the murine norovirus 1 capsid protein protruding domain. J. Virol.
84:5695–5705.http://dx.doi.org/10.1128/JVI.00316-10.
51. Wang X, Xu F, Liu J, Gao B, Liu Y, Zhai Y, Ma J, Zhang K, Baker TS, Schulten K, Zheng D, Pang H, Sun F.2013. Atomic model of rabbit hemorrhagic disease virus by cryo-electron microscopy and crystallogra-phy. PLoS Pathog. 9:e1003132. http://dx.doi.org/10.1371/journal.ppat .1003132.
52. Hutson AM, Atmar RL, Marcus DM, Estes MK.2003. Norwalk virus-like particle hemagglutination by binding to H histo-blood group anti-gens. J. Virol. 77:405– 415. http://dx.doi.org/10.1128/JVI.77.1.405-415 .2003.