0022-538X/97/$04.00
1
0
Copyright
q
1997, American Society for Microbiology
Independently Cloned Halves of Cytomegalovirus Assemblin,
A
n
and A
c
, Can Restore Proteolytic Activity to Assemblin
Mutants by Intermolecular Complementation
MATTHEW R. T. HALL†
ANDWADE GIBSON*
Virology Laboratories, Department of Pharmacology and Molecular Sciences, The Johns Hopkins
University School of Medicine, Baltimore, Maryland 21205
Received 17 July 1996/Accepted 15 October 1996
Herpesviruses encode an essential serine proteinase called assemblin that is responsible for cleaving the
precursor assembly protein during the process of capsid formation. In cytomegalovirus (CMV), assemblin
undergoes autoproteolysis at an internal (I) site located near the middle of the molecule. I-site cleavage
converts the enzyme to an active two-chain form consisting of the subunits A
nand A
c. We have recently shown
that the recombinant A
nand A
csubunits can spontaneously associate within eukaryotic cells to yield active
two-chain proteinase. This finding indicates that the subunits are able to independently assume their correct
functional conformations and led us to test whether they are capable of intermolecular complementation. This
was done by coexpressing inactive mutant (point, deletion, and insertion) forms of assemblin together with the
wild-type subunit (either A
nor A
c) corresponding to the domain of assemblin that was mutated. Results of
these experiments showed that both A
nand A
care able to rescue the enzymatic activity of assemblin mutants.
I-site cleavage of the mutated assemblin occurred during complementation but was not absolutely required, as
shown by effective complementation of inactive assemblins with noncleavable I sites. We have also shown that
intermolecular complementation can rescue the activity of an inactive mutant full-length proteinase precursor
and can occur between different species of CMV (e.g., human CMV subunit can rescue activity of mutant
simian CMV assemblin). These results indicate that assemblin is able to form active multimeric structures that
may be of functional importance.
Herpesviruses encode a serine proteinase (5, 9, 15, 32, 39)
that is synthesized as a precursor (20, 27, 40) and is essential
for the production of infectious virus (10, 26). Both the
pre-cursor and its substrate, the prepre-cursor assembly protein (pAP)
(Fig. 1A), are needed during capsid assembly and have a direct
role in forming the icosahedral, outer capsid shell (7, 34, 35).
During the capsid assembly and maturation processes, the
pro-teinase cleaves pAP at its maturational (M) site, severing the
mature assembly protein (AP) from its carboxyl tail (Fig. 1A)
(20, 40). In addition, the precursor proteinase called pNP1
(Fig. 1A) or ACpra (11) in cytomegalovirus (CMV) undergoes
three major autoproteolytic cleavages: (i) one at its M site,
present due to the nested, overlapping relationship of the
pro-teinase and assembly protein precursors (21, 38); (ii) another
at its release (R) site near the middle of pNP1, which releases
the proteolytic amino half of the precursor, assemblin (NP1
n),
from the nonproteolytic carboxyl half (NP1
c) (Fig. 1A) (1, 8,
40); and (iii) a third at its internal (I) site (1, 2, 39), which
converts active single-chain assemblin into an active two-chain
form (15) composed of its amino half (A
n) and its carboxyl half
(A
c) (Fig. 1A).
It has recently been demonstrated that active two-chain
CMV assemblin can be formed from the independently cloned
and expressed subunits, A
nand A
c. This was shown in vitro by
expressing and purifying A
nand A
cfrom bacteria as separate
glutathione S-transferase fusion proteins, removing the
gluta-thione S-transferase portion, and then combining the
frag-ments and renaturing them together (24). We showed that
two-chain assemblin can also form spontaneously within cells
coexpressing the A
nand A
csubunits (14). This in vivo
forma-tion of two-chain assemblin occurs in plasmid-transfected
hu-man cells and in recombinant baculovirus (rBV)-infected
in-sect cells and was demonstrated for both the human CMV
(HCMV) and simian CMV (SCMV) enzymes and their
inter-typic pairs (e.g., HCMV A
nplus SCMV A
c) (14).
The ability of A
nand A
cto associate spontaneously within
cells to form two-chain assemblin indicated that they can
fold and function without first being synthesized as a
one-chain enzyme and suggested that each might be able to rescue
the enzymatic activity of mutant proteinases by
intermolec-ular complementation. To test this possibility, we have
co-expressed A
nor A
cwith a variety of inactive mutant
assem-blins in two expression systems: Spodoptera frugiperda Sf9
insect cells infected with rBVs, and human embryonal
kid-ney (HEK) cells transfected with recombinant expression
plasmids. The assemblin mutants included insertion,
dele-tion, and point mutations that affect domains and residues
critical to enzyme activity: insertion between the highly
con-served domains 1 and 2 (CD1 and CD2); deletion of the
carboxyl end (40); and replacement of the nucleophilic
serine (5, 9, 15, 32, 39) (e.g., Ser118 in SCMV), the
abso-lutely conserved histidine (19, 39) (e.g., His47 in SCMV), or
amino acids surrounding the I site (e.g., Ala127 in SCMV).
Results of our experiments show that intermolecular
com-plementation of assemblin can occur, indicating that the
en-zyme can form multimers that may be of functional
impor-tance. This conclusion is consistent with two recent reports that
* Corresponding author. Mailing address: Department of
Pharma-cology and Molecular Sciences, The Johns Hopkins University School
of Medicine, 725 N. Wolfe St., Baltimore, MD 21205. Phone: (410)
955-8680. Fax: (410) 955-3023. E-mail: [email protected]
.edu
† Present address: Department of Biological Chemistry, The Johns
Hopkins Medical Institutions, Baltimore, MD 21205.
956
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 1. Cleavage products, landmarks, and constructions. (A) SCMV strain Colburn cleavage products. Shown are the protein fragments generated by cleavage at the I, R, and M sites in the proteinase and by cleavage of pAP at its M site. The computer-predicted molecular mass is shown for each cleavage product. The shaded boxes labeled N2, C2, N1, and C1 indicate the peptide epitopes recognized by the anti-N2, anti-C2, anti-N1, and anti-C1 antisera, respectively. The highly conserved domains (CD1, CD2, and CD3) of assemblin and its herpesvirus homologs are also indicated by shaded boxes. (B) SCMV strain Colburn plasmid constructs and rBVs used. Names of plasmids and rBVs are given at the left, and the expected protein products are given at the right. Dashes indicate that no rBV was made. M1is the
start methionine for the full-length (APNG1 open reading frame product), and assemblin forms of the proteinase as well as An. Mpand M281are the start methionines
for Acand the pAP (product of the APNG.5 open reading frame), respectively. A127, A249, and A557represent the P1 position (29) of the I, R, and M sites. The box
labeled pvc indicates the position of the 45-bp insertion in the inactive assemblin proteinase, LM3.S. A179IQT indicates the final carboxyl residues of the inactive
assemblin deletion mutant AW5, A179is the final assemblin-specific residue, and I, Q, and T were added in the cloning process. Landmarks depicted are the same as
for panel A.
on November 9, 2019 by guest
http://jvi.asm.org/
the active form of bacterially synthesized, purified HCMV
as-semblin is a dimer (6, 23).
MATERIALS AND METHODS
Cells and antisera. Propagation and assay of rBVs was done in from S. frugiperda Sf9 cells (CRL 1711; American Type Culture Collection, Rockville, Md.). Sf9 cells were grown in suspension at 288C in 100-ml spinner flasks con-taining supplemented Grace’s medium (catalog no. 350-1605AJ; GIBCO, Grand Island, N.Y.) with 10% fetal calf serum (HyClone, Logan, Utah), 50mg of gentamicin (catalog no. 600-5750AD; GIBCO) per ml, and 125 ng of amphoter-icin B (Fungizone; catalog no. 600-5295AE; GIBCO) per ml.
HEK cells (CRL 1573, line 293; American Type Culture Collection) for trans-fection assays were grown in 24-well plates (catalog no. 3047; Becton Dickinson Labware, Lincoln Park, N.J.) containing Dulbecco’s modified Eagle’s medium (catalog no. 12100-061; GIBCO) supplemented with 10% fetal calf serum (Hy-Clone), 100 U of penicillin-streptomycin (catalog no. 15140-122, GIBCO) per ml, and 10 U of nystatin (catalog no. 153400-52; GIBCO) per ml. Each well con-tained approximately 106cells and 1 ml of medium.
Antisera were prepared by injecting rabbits with synthetic peptides conjugated to keyhole limpet hemocyanin (catalog no. 77100G; Pierce, Rockford, Ill.). The peptides used to make polyclonal antisera anti-N1, anti-N2, and anti-C2 have been described previously (14, 30, 39) and are mimics of the proteinase se-quences represented as shaded boxes in Fig. 1. Antisera were used at a dilution of 1:40 in a solution containing 10 mM Tris, 0.9% NaCl, and 5% bovine serum albumin (catalog no. 126602; Calbiochem, San Diego, Calif.), pH 7.4.
Construction of plasmids.Standard techniques were used to construct, clone, and propagate the plasmids (28). The new plasmids encoding SCMV proteins described here were derived from AW4, BJ1 (14), and AW1 (40) (Fig. 1B), which are constructs of the RSV.5(neo) expression vector (22) that contain, respec-tively, the SCMV Colburn APNG1 gene (encoding the full-length proteinase), the assemblin coding region, and the APNG.5 gene (encoding the assembly protein precursor) (Fig. 1B). Plasmids encoding altered forms of assemblin are referred to by their plasmid construction number (e.g., BJ14). Mutants of full-length proteinase constructs are denoted by the suffix .L.
The construction of MH28 and MH29, encoding SCMV Anand SCMV Ac,
respectively, has been described elsewhere (14).
BJ14 encodes S118A, a proteolytically inactive assemblin mutant in which the Ser118 nucleophile (39) has been changed to Ala. It was made by replacing the small SalI-to-BsrGI fragment of wild-type BJ1 with the corresponding mutated fragment from a plasmid, AW48 (39), that encodes S118A.L.
BJ20 encodes H47A and BJ21 encodes H47Q, two proteolytically inactive assemblin mutants in which His47 has been replaced with Ala and Gln, respec-tively. They were made by replacing the small SalI-to-SstII fragment of wild-type BJ1 with the corresponding fragment from plasmids AW44 and AW45, respec-tively (39), which encode H47A.Land H47Q.L.
BJ12 encodes I125-D1292, an assemblin mutant in which five I-site residues have been deleted (i.e., I125NA2AD129). It was made by replacing the small CelII-to-KspI fragment in wild-type BJ1 with the corresponding fragment from plasmid MH9 (39), which encodes I125-D1292.L
MH15 encodes A127Q.L, a point mutation at the I site of pNP1 in which the P1 (for nomenclature, see reference 29) Ala127 was changed to Gln by site-directed mutagenesis, essentially as described elsewhere for the construct MH14 (14). The oligonucleotide used for mutagenesis was 59-CGAGATATAAACCA GGCCGATGGCG-39(the underlined portion encodes the Gln change).
BJ13 encodes A127Q, an Ala1273Gln point mutation at the I site of assem-blin. It was made by replacing the small CelII-to-KspI fragment of wild-type BJ1 with the corresponding fragment from MH15, which encodes A127Q.L (de-scribed above).
Three doubly mutated assemblin constructs were made. MH34 encodes H47A, I125-D1292, which combines the His473Ala point mutation and the I125-D1292 I-site deletion; MH35 encodes H47A, A127Q, which combines the His473Ala and Ala1273Gln point mutations; and MH36 encodes H47Q, A127Q, which combines the His473Gln and Ala1273Gln point mutations. These doubly mutated plasmids were made by replacing the small EcoRV-to-BamHI fragment of BJ20 (encodes H47A) or BJ21 (encodes H47Q) with the corresponding fragment of BJ12 (encodes I125-D1292) or BJ13 (encodes A127Q).
MH43 encodes LM3.S, assemblin with a 15-amino-acid insertion in its amino half. This mutation has been previously described in the full-length proteinase construct, LM3 (40), and was made by replacing the small SalI-to-SstII fragment of BJ1 with the corresponding mutated fragment of LM3. The .Ssuffix denotes the short, NP1nor assemblin form of the enzyme.
AW5 encodes the first 179 amino acids of assemblin, with the addition of Ile, Gln, Thr, and a stop codon at its carboxyl end; its construction has been de-scribed elsewhere (40).
Transfection assay.Calcium phosphate transfection of HEK cells (4) was done essentially as described previously (12, 40). The amount of DNA used in single plasmid transfections was 1mg for the pAP-encoding plasmid or 2 mg for plasmids encoding the proteinase or its mutated forms. In all multiple plasmid transfections, the amount of each plasmid encoding An, Ac, or a mutant form of
the proteinase was 0.5mg and the amount of plasmid encoding pAP was 1.0mg.
Each transfection was done with 0.2mg of a plasmid encoding the simian virus 40 (SV40) large T antigen added to enhance plasmid copy number (12).
Construction and assay of rBVs.rBVs were made with the Baculogold system (catalog no. 21001K; Pharmingen, La Jolla, Calif.) as instructed by the manu-facturer and are referred to by the construct numbers of their recombinant transfer plasmids. SCMV and HCMV genes were introduced into the BV ge-nome by homologous recombination mediated by sequences in the transfer plasmid, pVL1392 or pVL1393 (25).
Construction of the following rBVs has been described previously (14): MH30 encodes wild-type SCMV assemblin with an amino-terminal N9-MHWHWH-C9 peptide fusion added for chromatographic purification (31), BJ28 encodes SCMV An, MH51 encodes SCMV Ac, JB2 encodes SCMV pAP, MH50 encodes
HCMV An, and MH52 encodes HCMV Ac.
MH38 encodes a Ser1183Ala mutation in SCMV assemblin (S118A). This construct was made by removing the small CelII-BamHI fragment from the previously described BJ14 construct (see above) and using it to replace the corresponding wild-type fragment of MH30 (14).
MH40 encodes the same Ser1183Ala mutation as MH38 but in the full-length proteinase (S118A.L). This construct was made by replacing the small wild-type CelII-BamHI fragment of MH30 (14) with the corresponding fragment from AW48 (39).
rBVs were plaque purified twice (3, 33); high-titer stocks were prepared for each rBV (33) and stored at 48C protected from light. Sf9 cells ('2.53105
/well) in 24-well plates (Becton Dickinson) were infected at multiplicity of infection of 5 to 10 by adding 100ml of virus in single infections or 50ml of each virus in coinfections. Infected cells were harvested 3 days after infection with 70ml of 23 sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) sam-ple buffer containing 4% SDS (catalog no. 161-0300; Bio-Rad, Melville, N.Y.), 20%b-mercaptoethanol, 20% glycerol, 50 mM Tris (pH 7.0), and 0.02% bro-mophenol blue. Samples were heated for 3 min in a boiling water bath and stored at2808C until analyzed.
SDS-PAGE and Western immunoassay.SDS-PAGE was done essentially as described by Laemmli et al. (17); the ratio of N,N9-methylenebisacrylamide to acrylamide was 0.735:28.
Western immunoassays were done essentially as described by Towbin et al. (36). A semidry transfer unit with an Immobilon P membrane (Millipore, Bed-ford, Mass.) was used. The buffer was 50 mM Tris–20% methanol, and the time of transfer was calculated by the following formula: gel width3height32.55 mA per 30 min. Membranes were blocked in 10 mM Tris buffer (pH 7.4) containing 0.9% NaCl and 5% bovine serum albumin, reacted sequentially with appropriate antiserum and [125
I]protein A (catalog no. IM 144; Amersham, Arlington Heights, Ill.), and exposed to X-ray film, usually with a calcium tung-state intensifying screen (18).
RESULTS
The SCMV genes, proteins, and landmarks referred to in
this report are diagrammed in Fig. 1. Both plasmid number
and protein name are indicated for reference.
SCMV A
n, A
c, assemblin, pAP, and AP are expressed in
BV-infected insect cells.
As shown before (14) and reported
here for reference (Fig. 2), insect cells infected with the rBVs
used in this work express the expected protein products. When
pAP was expressed alone, no cleavage to AP was detected (Fig.
2, lane 5); when it was expressed with wild-type assemblin, both
expected cleavages (i.e., pAP
3
AP and A
3
A
n1
A
c) were
detected (Fig. 2, lane 6). Expression of pAP together with the
independently expressed subunits, A
nand A
c, resulted in
pAP
3
AP cleavage, due to the spontaneous association of the
A
nand A
csubunits to form a two-chain proteinase (Fig. 2, lane
7) (14). All of these proteins were detected in SCMV-infected
human foreskin fibroblast cells: pAP was detected primarily in
the Nonidet P-40 (NP-40) cytoplasmic fraction (Fig. 2, lane 1);
AP, A
n, and A
cwere found primarily in the NP-40 nuclear
fraction (Fig. 2, lane 2); and assemblin (A) was found to be
approximately evenly split between the two fractions.
SCMV and HCMV A
ncan complement SCMV S118A
mu-tant proteinase.
To determine whether the wild-type A
nand
A
csubunits of assemblin are able to rescue the proteolytic
activity of enzymatically inactive mutants by intermolecular
complementation, we expressed each subunit of HCMV (hA
nand hA
c) and of SCMV (sA
nand sA
c) with the S118A mutant
SCMV proteinase. This mutation eliminates the serine
nucleo-phile and renders the proteinase inactive (5, 39). We tested for
on November 9, 2019 by guest
http://jvi.asm.org/
complementation of both the mature (S118A) and precursor
(S118A.
L) forms of the mutant, as indicated by restoration of
pAP
3
AP processing.
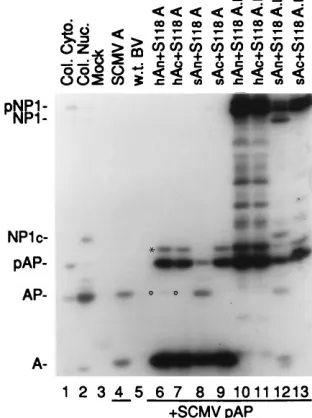
Results of this experiment are summarized in Fig. 3. As
expected, coexpression of pAP and wild-type assemblin
re-sulted in conversion of pAP
3
AP (Fig. 3, lane 4). When the
mutants S118A and S118A.
Lwere expressed with SCMV A
n,
the wild-type subunit that contains the serine nucleophile
mu-tated in the corresponding A
ndomain of the mutants,
conver-sion of pAP
3
AP was readily detected (Fig. 3, lanes 8 and 12,
respectively). Additionally, self-cleavage of the mutant
pro-teinase S118A.
Loccurred at both its M (indicated by
pNP1
3
NP1) and R (evidenced by NP1
3
NP1
c1
A) sites (Fig.
3, lane 12; also see Fig. 1A). Thus, wild-type SCMV A
ncan
rescue both the pAP and self-cleavage activities of the S118A
mutants whose lesion is in CD3 (Fig. 1A). The SCMV A
csubunit did not restore proteolytic activity to either S118A
(Fig. 3, lane 9) or S118A.
L(Fig. 3, lane 13), as indicated by the
absence of pAP
3
AP cleavage.
We have shown previously that the A
nand A
csubunits of
SCMV and HCMV can be interchanged and still yield active
two-chain proteinase (14). For example, a two-chain
protein-ase consisting of the heterologous subunit pair, HCMV A
n/
SCMV A
c, has nearly the same proteolytic activity as one
composed of the homologous pair (e.g., SCMV A
n/SCMV A
c)
(14). We tested the interchangeability of HCMV and SCMV
A
nsubunits in complementing the SCMV proteinase mutants
and found that HCMV A
nis able to substitute for SCMV A
nin complementing the S118A mutant, as evidenced by
pAP
3
AP (Fig. 3, lane 6, open circle), but the efficiency of
heterologous complementation was far below that of the
ho-mologous SCMV A
n(Fig. 3; compare lanes 6 and 8).
Comple-mentation of the mutant precursor enzyme, S118A.
L, by the
HCMV A
nsubunit was below the limits of detection in this
assay system (Fig. 3, lane 10; no pAP
3
AP and no pNP1
3
NP1
1
A
1
NP1
c). The finding that the cross-species HCMV
A
csubunit does not complement the S118A mutants (Fig. 3,
lanes 7 and 11) was expected because the SCMV A
csubunit
did not complement (Fig. 3, lanes 9 and 13).
SCMV A
nand A
csubunits complement other assemblin
mutants.
To examine the intermolecular complementation of
assemblin in greater detail, we used a set of plasmids encoding
inactive assemblins that had been previously made and
char-acterized in transient transfection assays of HEK cells (39).
Expression and detection of assemblin, of its A
nand A
csub-units, and of pAP and AP in transfected HEK cells has been
described elsewhere (14, 40).
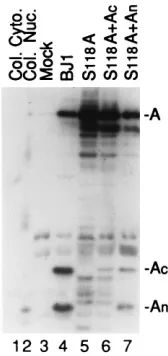
[image:4.612.133.223.70.272.2]In addition to the S118A mutation described above that
inactivates the enzyme by eliminating the nucleophile,
muta-tion of the absolutely conserved histidine in CD2 (His47 in the
SCMV A
ndomain) (19, 39) or deletion of the carboxy-terminal
70 residues of the A
cdomain of SCMV assemblin (construct
AW5) (40) also renders the enzyme inactive. Thus, when the
SCMV assemblin mutant H47A, H47Q, S118A, or AW5 was
coexpressed with pAP in transfection assays, no pAP
3
AP
processing was detected (Fig. 4, lanes 3 to 6). However, when
the three constructs that contain changes in their A
ndomain
(i.e., H47A, H47Q, and S118A) were expressed together with
the SCMV A
nsubunit, each yielded pAP
3
AP cleavage (Fig. 4,
lanes 7, 8, and 10). Thus, point mutations of the essential His47
in SCMV assemblin were also complemented by the SCMV A
nsubunit. Consistent with the results obtained for the S118A
mutants expressed in insect cells (Fig. 3, lanes 9 and 13), the
SCMV A
csubunit did not complement either the His47 or
S118A mutant in these transfection assays (i.e., no pAP
3
AP)
(Fig. 4, lanes 11, 12, and 14). Conversely, when the deletion
FIG. 2. Expression and detection of rBV-expressed proteins. A Western [image:4.612.356.512.426.635.2]im-munoblot probed with a mixture of the anti-N2, anti-N1, and anti-C2 sera is shown. Insect cells were infected with rBVs as described in Materials and Meth-ods and harvested 3 days later. Lysates were separated by SDS-PAGE in a 14% polyacrylamide gel and subjected to Western immunoassay as described in Ma-terials and Methods. pAP was expressed in the preparations contained in lanes 5 to 7; proteins of interest are indicated to the right, abbreviated as in Fig. 1A; the asterisk denotes one of several modified forms of pAP that are detected (12). Mock and w.t. BV are lysates of noninfected Sf9 cells and wild-type BV-infected cells, respectively. NP-40 cytoplasmic and nuclear fractions (Col. Cyto. and Col. Nuc.) of SCMV strain Colburn-infected human foreskin fibroblasts are shown for reference.
FIG. 3. Intermolecular complementation of the inactive S118A mutated forms of the full-length proteinase and assemblin expressed in insect cells. Shown is a fluorogram of a Western immunoblot probed with a mixture of anti-N2, anti-C2, and anti-N1. Samples were harvested and processed as described for Fig. 2; proteins were separated by SDS-PAGE in a 10% polyacrylamide gel and analyzed by Western immunoassay. SCMV pAP was expressed in the prepara-tions in lanes 4 and 6 to 13. Mock and w.t. BV are lysates of noninfected and wild-type BV-infected Sf9 cells; the prefix h or s indicates HCMV or SCMV origin of protein. Proteins of interest are labeled on the left, abbreviated as in Fig. 1A; the asterisk is explained in the legend to Fig. 2; open circles indicate the position of AP. Col. Cyto. and Col. Nuc. are described in the legend to Fig. 2.
on November 9, 2019 by guest
http://jvi.asm.org/
mutant AW5, which lacks 43% of its A
cdomain (i.e., Leu180
to Ala249), was coexpressed with the SCMV A
csubunit,
pAP
3
AP cleavage was detected (Fig. 4, lane 13). As expected,
activity of AW5 was not rescued by the A
nsubunit (Fig. 4, lane
9).
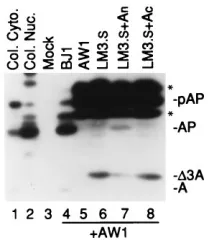
We also tested the ability of the A
nsubunit to complement
an insertion mutation. Previous experiments had shown that
LM3, a full-length proteinase containing a 15-amino-acid
in-sertion between CD1 and CD2 in its A
ndomain, has only
minimal activity (40). LM3.
Scontains the same insertion in
assemblin and is also essentially inactive (i.e., no pAP
3
AP
cleavage) (Fig. 5, lane 6). The insertion also makes the mutant
proteinases larger than the wild-type forms (Fig. 5; compare
D
3A in lane 6 with A in lane 4). Coexpression of LM3.
Swith
the A
nsubunit resulted in rescue of the mutation and cleavage
of pAP to AP (Fig. 5, lane 7). As expected, coexpression of
LM3.
Swith the noncomplementing A
csubunit did not restore
cleavage activity (Fig. 5, lane 8).
Several other inactive SCMV assemblin mutants were
sim-ilarly tested in complementation assays. The results of these
assays are presented in Table 1. Three inactive glycine point
mutations in assemblin (obtained from Carlos Lopez and
El-cira Villarreal, Lilly Research Laboratories), one (G116V) in
the A
ndomain and two (G149V and G216V) in the A
cdomain,
were not complemented by the appropriate wild-type subunit
(e.g., A
ndid not complement G116V). These Gly mutants
were originally made to test the possibility that they are
con-formationally critical residues, and we suspect that their
inabil-ity to be complemented by the A
nor A
csubunit is due to the
fact that they are. But this has not been proven, and other
explanations are possible. We also tried to demonstrate
blin multimer formation directly by testing whether one
assem-blin mutant could complement another. This was done with the
pairs shown in Table 1; we found that (i) two mutant
assem-blins (S118A and H47A), both having changes in their A
ndomain, did not complement one another, whereas (ii) two
mutants with changes in their A
ndomain (H47A) and (S118A)
showed complementation and rescue of their proteolytic
activ-ity when coexpressed with an assemblin mutant having an A
cdomain change (AW5); (iii) no evidence of complementation
was detected between the A
cdomain mutant G216V and
ei-ther of the A
ndomain mutants, S118A or H47A, consistent
with the noncomplementability of the Gly mutants with the A
csubunit.
[image:5.612.384.489.69.189.2]A possible explanation for intermolecular complementation
in these experiments could be homologous recombination
be-tween the plasmids to generate a wild-type assemblin coding
sequence during the transfection. However, because each
mu-tant would have a similar chance of rescue by genetic
recom-bination with the complementing plasmid (particularly the
G116V and S118A mutations, which are within 6 bp), the
failure of several inactive assemblin mutants (e.g., G116V,
G129V, and G216V) to be complemented provides strong
ev-idence that the intermolecular complementation observed
does not result from homologous recombination. The fact that
FIG. 4. Intermolecular complementation of H47, AW5, and S118 mutantsexpressed by transfection of human 293 cells. Shown is a fluorogram prepared from a Western immunoblot probed with a mixture of anti-N2, anti-C2, and anti-N1. Cells were transfected with the indicated plasmids, with or without An
or Ac, and harvested 3 days later. Lysates were subjected to SDS-PAGE in 10%
polyacrylamide gels and analyzed by Western immunoassay as described in Materials and Methods. BJ1 expresses wild-type assemblin and coexpression of An1Acresults in formation of an active two-chain assemblin (14). AW1
[image:5.612.60.298.70.212.2](en-codes the SCMV pAP) was present in all of the transfections. H47 is an abso-lutely conserved histidine found in CD2 (Fig. 1A) that is required for proteolytic activity (39); H47A and H47Q are two inactive mutants. S118 is the serine nucleophile of SCMV assemblin, and the S118A mutant is proteolytically inac-tive (39). AW5 encodes an assemblin mutant that lacks its carboxy-terminal 70 amino acids and is proteolytically inactive (40). The positions of the assembly protein precursor (pAP) and product (AP) and of assemblin (A) are indicated in the right margin and were determined by correspondence with marker proteins (“A” in lane 1; pAP and AP in a lysate of AW1-transfected and NP-40 nuclei of CMV-infected cells, as shown in Fig. 5, lanes 1, 2, 3, and 5). Trace assemblin band in lane 9 is due to spillover from adjacent lane.
FIG. 5. Intermolecular complementation of insertion mutation, LM3.S. Shown is a fluorogram of a Western immunoblot probed with a mixture of anti-N2, anti-C2, and anti-N1. Transfected cells were prepared and processed as described for Fig. 4; lysates were subjected to SDS-PAGE in a 10% polyacryl-amide gel and subjected to Western immunoassay. LM3.Sincludes a 15-amino-acid insertion in assemblin that results in loss of proteolytic activity and a shift in electrophoretic mobility (indicated byD3A). Proteins of interest are indicated to the right, abbreviated as in Fig. 1A; asterisks are explained in the legend to Fig. 2. BJ1 encodes SCMV wild-type assemblin; Mock is a lysate of cells transfected with a plasmid encoding the SV40 large antigen T alone. Preparations in lanes 4 to 8 contained SCMV pAP (encoded by AW1).
TABLE 1. Complementation of SCMV assemblin mutants
aAssemblin mutant
Coexpressed protein
Substrate cleavageb
G116V
cA
n
2
G149V
cA
c
2
G216V
cA
c
2
G216V
cS118A
2
G216V
cH47A
2
S118A
H47A
2
S118A
AW5
1
H47A
AW5
1
aSummary of results from at least three intermolecular complementation
experiments per coexpressed protein pair. Plasmids encoding mutant proteins were cotransfected into HEK cells along with a plasmid encoding SCMV pAP. Cells were harvested, processed, and analyzed by immunoassay with anti-N1 to detect pAP3AP cleavage.
b“1”, pAP3AP cleavage detected;2, no pAP3AP cleavage detected. cAssemblin mutants obtained from Carlos Lopez and Elcira Villarreal, Eli
Lilly Research Laboratories.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.317.555.559.661.2]neither complementation of the LM3.
Sinsertion mutant by A
n(Fig. 5, compare lane 6 with lane 4) nor complementation of
the AW5 deletion mutant by A
cyielded detectable
wild-type-size assemblin (Fig. 4, compare lane 1 with lane 13), even after
much longer exposures, also argues against homologous
re-combination between plasmids as the mechanism of the
inter-molecular complementation observed here.
I-site cleavage is not required for intermolecular
comple-mentation.
Two additional experiments were done to examine
the requirement for I-site cleavage in intermolecular
comple-mentation of assemblin. First, the S118A mutant was
coex-pressed with its complementing subunit, A
n, to determine
whether intermolecular complementation resulted in I-site
cleavage of the mutated enzyme. I-site cleavage was clearly
evidenced in this experiment by the presence of A
c(Fig. 6, lane
7), which could have come only from assemblin cleaved at its I
site. The presence of A
nin this lane is due in part to its
synthesis from plasmid MH28 and in part to assemblin
cleav-age; other bands are either degradation products of the
inac-tive assemblin (also present in Fig. 6, lane 5) or
immunologi-cally cross-reactive host proteins (e.g., two bands above A
care
also present in (Fig. 6, lane 3). Controls showed that there was
no I-site cleavage by the inactive S118A mutant alone (i.e., no
A
nor A
c) (Fig. 6, lane 5) or when it was coexpressed with the
noncomplementing A
csubunit (i.e., no A
nseen in Fig. 6, lane
6; A
cdetected is expressed from noncomplementing MH29
plasmid). Thus, complementation of the S118A assemblin
mu-tant by the A
nsubunit does result in I-site cleavage, yielding
mutant A
nand wild-type A
c. I-site cleavage also occurred
dur-ing complementation of LM3.
Sby the A
nsubunit and explains
the lower amount of
D
3A in lane 7 of Fig. 5 than in lanes 6
(enzyme alone) and 8 (noncomplementing A
csubunit) (data
not shown from Western immunoassays for A
nand A
cdone
with 14% polyacrylamide gels).
The occurrence of I-site cleavage during intermolecular
complementation of assemblin mutants raised the question of
whether this cleavage might be required for the restoration of
proteolytic activity observed. We tested this possibility by
con-structing assemblin mutants that are both proteolytically
inac-tive and noncleavable at their I sites. The H47A and H47Q
assemblin mutants were combined with one of two I-site
mu-tations. The first I-site mutation was a deletion of five I-site
residues. We have shown before that deleting residues Ile125
to Asp129 in the full-length proteinase (I125-D129
2.
L)
elimi-nates I-site cleavage without loss of proteolytic activity (39).
When this deletion was subcloned into assemblin (BJ12), I-site
cleavage was eliminated (data not shown); however, a new
cleavage product was observed (Fig. 7, lane 15, open circle),
which has a counterpart that is detected when the I site is
mutated in HCMV assemblin (16). In HCMV assemblin, this
[image:6.612.136.220.69.247.2]'
24-kDa fragment arises from cleavage at a cryptic (C)
cleav-age site in the sequence VDA209
2
SG (16). A potentially
FIG. 6. Intermolecular complementation of S118A leads to I-site cleavage.Shown is an autoradiogram of a Western immunoblot probed with a mixture of anti-N2 and anti-C2. Transfected cells were prepared and processed as described for Fig. 4; lysates were subjected to SDS-PAGE in a 14% polyacrylamide gel and analyzed by Western immunoassay as described in Materials and Methods. BJ1 encodes SCMV wild-type assemblin. Mock, Col. Cyto., and Col. Nuc. are defined in the legends to Fig. 4 and 5. Proteins of interest are indicated on the right by abbreviations used in Fig. 1A.
FIG. 7. Intermolecular complementation in the absence of I-site cleavage. Shown is an autoradiogram of a Western immunoblot probed with a mixture of anti-N2, anti-C2, and anti-N1. Transfected cells were prepared and processed as described for Fig. 4; lysates were subjected to SDS-PAGE in a 14% polyacrylamide gel, and analyzed by Western immunoassay as described in Materials and Methods. BJ1, Mock, Col. Cyto., and Col. Nuc. are defined in the legends to Fig. 4 and 5. The presence (1) or absence (2) of plasmids encoding SCMV pAP (AW1), An, and Acis indicated below each lane number. I125-D1292indicates the deletion of those residues
that constitute the I site in SCMV assemblin (39). A127 is the P1 (29) residue of the SCMV I site (39). H47 is an absolutely conserved histidine that is essential for proteolytic activity (39). An1Acis a cotransfection of Anand Acthat yields active two-chain assemblin (14). Proteins of interest are indicated to the right, abbreviated
as in Fig. 1A. An open circle is used to mark the position of the new cleavage product resulting from self-cleavage of an I-site mutant at its cryptic site; asterisks are explained in the legend to Fig. 2.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.138.481.493.658.2]homologous sequence in SCMV assemblin, ESSA200
2
AA,
could be the corresponding cryptic site. The cryptic site
cleav-age product is not easily detected when wild-type assemblin is
expressed alone (Fig. 7, lane 2, and reference 16) or in
SCMV-infected human foreskin fibroblasts (Fig. 2, lanes 1 and 2).
The second I-site mutant was a point mutation at the P1 (29)
residue of the I site (i.e., A127Q in SCMV) and had a
pheno-type similar to that of the deletion mutant, that is, no detected
I-site cleavage (data not shown) and the appearance of a new
self-cleavage product (Fig. 7, lane 16, open circle), presumably
resulting from cleavage at the same cryptic site revealed by the
I125-D129
2deletion mutant. The smaller size of the fragment
from the deletion mutant is consistent with the loss of five
amino acids. Both of these I-site cleavage mutants still
pro-cessed pAP
3
AP (Fig. 7, lanes 32 and 33).
The two I-site mutations were combined with the H47A and
H47Q mutations described above to create three double
mu-tations: one in which the I-site residues were deleted from
H47A (i.e., H47A, I125-D129
2), and two in which the I-site
point mutation, A127Q, was combined with either the H47A or
H47Q mutation (H47A, A127Q and H47Q, A127Q,
respec-tively). When each of the doubly mutated assemblins was
ex-pressed alone, neither I-site cleavage (i.e., no A
nor A
cpro-duced) (data not shown) nor C-site cleavage (Fig. 7; compare
lanes 10 to 12 with lanes 15 and 16) occurred, as expected
because of the inactivating H47 mutations. For the same
rea-son, no processing to AP was observed when each mutant was
coexpressed in transfections with pAP (Fig. 7, lanes 17 to 19).
Controls demonstrated pAP
3
AP cleavage by wild-type
assem-blin (Fig. 7, lane 8) and recombinant two-chain assemassem-blin (Fig.
7, lane 9). The presence of a band at the position of assemblin
in the two-chain proteinase sample must be due to spillover
from the adjacent lane, as no corresponding band has been
observed in other experiments (Fig. 2, lane 7, and Fig. 4, lane
2) as well as previously published experiments (see Fig. 3, 4, 6,
7, and 9 in reference 14). Controls also showed that the H47A
and H47Q single mutants were unable to cleave pAP
3
AP
(Fig. 7, lanes 20 and 21) unless they were complemented with
by the A
nsubunit (Fig. 7, lanes 30 and 31).
When the new doubly mutated assemblins were coexpressed
with the A
nsubunit, pAP
3
AP cleavage occurred (Fig. 7, lanes
27 to 29), indicating that intermolecular complementation of
inactive assemblin mutants by the wild-type A
nsubunit does
not require I-site cleavage of the mutant. Interestingly, neither
of these mutants showed C-site cleavage during
complemen-tation, even though both parental I-site mutants did. No
pAP
3
AP cleavage was detected when these mutants were
coexpressed with the A
csubunit (Fig. 7, lanes 22 to 24).
Al-though, in complementations with A
n, the mutants having a
cleavable I site (I site positive) showed more pAP
3
AP than
the double mutants lacking a cleavable I site (I site negative),
they also showed correspondingly more inactive assemblin
(compare intensities of AP and A bands in lanes 30 and 31 of
Fig. 7 with those in lanes 27, 28, and 29). Thus, the enzymatic
efficiency of the complementation reaction, as estimated from
the amount of pAP
3
AP relative to mutant assemblin, was not
appreciably enhanced by the presence of a cleavable I site.
DISCUSSION
We have found that the proteolytic activity of an inactive
CMV assemblin mutant (e.g., S118A) can be rescued by
coex-pressing it with an appropriate fragment or subunit (e.g., A
n)
of wild-type assemblin. Subunit-specific rescue of three point
mutations, an insertion mutation, and a deletion mutation was
demonstrated and shown to occur in both BV-infected insect
cells and plasmid-transfected human cells. These results
indi-cate that the independently cloned A
nand A
csubunits of
assemblin can interact with their partner domains in a mutant
form of the enzyme to restore its proteolytic activity by
inter-molecular complementation.
Intermolecular complementation is characteristic of
pro-teins that form multimers, such as
b
-galactosidase (37, 41) and
interleukin-converting enzyme (13), and its occurrence in the
experiments reported here suggests that assemblin is able to
form multimers. This conclusion, based on interactions of the
proteins in vivo, is consistent with recent evidence from in vitro
experiments that the active form of HCMV assemblin is a
dimer (6, 23). Intermolecular complementation was also
ob-served with an inactive mutant form of the full-length
assem-blin precursor, pNP1 (Fig. 1 and 3), demonstrating that
inter-action of the complementing subunit with assemblin does not
require that the nonproteolytic carboxyl end of the precursor
(e.g., NP1
c) first be shortened or removed by M- or R-site
cleavage (Fig. 1A).
Although the stoichiometry of the wild-type subunit and
mutant assemblin in the complex formed during
intermolecu-lar complementation is unknown, the minimal structure would
be a heterodimer composed of one complementing subunit
and one mutant assemblin molecule, as suggested in the
hypo-thetical model presented in Fig. 8. If assemblin absolutely
requires dimerization to become active (6, 23), then the
prod-ucts of A
nplus mutant assemblin (Fig. 8, line 1) could include
(i) the heterodimer shown, (ii) a homodimer composed of two
mutant assemblin molecules, and (iii) a possible heterotrimer
composed of two mutant assemblins and a complementing
wild-type subunit. If still higher-order complexes can form, the
potential products would be more numerous and complicated.
We demonstrated that formation of the complementation
complex (e.g., mutant assemblin/A
nsubunit shown in Fig. 8,
line 1) results in I-site cleavage of the assemblin mutant (Fig.
6, lane 7) but that this cleavage is not required for proteolytic
activity (Fig. 7, lanes 27 to 29).
When I-site cleavage can occur during complementation, it
could lead to formation of wild-type two-chain enzyme in at
least two ways (Fig. 8). One would be through I-site cleavage of
the active complementation complex, either by a similar
com-plex (Fig. 8, arrow a) or by a wild-type two-chain assemblin that
resulted from complementation (Fig. 8, line 3). A second
would be through (i) sequential cleavage of inactive mutant
assemblin (Fig. 8, line 1), either by a complementation
com-plex (Fig. 8, arrow b) or by a previously formed wild-type
two-chain enzyme, to yield two-chain mutant assemblin (Fig. 8,
line 2), followed by (ii) direct or indirect displacement of the
mutant A
nsubunit by a wild-type recombinant subunit to form
wild-type two-chain assemblin (Fig. 8, arrow c). Although this
figure depicts the hypothetical interactions between monomers
for simplicity of illustration, analogous interactions could take
place between complexes. Our finding that I-site cleavage did
not noticeably enhance the efficiency of pAP
3
AP (Fig. 7)
indicates either that little wild-type two-chain enzyme is
formed in these complementation reactions or that the
enzy-matic activity of the complementation complex is comparable
to that of the wild-type two-chain enzyme. Because rescue of
CMV assemblin by intermolecular complementation did not
require I-site cleavage, similar results would be expected with
the assemblin homologs of non-CMV herpesviruses that have
no I site.
If the complementation complex evidenced here is reflective
of an assemblin dimer structure, then our results make several
predictions. First, because a heterodimer composed of S118A
and an A
nsubunit would have just one S118 nucleophile (i.e.,
on November 9, 2019 by guest
http://jvi.asm.org/
in the complementing subunit) and therefore only one
func-tional active site, our results indicate that the wild-type enzyme
does not require two functional active sites for proteolytic
activity. Second, the inability of two probable active-site
mu-tants, S118A and H47A, to complement one another by
form-ing an active dimer indicates that these putative catalytic
res-idues are not shared in a single active site formed between two
assemblin monomers.
We also showed in this report that intermolecular
comple-mentation can occur between HCMV and SCMV assemblins,
as HCMV A
nrescued the activity of an SCMV assemblin
mutant (Fig. 3). This finding is consistent with our previous
report that HCMV and SCMV assemblin subunits can
com-bine to form active intertypic two-chain proteinase (e.g.,
HCMV A
n/SCMV A
c) (14). However, the extent of SCMV
pAP processing was reduced when HCMV A
nwas used in
place of SCMV A
nto complement mutant SCMV assemblin
(Fig. 3). This contrasted with our finding that pAP cleavage by
an intertypic two-chain assemblin (e.g., HCMV A
n/SCMV A
c)
was comparable to that of the homotypic two-chain enzyme
(i.e., SCMV A
n/A
c) (Fig. 10 in reference 14). We interpret
these results as indicating that intertypic complementation
in-volves competition (by displacement or simple occupancy) of
the complementing subunit (e.g., HCMV A
n) for access to its
partner domain within the assemblin mutant (e.g., SCMV A
c)
and that the decreased pAP processing in these intertypic
complementation experiments may result from the HCMV A
nsubunit being less able than the SCMV A
nsubunit to compete
for SCMV A
cinteractions. This finding implies that there are
differences in the interactions between HCMV and SCMV
subunits. These differences were not apparent in our earlier
studies (14) on the formation of cross-species two-chain
re-combinant assemblins, presumably because no competing
same-species domains were present.
The in vivo intermolecular complementation of CMV
as-semblin mutants by wild-type asas-semblin subunits reported
here, and the recent reports that purified HCMV assemblin
functions as a dimer in vitro (6, 23), indicates that the
protein-ase can form active multimers. Although the biological
sig-nificance of these higher-order structures remains to be
determined, the finding that the pAP substrate of assemblin
interacts with itself to form multimers (42) raises the possibility
that assemblin dimerization both activates it and promotes
more efficient recognition and cleavage of its multimeric
sub-strate. A better understanding of the structure of these
assem-blin complexes and how they form is likely to contribute to our
understanding of how this novel proteinase works.
ACKNOWLEDGMENTS
We thank Jenny Borchelt, Kendra Clopper, and Becky Magno for
excellent technical assistance, and we thank Carlos Lopez and Elcira
Villarreal, Eli Lilly Research Laboratories, for help in obtaining the
antipeptide antisera anti-N2 and anti-C2 as well as for providing the
three glycine mutants of SCMV assemblin (G116V, G149V, and
G216V).
M.R.T.H. was a fellow in the Biochemistry, Molecular, and Cell
Biology Training Program. This work was supported by Public Health
Service research grants AI13718 and AI32957.
REFERENCES
1. Baum, E. Z., G. A. Bebernitz, J. D. Hulmes, V. P. Muzithras, T. R. Jones, and Y. Gluzman.1993. Expression and analysis of the human cytomegalovirus UL80-encoded protease: identification of autoproteolytic sites. J. Virol. 67: 497–506.
2. Burck, P. J., D. H. Berg, T. P. Luk, L. M. Sassmannshausen, M. Wakulchik, G. W. Becker, D. P. Smith, H. M. Hsiung, W. Gibson, and E. C. Villarreal. 1994. Human cytomegalovirus proteinase: purification of the enzyme and determination of its activity using peptide substrates that mimic its native cleavage sites. J. Virol. 68:2937–2946.
3. Carrascosa, A. L. 1994. Enhancement of baculovirus plaque assay in insect cell monolayers by DEAE-dextran. BioTechniques 16:1078–1081, 1083–1085.
4. Chen, C., and H. Okayama. 1987. High-efficiency transformation of mam-FIG. 8. Hypothetical model of intermolecular complementation for CMV assemblin. For simplicity of illustration, heterodimers are used to illustrate the multimeric forms. Line 1 depicts the interaction of a wild-type assemblin subunit (i.e., An) with an enzymatically inactive assemblin mutant, having a genetic lesion (indicated by
the lollipop) in its Andomain, to form an active complementation complex. The molecular mechanism by which the wild-type subunit interacts with its partner domain
(i.e., Acdomain in this diagram) remains to be established. Once formed, the active complementation multimer could cleave other heterodimers (path a) at their I site
(filled square) to form wild-type two-chain enzyme plus mutant subunit (line 3). Alternatively, it could cleave one-chain mutant assemblin (path b) to form two-chain mutant assemblin (line 2), which could then interact with wild-type An(path c) to form wild-type two-chain assemblin plus mutant subunit (line 3). Whether, and how,
wild-type two-chain assemblin is formed in this complementation remains to be determined. The active enzymes generated by this complementation then carry out assemblin-specific cleavages.
on November 9, 2019 by guest
http://jvi.asm.org/
malian cells by plasmid DNA. Mol. Cell. Biol. 7:2745–2752.
5. Cox, G. A., M. Wakulchik, L. M. Sassmannshausen, W. Gibson, and E. C. Villarreal.1995. Human cytomegalovirus proteinase: candidate glutamic acid identified as third member of putative active site triad. J. Virol. 69: 4524–4528.
6. Darke, P. L., J. L. Cole, L. Waxman, D. L. Hall, M. K. Sardana, and L. C. Kuo.1996. Active human cytomegalovirus protease is a dimer. J. Biol. Chem. 271:7445–7449.
7. Desai, P., S. C. Watkins, and S. Person. 1994. The size and symmetry of B capsids of herpes simplex virus type 1 are determined by the gene products of the UL26 open reading frame. J. Virol. 68:5365–5374.
8. DiIanni, C. L., D. A. Drier, I. C. Deckman, P. J. McCann, F. Liu, B. Roizman, R. J. Colonno, and M. G. Cordingley.1993. Identification of the herpes simplex virus type 1 protease cleavage sites by direct sequence analysis of autoproteolytic cleavage products. J. Biol. Chem. 268:2048–2051. 9. DiIanni, C. L., J. T. Stevens, M. Bolgar, D. R. O’Boyle, S. P. Weinheimer,
and R. J. Colonno.1994. Identification of the serine residue at the active site of the herpes simplex virus type I protease. J. Biol. Chem. 269:12672–12676. 10. Gao, M., L. Matusick-Kumar, W. Hurlburt, S. F. DiTusa, W. W. Newcomb, J. C. Brown, P. J. McCann, I. Deckman, and R. J. Colonno.1994. The protease of herpes simplex virus type 1 is essential for functional capsid formation and viral growth. J. Virol. 68:3702–3712.
11. Gibson, W., A. R. Welch, and M. R. T. Hall. 1995. Assemblin, a herpes virus serine maturational proteinase and new molecular target for antivirals. Per-spect. Drug Discov. Des. 2:413–426.
12. Gibson, W., A. R. Welch, and J. Ludford. 1994. Transient transfection assay of the herpesvirus maturational proteinase, assemblin. Methods Enzymol. 244:399–411.
13. Gu, Y., J. Wu, C. Faucheu, J.-L. Lalanne, A. Diu, D. J. Livingston, and M. S.-S. Su.1995. Interleukin-1bconverting enzyme requires oligomeriza-tion for activity of processed forms in vivo. EMBO J. 14:1923–1931. 14. Hall, M. R. T., and W. Gibson. 1996. Cytomegalovirus assemblin: the amino
and carboxyl domains of the proteinase form active enzyme when separately cloned and coexpressed in eukaryotic cells. J. Virol. 70:5395–5404. 15. Holwerda, B. C., A. J. Wittwer, K. L. Duffin, C. Smith, M. V. Toth, L. S. Carr,
R. C. Wiegand, and M. L. Bryant.1994. Activity of two-chain recombinant human cytomegalovirus protease. J. Biol. Chem. 269:25911–25915. 16. Jones, T. R., L. Sun, G. A. Bebernitz, V. P. Muzithras, H.-J. Kim, S. H.
Johnston, and E. Z. Baum.1994. Proteolytic activity of human cytomegalo-virus UL80 proteinase cleavage site mutants. J. Virol. 68:3742–3752. 17. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of
the head of bacteriophage T4. Nature (London) 227:680–685.
18. Laskey, R. A., and A. D. Mills. 1977. Enhanced autoradiographic detection of32
P and125
I using intensifying screens and hypersensitized film. FEBS Lett. 82:314–316.
19. Liu, F., and B. Roizman. 1992. Differentiation of multiple domains in the herpes simplex virus 1 protease encoded by the UL26 gene. Proc. Natl. Acad. Sci. USA 89:2076–2080.
20. Liu, F., and B. Roizman. 1991. The herpes simplex virus 1 gene encoding a protease also contains within its coding domain the gene encoding the more abundant substrate. J. Virol. 65:5149–5156.
21. Liu, F., and B. Roizman. 1991. The promoter, transcriptional unit, and coding sequence of herpes simplex virus 1 family 35 proteins are contained within and in frame with the UL26 open reading frame. J. Virol. 65:206–212. 22. Long, E. O., S. Rosen-Bronson, D. R. Karp, M. Malnati, R. P. Sekaly, and D. Jaraquemada.1991. Efficient cDNA expression vectors for stable and tran-sient expression of HLA-DR in transfected fibroblast and lymphoid cells. Hum. Immunol. 31:229–235.
23. Margosiak, S. A., D. L. Vanderpool, W. Sisson, C. Pinko, and C. C. Kan. 1996. Dimerization of the human cytomegalovirus protease—kinetic and biochemical characterization of the catalytic homodimer. Biochemistry 35: 5300–5307.
24. O’Boyle, D. R., K. Wager-Smith, J. T. Stevens, and S. P. Weinheimer. 1995.
The effect of internal autocleavage on kinetic properties of the human cytomegalovirus protease catalytic domain. J. Biol. Chem. 270:4753–4758. 25. O’Reilly, D. R., L. K. Miller, and V. A. Luckow. 1992. Baculovirus expression
vectors: a laboratory manual. W. H. Freeman and Company, New York, N.Y.
26. Preston, V. G., J. A. Coates, and F. J. Rixon. 1983. Identification and characterization of a herpes simplex virus gene product required for encap-sidation of virus DNA. J. Virol. 45:1056–1064.
27. Preston, V. G., F. J. Rixon, I. M. McDougall, M. McGregor, and M. F. Al Kobaisi.1992. Processing of the herpes simplex virus assembly protein ICP35 near its carboxy terminal end requires the product of the whole of the UL26 reading frame. Virology 186:87–98.
28. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
29. Schechter, I., and A. Berger. 1967. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27:157–162.
30. Schenk, P., A. S. Woods, and W. Gibson. 1991. The 45-kDa protein of cytomegalovirus (Colburn) B-capsids is an amino-terminal extension form of the assembly protein. J. Virol. 65:1525–1529.
31. Smith, M. C., T. C. Furman, T. D. Ingolia, and C. Pidgeon. 1988. Chelating peptide immobilized metal ion affinity chromatography. A new concept in affinity chromatography for recombinant proteins. J. Biol. Chem. 263:7211– 7215.
32. Stevens, J. T., C. Mapelli, J. Tsao, M. Hail, D. O’Boyle, S. P. Weinheimer, and C. L. DiIanni.1994. In vitro proteolytic activity and active-site identifi-cation of the human cytomegalovirus proteinase. Eur. J. Biochem. 226:361– 367.
33. Summers, M. D., and G. E. Smith. 1987. A manual of methods for baculo-virus vectors and insect cell culture procedures. Texas Agricultural Experi-ment Station, College Station, Tex.
34. Tatman, J. D., V. G. Preston, P. Nicholson, R. M. Elliott, and F. J. Rixon. 1994. Assembly of herpes simplex virus type 1 capsids using a panel of recombinant baculoviruses. J. Gen. Virol. 75:1101–1113.
35. Thomsen, D. R., L. L. Roof, and F. L. Homa. 1994. Assembly of herpes simplex virus (HSV) intermediate capsids in insect cells infected with re-combinant baculoviruses expressing HSV capsid proteins. J. Virol. 68:2442– 2457.
36. Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350–4354.
37. Ullmann, A., F. Jacob, and J. Monod. 1967. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal seg-ment of theb-galactosidase structure gene of Escherichia coli. J. Mol. Biol. 24:339–343.
38. Welch, A. R., L. M. McNally, and W. Gibson. 1991. Cytomegalovirus assem-bly protein nested gene family: four 39-coterminal transcripts encode four in-frame, overlapping proteins. J. Virol. 65:4091–4100.
39. Welch, A. R., L. M. McNally, M. R. T. Hall, and W. Gibson. 1993. Herpes-virus proteinase: site-directed mutagenesis used to study maturational, re-lease, and inactivation cleavage sites of precursor and to identify a possible catalytic site serine and histidine. J. Virol. 67:7360–7372.
40. Welch, A. R., A. S. Woods, L. M. McNally, R. J. Cotter, and W. Gibson. 1991. A herpesvirus maturational protease, assemblin: identification of its gene, putative active site domain, and cleavage site. Proc. Natl. Acad. Sci. USA 88:10792–10796.
41. Welply, J. K., A. V. Fowler, and I. Zabin. 1981.b-Galactosidasea -comple-mentation. J. Biol. Chem. 256:6811–6816.
42. Wood, L. J., M. K. Baxter, S. M. Plafker, and W. Gibson. 1997. Human cytomegalovirus capsid assembly protein precursor (pUL80.5) interacts with itself and with the major capsid protein (pUL86) through two different domains. 71:179–190.