0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
Serial Passage of Tobacco Rattle Virus under Different
Selection Conditions Results in Deletion of Structural and
Nonstructural Genes in RNA 2
CARMEN HERNANDEZ,1JAN E. CARETTE,1DEREK J. F. BROWN,2
ANDJOHN F. BOL1*
Institute of Molecular Plant Sciences, Gorlaeus Laboratories, Leiden University, 2333 CC Leiden, The Netherlands,1 and Scottish Crop Research Institute, Invergowrie, Dundee DD2 5DA, Scotland2
Received 3 January 1996/Accepted 29 April 1996
The RNA genome of tobacco rattle virus (TRV) is bipartite. RNA 2 of the nematode-transmissible TRV isolate PPK20 encodes the viral coat protein (cp) and proteins with molecular weights of 29,400 and 32,800 (29.4K and 32.8K proteins). When this isolate was serially passaged in tobacco by using phenol-extracted RNA as the inoculum in each transfer, defective interfering (DI) RNAs rapidly accumulated. A number of these DI RNAs were cloned. Six DI RNAs had single internal deletions in RNA 2 that removed most of the cp gene, the
29.4K gene, and the 5*half of the 32.8K gene. The borders of the deletions in these DI RNAs were found to be
flanked in the genomic RNA 2 by short nucleotide repeats or sequences resembling the 5*end of TRV genomic
and subgenomic RNAs. Two DI RNAs were found to be recombinants containing a 5*sequence derived from
RNA 2 and a 3*sequence derived from RNA 1. When serial passage of TRV isolate PPK20 was carried out by
using leaf homogenates as inocula in each transfer, accumulation of a DI RNA (designated D7) with a
functional cp gene was observed. The deletion in D7 covered the 3*end of the cp gene, the 29.4K gene, and the
5*half of the 32.8K gene. An infectious cDNA clone of D7 RNA was made. In mixed infections, D7 RNA rapidly
outcompeted RNA 2 but did not compete with RNA 1. The deletion in D7 RNA abolished the nematode transmissibility of the PPK20 isolate. These results may explain the observation that many laboratory isolates of tobraviruses have lost their nematode transmissibility and contain RNA 2 molecules of widely different lengths.
Although the presence of defective interfering (DI) RNAs was initially associated with animal RNA virus groups (16), several subsequent reports indicate that the generation of DI RNAs is also a general phenomenon among plant RNA vi-ruses. The DI RNAs are derived from the parental virus ge-nome by internal deletions, sometimes accompanied by com-plex rearrangements, and maintain the cis-acting elements required for replication. The accumulation of the parental virus is affected negatively by the DI RNAs, although defective RNAs with no apparent effect on the genomic RNAs have also been reported. Examples of DI or defective RNAs have been found for members of the carmoviruses (22, 23), tombusviruses (4, 15), potexviruses (38), hordeiviruses (18), rhabdoviruses (17), furoviruses (3, 6), closteroviruses (27), bromoviruses (34), and, recently, cucumoviruses (10). In some cases, the presence of DI RNAs notably influenced symptom development (6, 22, 34).
Tobacco rattle virus (TRV), the type member of the tobra-virus group, has a bipartite genome consisting of single-stranded, positive-sense RNAs. The larger genomic RNA, des-ignated RNA 1, is highly homologous between isolates and contains four open reading frames (ORFs) (11). The first two ORFs encode a protein with a molecular weight of 134,000 (134K protein) and a 194K protein, which is produced by read-through translation of the 134K gene. The two products are expressed directly from the genomic RNA and have a putative function in viral RNA replication. The third ORF
encodes a 29K protein with a possible role in cell-to-cell move-ment of the virus. Finally, the 39-proximal ORF encodes a small cysteine-rich 16K protein, the function of which is un-known. The 29K and 16K proteins are translated from sub-genomic messengers, referred to as 1a (1.5 kb) and 1b (0.7 kb), respectively.
The genomic RNA 2 contains the coat protein (cp) gene, which is expressed via a subgenomic mRNA (RNA 2a). In contrast to the similarity in nucleotide sequence and length between RNA 1 molecules of different TRV isolates, RNA 2 molecules show a high degree of variation due to (i) the pos-sible presence of additional ORFs of unknown function down-stream from the cp gene and (ii) a variably sized 39-terminal region homologous to that of the corresponding RNA 1 mol-ecule (26). RNA 1 and RNA 2 are separately encapsidated in long and short rod-shaped particles, respectively (12). As a consequence of the TRV genomic organization, RNA 1 can replicate and spread in plants in the absence of RNA 2, giving rise to NM (nonmultiplying) infections, in which particles are not formed, in contrast to M (multiplying) infections, in which both genomic components are present and, thus, viral particles are generated.
TRV is transmitted in the field by soil-inhabiting nematodes of the genera Trichodorus and Paratrichodorus (29). The factor determining vector transmissibility is encoded by RNA 2 (31). Recently, we have determined the sequence of RNA 2 of TRV isolate PPK20 (14), which is transmitted by Paratrichodorus pachydermus. In addition to the cp gene, PPK20 RNA 2 (3,856 nucleotides [nt]) contains two ORFs potentially encoding 29.4K and 32.8K proteins. Available evidence indicates that these ORFs may have a role in transmission of the virus by nematodes (25).
Studies done in the sixties (13, 24) showed that some TRV
* Corresponding author. Mailing address: Gorlaeus Laboratories, Leiden University, Einsteinweg 55, 2333 CC Leiden, The Netherlands. Phone: 31-71-5274749. Fax: 31-71-5274340. Electronic mail address: [email protected].
4933
on November 9, 2019 by guest
http://jvi.asm.org/
quences whose properties are consistent with those of DI RNAs. Different types of DI RNAs were generated when TRV isolate PPK20 was serially passaged in tobacco by mechanical inoculation using phenol extracts or leaf homogenates from infected plants as inocula. These DI RNAs contained internal deletions in RNA 2 or were generated by recombination be-tween RNAs 1 and 2. An infectious clone of one of the DI RNAs was constructed and used to analyze the effect of a deletion in RNA 2 on nematode transmissibility of the TRV isolate.
MATERIALS AND METHODS
Virus isolate, clones, and plant inoculations.TRV isolate PPK20 (30) was propagated in a greenhouse by mechanical sap inoculation onto Nicotiana
taba-cum (cv. Samsun NN) plants. Purified RNA 1 of isolate PPK20 was used to
produce nonmultiplying infections on tobacco plants, and this type of infection was maintained by using total RNA extracts from the primary infected leaf material as inocula.
The construction of the clone pCaK20-2T7, containing an infectious cDNA of PPK20 RNA 2 inserted between the cauliflower mosaic virus 35S promoter and the terminator sequence for the nopaline synthase gene (nos), has been de-scribed previously (14). An additional infectious full-length cDNA clone of PPK20 RNA 2, designated pCaK20-2T16, was used in this study. This clone was made by replacing an internal region of the insert in pCaK20-2T7 (a BglII/NruI fragment spanning positions 1197 to 3066) by the corresponding one of a partial-length PPK20 RNA 2 cDNA. As a consequence of this replacement, the se-quence of the viral cDNA inserted into pCaK20-2T16 differs from that of pCaK20-2T7 at two nucleotide (G-to-A changes at positions 1782 and 1884).
pCaK20-2T7 or pCaK20-2T16 was digested with BglI, which cleaves at posi-tions 1,380 bp upstream of the 35S promoter and 150 bp downstream of the nos terminator. After digestion, 20mg of either plasmid was combined with 300mg of total RNA extracted from tobacco leaves infected with PPK20 RNA 1 in 300
ml of 18 mM sodium phosphate buffer (pH 7.0) and these mixtures were used as inocula. Three half-leaves of four N. tabacum (cv. Samsun NN) plants were dusted with Carborundum and inoculated with 20ml of each mixture. At 3 days after inoculation virus infection symptoms became apparent and leaf samples were harvested for further analysis.
Serial passage experiments were performed with the following as inocula: (i) portions (25mg per half-leaf) of total RNA extracted from infected tissue or (ii) homogenates prepared by grinding in a mortar 0.5 g of infected leaf material together with 1 ml of 18 mM sodium phosphate buffer (pH 7.0).
RNA analysis.Total RNA from inoculated leaves was isolated by phenol extraction and lithium precipitation (36). Four micrograms of RNA was dena-tured by glyoxal-dimethyl sulfoxide treatment, electrophoresed in 1% agarose gels, blotted to Hybond-N1membranes (Amersham), cross-linked by UV irra-diation, and hybridized with32P-labelled cDNA fragments. The filters were
pretreated with hybridization buffer (0.5 M sodium phosphate buffer [pH 7.2], 1% bovine serum albumin, 1 mM EDTA, 7% sodium dodecyl sulfate [SDS]) at 658C for 30 min (37), and radioactive probes, prepared by random oligonucle-otide priming (8), were added in the same buffer at 658C. The probes are described in the legends to Fig. 1 and 5.
Protein analysis.Total plant protein preparations were obtained by grinding 0.4 g of leaf tissue in a mortar with 800ml of PEN buffer (10 mM NaH2PO4, 1
mM EDTA, 1 mM sodium azide [pH 7.0]). Five hundred microliters of Laemmli loading buffer (50 mM Tris-HCl [pH 6.8], 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol) was added to 250ml of the leaf homogenate, and insoluble material was eliminated by centrifugation for 5 min at 6,500 rpm in an Eppendorf centrifuge. The proteins were separated by SDS-polyacrylamide gel electrophoresis in 11% polyacrylamide gels and analyzed by the Western blot (immunoblot) technique (35) using antiserum raised against the cp of TRV isolate PSG, which is serologically related to isolate PPK20.
Reverse transcriptase PCR (RT-PCR) analysis of infected plants.First-strand cDNA synthesis was done by using 1mg of total RNA from infected plants as a template and oligonucleotide TRV3 (59-ACTCGAGGGCGTAATAACGC-39) as a primer. The 39-terminal 14 nucleotides of this primer are complementary to the 39-terminal sequence of all known TRV RNAs; the 59-terminal 7 nucleotides of the primer contain an XhoI-restriction site. First-strand cDNA synthesis was carried out at 428C for 60 min in a 20-ml reaction mixture containing 13avian myeloblastosis virus reaction buffer (Promega); dATP, dCTP, dGTP, and dTTP at 1 mM each; 10 U of RNasin; 10 U of avian myeloblastosis virus reverse transcriptase (Promega); and 100 pmol of oligonucleotide TRV3. After
inacti-9 9
end of TRV RNA 1; the 59-terminal 7 nt contain an XbaI restriction site. PCR was carried out under the following conditions: 40 cycles of 1 min at 948C, 1 min at 508C, and 2.5 min at 728C. The products of the reactions were analyzed by agarose gel electrophoresis.
Cloning and sequencing of DI RNA cDNAs.Several of the RT-PCR products, corresponding to double-stranded cDNAs of DI RNAs, were gel purified and ligated into the pGEM-T vector (Promega) under the conditions recommended by the manufacturer. Restriction enzyme digestion of cDNA clones was used to provisionally map the sizes and locations of deletions. Subsequently, primers were designed to determine the precise deletion junction within each clone by sequence analysis using the T7 DNA polymerase kit from Pharmacia.
Nematode transmission studies.A virus-free, mixed population of P.
pachy-dermus and Trichodorus primitivus, at a ratio of 2:1, from Woodhill, Scotland, was
used to determine the vector transmissibility of RNA D7. The virus transmission experiments were done, with wild-type PPK20 as the control virus, by standard procedures (25) using Nicotiana clevelandii source and bait plants grown singly in 25-cm3
plastic pots. Groups of 60 nematodes were placed into each pot, and an
N. clevelandii source plant which, 2 days previously, had been mechanically
inoculated with virus was added. The nematodes were recovered after 4 weeks of access to the roots of the virus-infected plants and added to a new pot containing a healthy N. clevelandii plant. After a further period of 4 weeks, the plants were removed from the pots and their roots were thoroughly washed and then tritu-rated with a mortar and pestle. The resultant suspension was rubbed by finger onto Corundum-dusted leaves of Chenopodium quinoa and Chenopodium
ama-ranticolor virus indicator plants. RNA was recovered from leaves of plants
show-ing symptoms of virus infection and examined by Northern (RNA) blottshow-ing to confirm the identity of the transmitted virus.
RESULTS
Detection of DI RNAs after serial passages using total RNA
extracts as inocula.Initially, four tobacco plants were
inocu-lated with the wild-type PPK20 virus and the infection was subjected to three serial passages. The inoculum for each pas-sage was a total RNA extract obtained from 10 g of infected leaf material from the preceding passage. The presence of viral RNA molecules was analyzed by Northern blot hybridization as shown in lanes 1 to 4 of Fig. 1. The gel shown in Fig. 1A was hybridized with a cDNA probe corresponding to a region of the 16K gene present at the 39end of the genomic RNA 1. This probe detects RNA 1 and the subgenomic RNAs 1a and 1b. The accumulation of RNA 2-specific molecules is shown in Fig. 1B to D, whose gels were probed with cDNA fragments de-rived from the cp gene, the 29.4K gene, and the 32.8K gene of PPK20-RNA 2, respectively.
In the plants infected with the wild-type virus, the expected pattern of genomic and subgenomic RNAs for TRV isolate PPK20 (14) was detected (Fig. 1, lane 1). However, after sev-eral passages differently sized viral RNAs became detectable. The probe derived from the 16K gene detected molecules migrating between the subgenomic RNAs 1a and 1b after the second and third passages (Fig. 1A, lanes 3 and 4). Moreover, the use of cDNA probes derived from different regions of PPK20 RNA 2 showed the presence of new molecules contain-ing RNA 2-specific sequences after a scontain-ingle passage (Fig. 1B, lane 2). When a second passage was carried out, these mole-cules became predominant (Fig. 1B and D, lanes 3), and no full-length genomic RNA 2 was detectable after a third passage (Fig. 1B to D, lanes 4). The patterns obtained with the three RNA 2-specific probes indicated that (i) some of the newly formed RNA 2-derived molecules contained at least part of the cp gene (Fig. 1B, lanes 2 to 4); (ii) a high percentage of these molecules, and particularly those with a size of approx-imately 1.3 kb, were carrying sequences corresponding to that of the 32.8K gene (Fig. 1D, lanes 2 to 4); and (iii) most of the
on November 9, 2019 by guest
http://jvi.asm.org/
new RNAs were lacking at least part of the 29.4K gene, since they were not detectable with the probe corresponding to this gene (Fig. 1C). Together, the results from Fig. 1B to D indicate that when phenol-extracted RNA is used as the inoculum for serial passages, RNA 2 molecules carrying internal deletions accumulate quickly. In addition, the results with the probe corresponding to the 16K gene (Fig. 1A) indicate that new RNA molecules with RNA 1-derived sequences were gener-ated in this experiment.
In a second set of experiments, the infection was initiated with a combination of PPK20 RNA 1 and the infectious cDNA clone of PPK20 RNA 2, pCaK20-2T16 (Fig. 1, lanes 5). After transfers using total RNA extracts as inocula, the presence of small RNA 2-derived molecules was observed (Fig. 1B and D, lanes 6 to 8). These newly formed molecules were more ho-mogeneous in size than those observed in the first set of ex-periments, in which the primary plants were infected with the wild-type virus (Fig. 1B; compare lanes 2 to 4 with lanes 6 to 8). As in the first experiment, molecules migrating between RNAs 1a and 1b were detected in the second experiment when the 16K probe was used (Fig. 1A, lanes 6 to 8).
RT-PCR analysis of the defective RNAs.Total RNA extracts
from the plants analyzed for Fig. 1 were used as templates to perform an RT-PCR analysis using the oligonucleotides TRV3 (corresponding to the 39 end of TRV RNAs 1 and 2) and TRV5 (corresponding to the 59end of TRV RNA 2) as down-stream and updown-stream primers, respectively. The products of each reaction were analyzed on agarose gels, and the results
are shown in Fig. 2. As expected, products were not generated when total RNA extracts from plants infected only with the genomic RNA 1 were used for the RT-PCRs (Fig. 2, lane 0). When the analysis was done on plants infected with the wild-type RNAs 1 and 2, a DNA fragment corresponding to the genomic RNA 2 was amplified. However, at least two addi-tional bands migrating faster than the full-length RNA 2 prod-uct (with estimated sizes of 3.0 and 1.5 kb) were also generated (Fig. 2, lane 1). This indicates the accumulation of small RNA 2-derived molecules in the primary infected plants at low levels that were not detectable by Northern blot hybridization (Fig. 1B to D, lanes 1). After the first passage, three major cDNA products with sizes of about 1.5, 1.3, and 1.0 kb were generated by RT-PCR (Fig. 2, lane 2), although faint bands correspond-ing to longer products were also present. Despite the presence of genomic RNA 2 in the first passage as revealed by Northern blot analysis, no DNA copies of this RNA were generated by RT-PCR. Apparently, under the conditions used, the DNA copies of the smaller RNA molecules are preferentially ampli-fied. Similar electrophoretic patterns were observed for the second and third passages of the infection (Fig. 2, lanes 3 and 4).
For plants infected by inoculation with a mixture of PPK20 RNA 1 and the full-length cDNA clone of PPK20 RNA 2, pCaK20-2T16, the analysis on agarose gels of the RT-PCR products revealed the presence of molecules smaller than the genomic RNA 2 (Fig. 2, lane 5). After serial passage, these small molecules accumulated at high levels (Fig. 2, lanes 6 to 8), in agreement with the results of the Northern blot analysis (Fig. 1B and D, lanes 6 to 8). After three passages a predom-inant RT-PCR product of about 1.2 kb, probably correspond-ing to the major defective RNA in the population, was ob-tained (Fig. 2, lane 8). Apparently, the defective RNAs derived from the inoculum containing the infectious clone of RNA 2 were more homogeneous in size (Fig. 2, lane 8) than those generated with the inoculum containing wild-type RNA 2 (Fig. 2, lane 4).
When a similar RT-PCR analysis was performed with the downstream primer TRV3 and the upstream primer TRV51 (corresponding to the 59 end of RNA 1), products were not generated from any of the passages (data not shown). This indicates that small defective RNAs with the 59-terminal se-quence of RNA 1 do not accumulate during passage at a level detectable by the PCR technique. Under the conditions used, no full-length copies of RNA 1 were amplified by RT-PCR with the primer set TRV3-TRV51 (data not shown).
Cloning and sequencing of DI RNAs.In order to determine
[image:3.612.61.294.71.244.2]which regions of the PPK20 RNA 2 have been lost in DI RNAs
FIG. 1. Accumulation of defective RNAs after serial passage of TRV PPK20 in tobacco plants using phenol-extracted RNA as the inoculum. Northern blots were loaded with total RNA extracted from plants infected with RNA 1 only (lanes 0), wild-type RNAs 1 and 2 (lanes 1 to 4), and wild-type RNA 1 plus progeny RNA of the cDNA 2 clone pCaK20-2T16 (lanes 5 to 8). Lanes 1 and 5 were loaded with RNA extracted from primary inoculated plants. Lanes 2, 3, and 4 show the accumulation of RNAs after one, two, and three mechanical transfers, respectively, of the infection initiated by wild-type RNAs 1 and 2. Lanes 6, 7, and 8 show the accumulation of RNAs after one, two, and three mechanical transfers, respectively, of the infection initiated by the mixture of wild-type RNA 1 and pCaK20-2T16. For each transfer, phenol-extracted RNA was used as the inoc-ulum. The blot was hybridized with32
P-labelled probes corresponding to nt 1528 to 1789 of RNA 2 of TRV isolate PLB (16K gene-derived cDNA probe) (A), nt 596 to 1148 of PPK20 RNA 2 (cp gene-derived cDNA probe) (B), nt 1555 to 2192 of PPK20 RNA 2 (29.4K gene-derived cDNA probe) (C), and nt 3066 to 3417 of PPK20 RNA 2 (32.8K gene-derived cDNA probe) (D). The positions of RNAs 1, 1a, and 1b are indicated in the left margin of panel A. The positions of wild-type RNA 2 and RNA 2a (the subgenomic cp mRNA) are indicated in the left margins of panels B to D. The arrowhead in the left margin of panel D indicates the position of the putative subgenomic mRNA involved in expression of the 32.8K gene. Note: as the 16K gene is not present in RNA 2 of TRV isolate PPK20, only RNAs with RNA 1-specific sequences were detected for panel A.
FIG. 2. RT-PCR analysis of TRV-infected tobacco plants. For lanes 0 to 8, total RNA extracts contained in lanes 0 to 8, respectively, of the Northern blots shown in Fig. 1 were used as templates in RT-PCRs using primers TRV3 and TRV5. The double-stranded cDNA products were electrophoresed in a 1.2% agarose gel and stained with ethidium bromide. Lane M, DNA size markers (in kilobase pairs) corresponding tolDNA digested with EcoRI and HindIII. The arrowhead on the left indicates the position of the full-length PPK20 cDNA 2.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.340.528.74.170.2]derived from this molecule, RT-PCR products generated with primers TRV3 and TRV5 were eluted from the gel and in-serted in the pGEM-T vector. The viral cDNAs that were cloned corresponded to those observed in lanes 4 and 5 of Fig. 2, with estimated sizes of 1.5, 1.3, and 1.0 kb, and the major product of lane 7, with a size of around 1.2 kb. Although these RT-PCR products migrated as more or less discrete bands on agarose gels, some variation in the size of the inserts was observed after cloning, indicating the presence of a heteroge-neous population of cDNAs in the eluted bands.
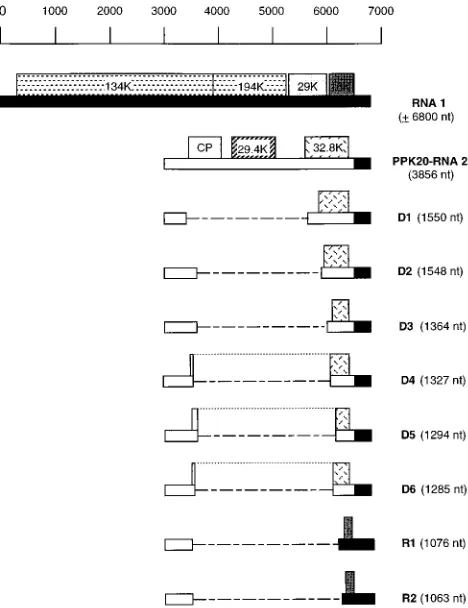
Several clones were chosen for further characterization. Re-striction enzymes with target sequences inside the full-length cDNA clone of RNA 2 were used to perform a restriction map analysis of the inserts. The results of these analyses allowed the choice of proper oligonucleotides to determine the deletion junction within each clone by partial sequencing. The charac-terization of 30 inserts showed that they were derived from eight different RNA species. Six of these RNAs (D1 to D6) were derived from RNA 2 and contained a single internal deletion. The two other RNA molecules (R1 and R2) repre-sented recombinants containing the 59-terminal region of RNA 2 and the 39-terminal region of RNA 1. Figure 3 shows a schematic representation of these RNAs; the deletion junction sequences are shown in Fig. 4. The extent of the deleted region varied from 2,307 nt (D1) to 2,574 nt (D6). Examination of the RNA 2 sequences upstream and downstream of the deleted
repeat) and D4 and in the recombinants R1 and R2 (Fig. 4). This stretch resembles the 59 sequence of TRV genomic and subgenomic RNAs. RNA D6 was found to have a 2-nt inser-tion (AC) at the juncinser-tion site, since such residues are not present at that position in the genomic RNA 2.
[image:4.612.60.294.73.377.2]It is interesting that in three of the defective RNAs (D4, D5, and D6), the juxtaposition of 59and 39sequences at the junc-tion has created an in-frame fusion between the coat protein gene and the 32.8K gene which is different in each case. The potential protein encoded by D4 results from the fusion of the N-terminal 5 amino acids (aa) of the cp with the C-terminal 121 aa of the 32.8K protein. D5 contains an ORF potentially encoding a protein resulting from the fusion of the N-terminal 12 aa of the cp with the C-terminal 103 aa of the 32.8K protein. D6 potentially encodes a fusion protein composed by the N-terminal 3 aa of the cp and the C-N-terminal 109 aa of the 32.8K protein. Moreover, the 2-nt insertion present at the junction site of this form maintains the reading frame of the cp into the 32.8K sequence. For RNAs D1, D2, and D3, in-frame fusions were not created as a consequence of the deletion. However, these RNAs maintained an ORF starting at an internal AUG codon in the truncated 32.8K gene (AUG codons at positions
[image:4.612.319.548.374.619.2]FIG. 3. Schematic representation of PPK20 DI RNAs and wild-type PPK20 genomic RNAs. RNA 1- and RNA 2-specific sequences are represented by solid and open bars, respectively. The relative positions of the ORFs are shown (numbers indicate the molecular masses [in kilodaltons {K}] of the potential products encoded by the genomic RNAs). The dashed lines represent the deleted sequences. The dotted lines join the N-terminal and C-terminal regions of the fusion proteins potentially encoded by D4, D5, and D6.
FIG. 4. Junction sequences of DI RNAs. Sequences surrounding the deletion junction sites in TRV PPK20 DI RNAs. Uppercase letters correspond to se-quences which are present in the DI RNA upstream and downstream of the junction site, whereas lowercase letters correspond to sequences which are ab-sent in the DI RNAs but are preab-sent in the corresponding parental RNA molecules. Direct repeats surrounding the 59- and 39-deletion borders in some DI RNAs are underlined. The 2-nt insertion in D6 is shown in italics. The nucleotide positions correspond to those in the genomic RNA 2 with the exception of those indicated for the sequences downstream of the 39-proximal junction in R1 and R2, which correspond to those in the genomic RNA 1. The clones corresponding to DI RNAs generated by serial passage of infections initiated with the wild-type isolate PPK20 and a combination of PPK20 RNA1 and pCaK20-2T16 are indi-cated by a and b, respectively.
on November 9, 2019 by guest
http://jvi.asm.org/
2846 to 2848, 2930 to 2932, and 3095 to 3097 of RNA 2 for D1, D2, and D3, respectively. The recombinants R1 and R2 con-tain the 39-terminal 321 and 306 nt, respectively, of the 16K gene and encode a small peptide of 45 aa that could be initi-ated at an in-frame internal AUG codon in the 16K gene.
Characterization of a deletion form of PPK20 RNA2 de-tected after serial passages using leaf homogenates as inocula.
Tobacco plants were inoculated with a mixture containing PPK20 RNA 1 plus the full-length clone of PPK20 RNA 2, pCaK20-2T7, and the infection was transferred by serial pas-sages using leaf homogenates as inocula. Total RNA samples from each passage were analyzed by Northern blot hybridiza-tion using an RNA 2-specific probe derived from the 32.8K gene (Fig. 5). The Northern blot indicated that the primary infected plants contained only full-length RNA 2 (Fig. 5, lane 1) and the expected subgenomic RNAs derived from it. How-ever, after a single mechanical transfer, two new RNA 2-spe-cific bands, with estimated sizes of 2.0 and 1.5 kb, appeared (Fig. 5, lane 2). Such bands were also detectable when a cDNA probe derived from the cp gene was used but not with a probe derived from the 29.4K gene (data not shown), suggesting that these new bands could correspond to a deletion form of RNA 2 (designated D7) and to a subgenomic RNA derived from it. This deletion form became predominant upon repeated me-chanical transfer (Fig. 5, lanes 3 to 8), and after the eighth passage no full-length RNA 2 was detectable (Fig. 5, lane 9). The experiment whose results are shown in Fig. 5 was carried out three times, but in only one of these experiments was the generation of a defective RNA detectable. In the other two experiments, the pattern of genomic and subgenomic RNAs did not change.
The RT-PCR and cloning strategy described above was used to determine the structure of the D7 RNA detectable in the experiment for Fig. 5. Figure 6 shows the products obtained in RT-PCRs using the total RNA extracts of the primary infected plants (lane 1) and the third passage (lane 2) as templates. In addition to a faint band corresponding to the full-length DNA copy of RNA 2, a major product with an estimated size of 1.2 kb was generated from the samples corresponding to the pri-mary infected material (Fig. 6, lane 1). This indicates the pres-ence of DI RNAs in these plants at a level that was not detectable by Northern blot analysis (Fig. 5, lane 1). After several passages the pattern of RT-PCR products changed and a major cDNA of about 2.0 kb, corresponding in size to the D7 RNA detected by Northern blot analysis (Fig. 5, lanes 2 to 9),
was obtained (Fig. 6, lane 2). This cDNA was eluted from the agarose gels and inserted into the pGEM-T vector. The precise position of the deletion in D7 was identified by partial dideoxy sequencing of five clones using a proper synthetic primer. The five clones corresponded to the defective RNA molecule of which the sequence is schematically shown in Fig. 7. Like the defective RNAs D1 to D6, D7 corresponds to a deletion mu-tant of RNA 2, but in this case small sequence rearrangements have occurred at the junction site. D7 contains the 59-terminal 1,126 nt and the 39-terminal 894 nt of RNA 2 joined by a sequence of 18 nt. The first 10 nt of this sequence are found at positions 2385 to 2395 of the genomic RNA 2, and the last 8 nt are found at positions 330 to 338. The deletion in D7 affects the 39-terminal 46 nt of the cp gene, but a new stop codon was created at the junction site. Because of this, D7 potentially encoded a mutant cp lacking the C-terminal 15 aa of the wild-type cp and carrying, instead, three non-wild-type amino acids (LRL). In addition, D7 contains a truncated 32.8K gene, but the expression of the resulting small ORF seems unlikely since the sequences for the generation of the corresponding subgenomic RNA are absent (Fig. 7).
[image:5.612.370.499.71.194.2]To verify the expression of the D7-encoded cp and the func-tion of this mutant cp in encapsidafunc-tion, D7 cDNA was cut out from the pGEM-T vector by cleavage at the XbaI and XhoI restriction sites present in the PCR primers TRV5 and TRV3,
FIG. 5. Accumulation of defective RNAs after serial passage of TRV PPK20 in tobacco plants using leaf homogenates as inocula. A Northern blot was loaded with total RNA extracts from inoculated plants. The inoculations were as follows: lane 1, RNA 1 plus plasmid pCaK20-2T7 (corresponding to wild-type RNA 2); lanes 2 to lane 9, eight mechanical transfers of the infection analyzed in lane 1; lane 10, RNA 1 plus plasmid pCaTD7 (corresponding to the defective RNA D7); and lanes 11 to 13, RNA 1 plus a mixture of plasmids pCaK20-2T7 and pCaTD7 at molar ratios of 1:1, 3:1, and 9:1, respectively. For the mechanical transfers analyzed in lanes 2 to 9, leaf homogenates were used as inocula. The blot was hybridized with a32
[image:5.612.86.269.73.140.2]P-labelled probe corresponding to the 32.8K gene (nt 3066 to 3417 of PPK20 RNA 2). The positions of RNAs 2 and 2a are indicated on the left. The arrowhead on the left indicates the putative subgenomic mRNA in-volved in expression of the 32.8K gene.
FIG. 6. RT-PCR analysis of infected tobacco plants. The total RNA extracts analyzed in lanes 1 and 3 of the Northern blot of Fig. 5 were used to perform RT-PCRs, and the products are shown in lanes 1 and 2, respectively. RT-PCR was done by using primers TRV3 and TRV5. Amplified fragments were sepa-rated by agarose gel electrophoresis and stained with ethidium bromide. The arrowhead on the left indicates the position of full-length PPK20 cDNA 2.
FIG. 7. Schematic representation of RNA 2 of TRV isolate PPK20 and the defective RNA D7. The RNA 1- and RNA 2-specific sequences are represented by solid and open bars, respectively. The relative positions of the ORFs are given (numbers indicate the molecular masses [in kilodaltons {K}] of the potential products). The mutated coat protein encoded by D7 has been designated mCP. The nucleotides at the junction site are indicated; the bolded triplet corresponds to the stop codon of the mutated cp gene created at the junction site.
on November 9, 2019 by guest
http://jvi.asm.org/
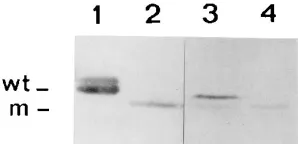
[image:5.612.334.536.550.669.2]respectively. The D7 cDNA was made blunt by filling with T4 DNA polymerase and inserted between the CaMV 35S pro-moter and the nos terminator as described previously for the full-length cDNA 2 (14). The resulting D7 cDNA clone was designated pCaTD7. Northern blot analysis of tobacco plants inoculated with a combination of PPK20 RNA 1 and pCaTD7 showed the accumulation of a D7-like molecule and the ex-pected subgenomic RNA for the expression of the cp derived from it (Fig. 5, lane 10). The progeny of the pCaTD7 clone could be transferred to tobacco by using a leaf homogenate that was incubated at 378C for 30 min as the inoculum (data not shown). This demonstrates that RNA 1 and D7 RNA molecules in the inoculum were properly encapsidated. West-ern blot analysis showed the presence of the mutated cp in the pCaTD7-infected plants (Fig. 8, lane 2), with this cp migrating faster than wild-type cp (Fig. 8, lane 1) as expected. Moreover, after the fourth passage of the wild-type infection in which D7 was generated, the wild-type and mutant cps were both detect-able (Fig. 8, lane 3), but after the eighth passage only the mutated cp was detectable (Fig. 8, lane 4). This is in agreement with the absence of genomic RNA 2 after this passage (Fig. 5, lane 9).
To compare the replication efficiencies of the genomic RNA 2 and D7 RNA, competition assays were performed. Tobacco plants were inoculated with PPK20 RNA 1 plus a mixture of plasmids pCaK20-2T7 (containing full-length cDNA 2) and pCaTD7 (containing D7 cDNA) at different molar ratios. The full-length RNA 2 accumulated at detectable levels only when the molar ratio of pCaK20-2T7 to pCaTD7 was 9:1 (Fig. 5, lane 13), but D7 was the only form detected at molar ratios of 3:1 and 1:1 (Fig. 5, lanes 11 and 12). This indicates that D7 inter-feres with the replication of RNA 2. The presence in lane 12 of Fig. 5 of an RNA molecule that comigrates with the sub-genomic RNA involved in expression of the 32.8K gene (po-sition indicated by the arrowhead in Fig. 5) suggests that at the 1:1 ratio of the inoculum cDNAs a low level of full-length RNA 2 still may be present.
Effect of a deletion in RNA 2 on transmissibility of the virus
by nematodes. Nematodes from a population mixture of P.
pachydermus and T. primitivus acquired the TRV PPK20 wild-type isolate during a 4-week period of access to the roots of N. clevelandii virus source plants and subsequently, during a fur-ther 4-week period, transmitted the virus to 8 of 10 healthy N. clevelandii bait plants. In a concurrent test nematodes from the same population were apparently unable to transfer the D7 isolate from virus source plants to any of 10 bait plants.
The defective RNAs outcompeted the genomic RNA 2 without affecting the accumulation of the genomic RNA 1. Although the DI RNAs accumulated at levels that became detectable by Northern blot analysis after one or two passages, the more sensitive RT-PCR technique showed the presence of these RNA species already in the plants initially inoculated with the wild-type PPK20 virus or a combination of PPK20 RNA 1 and a full-length cDNA clone of RNA 2. The results with the infection initiated with the cDNA 2 clone demonstrate that the DI RNAs are generated de novo. Similarly, detection of DI RNAs in primary plants inoculated with transcripts of tomato bushy stunt virus cDNA clones has been reported (21).
For the experiments in which plants were initially infected with wild-type virus, we cannot exclude the possibility that DI RNAs were already present in the inoculum. TRV isolate PPK20 has been propagated mechanically by sap inoculation for approximately 3 years in our laboratory, but DI RNAs generally did not reach levels detectable by Northern blot analysis. Sap inoculation selects for encapsidated genomic RNAs but permits the virus to lose nematode transmissibility. The generation of the D7 RNA illustrates that after mechan-ical transfer, functions involved in vector transmission may be lost. Our unpublished results indicate that the 29.4K gene in RNA 2 of TRV isolate PPK20 is essential for nematode trans-missibility. The loss of transmissibility when RNA 2 in the inoculum is replaced by D7 RNA may be due to the deletion of the 29.4K gene in this RNA and/or to the C-terminal trun-cation of its encoded cp. The observation that deletions can occur in RNA 2 after passage by sap inoculation may explain the observation that many laboratory isolates of TRV do not contain nonstructural genes in RNA 2 and are not transmissi-ble by any known nematode vector (1, 2, 7, 14).
Curiously, after a single mechanical transfer using total RNA extracts from infected plants as inocula, some DI RNAs accumulated at amounts detectable in a Northern blot (Fig. 1). There is no selection for encapsidation with this type of inoc-ulum, and apparently this permits the rapid accumulation of DI RNAs that have lost a functional cp gene. It should be noted that the RNA inocula were prepared from 10 g of leaf material, whereas 0.5 g of leaf material was used to prepare the sap inocula. As the DI RNAs are not very abundant in the initial infected plants and their distribution may be irregular in the local lesion host N. tabacum, the use of larger amounts of material to prepare the extracts may have contributed to trans-fer and spread of those DI RNAs during passages.
Replication competence is believed to be one of the main factors involved in DI RNA accumulation (40). In addition, the presence of an ORF was found to be important for the accu-mulation of a defective RNA of clover yellow mosaic potexvi-rus (39) and possibly other plant vipotexvi-ruses (10, 32, 34). The potential coding capacity of all DI RNAs characterized in this study suggests that a coupling between translation and repli-cation also may be important for the accumulation of TRV DI RNAs.
Although DI RNAs may substantially affect symptom pro-duction (6, 22, 34), significant changes were not present in the symptoms on N. tabacum of TRV infections accumulating DI RNAs or infections in which the wild-type RNA 2 was replaced by the defective RNA. This result is probably due to the fact that the symptomatology of a TRV infection is mainly
deter-FIG. 8. Western blot analysis of the expression of cp from wild-type TRV RNA 2 and defective RNA D7. Proteins were extracted from plants inoculated with the following: lane 1, wild-type TRV isolate PPK20; lane 2, PPK20 RNA 1 plus plasmid pCaTD7 (corresponding to D7 RNA); and lanes 3 and 4, the fourth and eighth passages, respectively, of the infection initiated with PPK20 RNA 1 plus pCaK20-2T7 (corresponding to wild-type RNA 2). Serial passage was done by using leaf homogenates as inocula. The protein samples shown in lanes 3 and 4 were extracted from the plants analyzed in lanes 5 and 9 of Fig. 5. The positions of wild-type cp (wt) and the mutated cp (m) are indicated on the left.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.100.249.71.143.2]mined by RNA 1 (12) and the accumulation of this RNA was not affected by the presence of DI RNAs. The observation that the DI RNAs outcompeted RNA 2 without a detectable effect on RNA 1 accumulation could suggest that RNAs 1 and 2 replicate by different mechanisms and may use separate pools of the viral replicase.
In addition to the DI RNAs derived by internal deletion from PPK20 RNA 2 (D1 to D7), we have identified small RNA molecules, R1 and R2, carrying the 59-terminal sequences of RNA 2 and the 39-terminal sequences of RNA 1. These RNAs probably represent the new bands shown in Fig. 1A. RNA 2 from laboratory isolates of tobraviruses has been found to contain 39-terminal RNA 1-derived sequences ranging in size from approximately 250 to 1,100 nt (26). The recombination events responsible for the generation of TRV DI RNAs may involve a “copy choice” mechanism as proposed for the gen-eration of other DI RNAs (20). This mechanism involves jumping of the replicase-nascent strand complex from one position on the template to another position on the same or another RNA molecule. The dissociation of the complex from its template is thought to be induced by strong secondary structures in the RNA. Folding of the sequences surrounding the deletion sites of the DI RNAs characterized in this study showed that, potentially, stem-loop structures can be formed (data not shown). Several features of the acceptor template have been proposed to aid reassociation of the detached rep-licase-nascent strand complex. These include the presence of short stretches of complementarity with the incomplete nas-cent strand (19, 20, 28, 32) and recognition sequences for the replicase or secondary structures in the template RNA (5). The presence of direct repeats and AU-rich sequences resembling the 59 end of genomic and subgenomic TRV RNAs at the junction sequences of TRV DI RNAs indicates that distinct mechanisms may function in TRV RNA recombination.
The results reported in this article indicate that the TRV genome is very flexible. Depending on the selection pressure that is applied, functions in nematode transmissibility and en-capsidation are rapidly deleted from RNA 2. In vegetatively propagated potatoes, so-called nonmultiplying infections, in which RNA 2 is completely lost, can be found (12). In the evolution of tobraviruses, recombination events are probably responsible for the acquisition of differently sized 39-terminal RNA 1 sequences by RNA 2 and for the exchange of cp genes between isolates of TRV and another tobravirus, pea early browning virus (1, 2, 9, 33). Our observation that TRV quickly adapts its genome structure to nonfield conditions stresses the requirement to have laboratory isolates of tobraviruses regu-larly transmitted by the nematode vector to maintain integrity of the viral genome.
ACKNOWLEDGMENTS
C.H. is a recipient of fellowships from the Consejo Superior de Investigaciones Cientificas (Spain) and from the European Commu-nity. J.F.B. and D.J.F.B. gratefully acknowledge financial assistance received under the NWO-British Council Joint Scientific Research Projects scheme. Research at the Scottish Crop Research Institute was aided by grants from the Scottish Office Agriculture and Fisheries Department (SOAFD).
Nonindigenous virus isolates were held and experiments with genet-ically modified viruses were done under licence from SOAFD.
REFERENCES
1. Angenent, G. C., H. J. M. Linthorst, A. F. van Belkum, B. J. C. Cornelissen,
and J. F. Bol.1986. RNA 2 of tobacco rattle virus strain TCM encodes an unexpected gene. Nucleic Acids Res. 14:4673–4682.
2. Angenent, G. C., E. Posthumus, F. T. Brederode, and J. F. Bol. 1989. Ge-nome structure of tobacco rattle virus strain PLB: further evidence on the
occurrence of RNA recombination among tobraviruses. Virology 171:271– 274.
3. Bouzoubaa, S., U. Niesbach-Klo¨sgen, I. Jupin, H. Guilley, K. Richards, and G. Jonard.1991. Shortened forms of beet necrotic yellow vein virus RNA-3 and -4: internal deletions and a subgenomic RNA. J. Gen. Virol. 72:259–266. 4. Burgyan, J., F. Grieco, and M. Russo. 1989. A defective interfering RNA molecule in cymbidium ringspot virus infections. J. Gen. Virol. 70:235–239. 5. Carpenter, C. D., J.-W. Oh, C. Zhang, and A. E. Simon. 1995. Involvement of a stem-loop structure in the location of junction sites in viral RNA recombination. J. Mol. Biol. 245:608–622.
6. Chen, J., S. A. MacFarlane, and T. M. A. Wilson. 1994. Detection and sequence analysis of a spontaneous deletion mutant of soil-borne wheat mosaic virus RNA 2 associated with increased symptom severity. Virology
202:921–929.
7. Cornelissen, B. J. C., H. J. M. Linthorst, F. T. Brederode, and J. F. Bol. 1986. Analysis of the genome structure of tobacco rattle virus strain PSG. Nucleic Acids Res. 14:2157–2169.
8. Feinberg, A. P., and B. Vogelstein. 1984. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem.
132:6–13.
9. Goulden, M. G., G. P. Lomonossoff, K. R. Wood, and J. W. Davies. 1991. A model for the generation of tobacco rattle virus (TRV) anomalous isolates: pea early browning virus RNA-2 acquires TRV sequences from both RNA-1 and RNA-2. J. Gen. Virol. 72:1751–1754.
10. Graves, M. V., and M. J. Roosinck. 1995. Characterization of defective RNAs derived from RNA 3 of the Fny strain of cucumber mosaic cucumo-virus. J. Virol. 69:4746–4751.
11. Hamilton, W. D. O., M. Boccara, D. J. Robinson, and D. C. Baulcombe. 1987. The complete nucleotide sequence of tobacco rattle virus RNA-1. J. Gen. Virol. 68:2563–2575.
12. Harrison, B. D., and D. J. Robinson. 1986. Tobraviruses, p. 339–369. In H. V. Van Regenmortel and H. Fraenkel-Conrat (ed.), The plant viruses, vol. 2. Plenum, New York.
13. Harrison, B. D., and R. D. Woods. 1966. Serotypes and particle dimensions of tobacco rattle viruses from Europe and America. Virology 28:610–620. 14. Herna´ndez, C., A. Mathis, D. J. F. Brown, and J. F. Bol.1995. Sequence of
RNA 2 of a nematode transmissible isolate of tobacco rattle virus. J. Gen. Virol. 76:2847–2851.
15. Hillman, B. I., J. C. Carrington, and T. J. Morris. 1987. A defective inter-fering RNA that contains a mosaic of a plant virus genome. Cell 51:427–433. 16. Holland, J. J. 1991. Defective viral genomes, p. 151–165. In B. N. Fields and
D. M. Knipe (ed.), Virology. Raven Press, New York.
17. Ismail, I. D., and J. J. Milner. 1988. Isolation of detective interfering parti-cles of sonchus yellow net virus from chronically infected plants. J. Gen. Virol. 69:999–1006.
18. Jackson, A. O., B. G. Hunter, and G. D. Gustafson. 1989. Hordeivirus relationships and genome organization. Annu. Rev. Phytopathol. 27:95–121. 19. Knorr, D. A., R. H. Mullin, P. Q. Hearne, and T. J. Morris. 1991. De novo generation of defective interfering RNAs of tomato bushy stunt virus by high multiplicity passage. Virology 181:193–202.
20. Lai, M. M. C. 1992. RNA recombination in animal and plant viruses. Mi-crobiol. Rev. 56:61–79.
21. Law, M. D., and T. J. Morris. 1994. De novo generation and accumulation of tomato bushy stunt virus defective interfering RNAs without serial host passage. Virology 198:377–380.
22. Li, X. H., L. A. Heaton, T. J. Morris, and A. E. Simon. 1989. Turnip crinkle virus defective interfering RNAs intensify viral symptoms and are generated
de novo. Proc. Natl. Acad. Sci. USA 86:9173–9177.
23. Li, X. H., and A. E. Simon. 1991. In vivo accumulation of a turnip crinkle virus defective interfering RNA is affected by alterations in size and se-quence. J. Virol. 65:4582–4590.
24. Lister, R. M., and C. E. Bracker. 1969. Defectiveness and dependence in three related strains of TRV. Virology 37:262–275.
25. MacFarlane, S. A., D. J. F. Brown, and J. F. Bol. 1995. The transmission by nematodes of tobraviruses is not determined exclusively by the virus coat protein. Eur. J. Plant Pathol. 101:535–539.
26. Mathis, A., and H. J. M. Linthorst. 1992. The tobraviruses, p. 1442–1446. In R. G. Webster and A. Granoft (ed.), Encyclopedia of virology. Academic Press, Ltd., London.
27. Mawassi, M., A. V. Karasev, E. Mietkiewska, R. Gafny, R. F. Lee, W. O.
Dawson, and M. Bar-Joseph.1995. Defective RNA molecules associated with citrus tristeza virus. Virology 208:383–387.
28. Pilipenko, E. V., A. P. Gmyl, and V. I. Agol. 1995. A model for rearrange-ments in RNA genomes. Nucleic Acids Res. 23:1870–1875.
29. Ploeg, A. T., D. J. F. Brown, and D. J. Robinson. 1989. Transmission of tobraviruses by trichodorid nematodes. EPPO Bull. 19:605–610. 30. Ploeg, A. T., D. J. F. Brown, and D. J. Robinson. 1992. The association
between species of Trichodorus and Paratrichodorus vector nematodes and serotypes of tobacco rattle virus. Ann. Appl. Biol. 121:619–630.
31. Ploeg, A. T., D. J. Robinson, and D. J. F. Brown. 1993. RNA 2 of tobacco rattle virus encodes the determinants of transmissibility by trichodorid nem-atodes. J. Gen. Virol. 74:1463–1466.
on November 9, 2019 by guest
http://jvi.asm.org/
broad bean mottle virus infections. Virology 194:576–584.
35. Towbin, H. T., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350–4354.
36. Verwoerd, T. C., B. M. M. Dekker, and A. Hoekema. 1989. A small-scale
determines in vivo accumulation of a defective RNA of clover yellow mosaic virus. J. Virol. 66:3069–3076.
40. White, K. A., and T. J. Morris. 1994. Nonhomologous RNA recombination in tombusviruses: generation and evolution of defective interfering RNAs by stepwise deletions. J. Virol. 68:14–24.