Structure-function analysis of soluble forms of herpes simplex virus glycoprotein D.
9
0
0
Full text
Figure
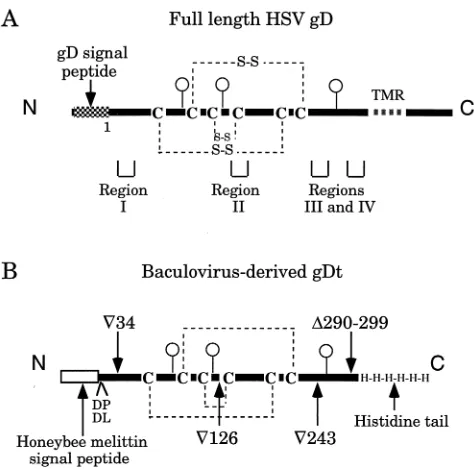

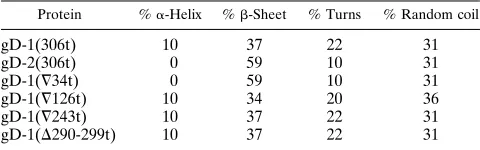
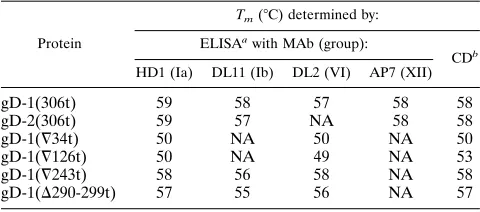
+3
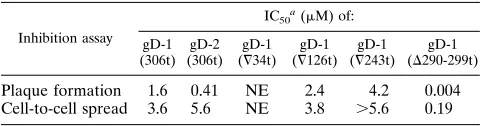
Related documents