0022-538X/95/$04.0010
Copyrightq1995, American Society for Microbiology
Cloning and Characterization of Herpes Simplex Virus Type 1 oriL:
Comparison of Replication and Protein-DNA Complex
Formation by oriL and oriS
MARY ANN HARDWICKEANDPRISCILLA A. SCHAFFER*
Division of Molecular Genetics, Dana-Farber Cancer Institute, and Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, Massachusetts 02115
Received 13 September 1994/Accepted 16 November 1994
The herpes simplex virus type 1 genome contains three origins of DNA replication: two copies of oriS and one copy of oriL. Although oriS has been characterized extensively, characterization of oriL has been severely limited by the inability to amplify oriL sequences in an undeleted form in Escherichia coli. We report the successful cloning of intact oriL sequences in anE. colistrain, SURE, which contains mutations in a series of genes involved in independent DNA repair pathways shown to be important in the rearrangement and deletion of DNA containing irregular structures such as palindromes. The oriL-containing clones propagated in SURE cells contained no deletions, as determined by Southern blot hybridization and DNA sequence analysis, and were replication competent in transient DNA replication assays. Deletion of 400 bp of flanking sequences decreased the replication efficiency of oriL twofold in transient assays, demonstrating a role for flanking sequences in enhancing replication efficiency. Comparison of the replication efficiencies of an 822-bp oriS-containing plasmid and an 833-bp oriL-oriS-containing plasmid demonstrated that the kinetics of replication of the two plasmids were similar but that the oriL-containing plasmid replicated 60 to 70% as efficiently as the oriS-containing plasmid at both early and late times after infection with herpes simplex virus type 1. The virus-specified origin-binding protein (OBP) and a cellular factor(s) (OF-1) have been shown in gel mobility shift experiments to bind specific sequences in oriS (C. E. Dabrowski, P. Carmillo, and P. A. Schaffer, Mol. Cell. Biol. 14:2545–2555, 1994; C. E. Dabrowski and P. A. Schaffer, J. Virol. 65:3140–3150, 1991). Although the nucleotides required for the binding of OBP to OBP binding site I in oriL and oriS are the same, a single nucleotide difference distinguishes OBP binding site III in the two origins. The nucleotides adjacent to oriS sites I and III have been shown to be important for the binding of OF-1 to oriS site I. Several nucleotide differences exist in these sequences in oriL and oriS. Despite these minor nucleotide differences, the protein-DNA complexes that formed with oriL and oriS sites I and III were indistinguishable when extracts of infected and uninfected cells were used as the source of protein. Furthermore, the results of competition analysis suggest that the proteins involved in protein-DNA complex formation with sites I and III of the two origins are likely the same.
The herpes simplex virus type 1 (HSV-1) genome is a linear double-stranded DNA molecule composed of two unique com-ponents, designated long (UL) and short (US), each of which is
flanked by inverted repeat sequences (Fig. 1A) (19, 28). Elec-tron microscopic examination of replicating HSV-1 DNA ini-tially suggested that the HSV-1 genome contains three origins of replication (8, 31). One origin, designated oriS, is located within the repeat regions flanking the USregion of the genome
and hence is diploid (Fig. 1A). The third origin, designated oriL, is present in one copy in the ULregion of the genome.
Further evidence for the existence of multiple origins in HSV DNA was suggested by the sequence composition and replica-tion competence of class I and class II defective genomes (13–15, 34). The functional significance of the existence of three origins of replication within HSV-1 DNA is not yet clear. Mutant viruses lacking either oriL or both copies of oriS are replication competent, indicating that the two types of origin can substitute for each other (22, 30). On the basis of this observation, it is likely that oriL or at least one copy of oriS is required for DNA replication and that oriL and oriS are func-tionally equivalent.
Deletion mutagenesis and transient DNA replication assays have demonstrated that the core origin of oriS consists of a 90-bp sequence which includes a 45-bp imperfect palindrome with an 18-bp AT-rich stretch at its center (25, 33). Also con-tained within the core are three binding sites (BS-I, -II, and -III) for the origin-binding protein (OBP), the product of the UL9 gene (Fig. 1B) (2, 4, 5, 11, 12, 23, 29, 35, 36). The limits of the 11-bp oriS OBP BS-I have been defined by DNase I footprinting of partially purified OBP, filter binding assays, and gel shift analysis using mutagenized oriS site I oligonucleotides (4, 9, 10, 11, 23, 25). The 11-bp sequence of OBP BS-II differs from that of OBP BS-I by two nucleotides, which leads to a 5-to 10-fold-reduced affinity of OBP for BS-II relative 5-to BS-I (Fig. 1B) (9, 11, 20, 27, 36). BS-I and II sequences are inverted with respect to each other within the left and right arms of the palindrome, respectively, with the AT-rich stretch between them. oriS BS-III differs from BS-I by four nucleotides and is located to the left of BS-I in an inverted orientation. OBP has been shown to bind to BS-III with low affinity relative to BS-I and II (9, 10).
The oriL palindrome is substantially larger than that of oriS, consisting of a 144-bp perfect repeat (37). Sequence analysis of oriL has revealed striking homology to oriS; of the 144 bp, 90 are common to both origins (37). Extensive homology exists between the two origins at and to the left of the center of * Corresponding author. Phone: (617) 8240. Fax: (617)
375-8255. Electronic mail address: [email protected].
1377
on November 9, 2019 by guest
http://jvi.asm.org/
symmetry of the oriS palindrome and includes the AT-rich region and the region including OBP BS-I and -III (Fig. 1B). In oriL, the sequence corresponding to BS-II is replaced by a second copy of the higher-affinity BS-I sequence. Because oriL contains two copies of BS-I- and BS-III-like sequences and no BS-II equivalent, the oriL palindrome is perfectly symmetrical. Although the nucleotides that comprise OBP BS-I are identi-cal in oriL and oriS, a single nucleotide distinguishes the BS-III-like sequence of oriL from its oriS counterpart (Fig. 1B). The nucleotides immediately adjacent to OBP BS-I in oriS have been shown to be important for the binding of a bipartite cellular DNA-binding factor(s), OF-1 (3). Thus, 8 of 11 nucle-otides in the 59half of the OF-1 binding site overlap OBP BS-I, whereas the 39half of the binding site is located immediately adjacent to the OBP BS-I (see Fig. 6). Nucleotide substitutions in the 39half of the OF-1 binding site immediately adjacent to OBP BS-I reduce DNA replication from an oriS-containing plasmid to less than 15% of the level of wild-type oriS in transient replication assays, suggesting a critical role for OF-1 in DNA replication from oriS (3). An additional minor differ-ence between the two origins involves the length of the AT-rich region: it is 18 bp long in oriS and 20 bp long in oriL.
Similar to the origins of other DNA viruses, the three origins of HSV-1 DNA replication are located between divergently transcribed genes (30, 33, 38, 40). oriS is located within the shared promoter regulatory region between the divergently transcribed immediate-early genes encoding regulatory pro-teins ICP4 and ICP22/ICP47 (33), whereas oriL lies in the regulatory region between two divergently transcribed early genes that are essential for viral DNA replication, the genes specifying the major DNA-binding protein, ICP8 (the product of the UL29 gene), and the DNA polymerase (the product of the UL30 gene) (30, 38, 40) (Fig. 1A). Significant evidence now exists to suggest that the binding of proteins to transcriptional
regulatory sequences flanking the oriS core origin as well as sequences flanking other well-characterized viral origins has a pronounced stimulatory effect on DNA replication (2a, 25, 39). Consequently, similar effects of protein interactions with cis-acting regulatory elements flanking oriL are likely to occur.
To date, functional characterization of HSV-1 origins has focused almost exclusively on oriS since the large oriL palin-drome is deleted upon cloning in bacteria (6, 24, 30, 37). Weller et al. successfully cloned a 2.3-kb fragment containing an intact copy of oriL in a yeast cloning vector (37). Propaga-tion in yeast cells proved to be an impractical source of oriL, however, as the yields were routinely very low. Weller et al. subsequently used this 2.3-kb fragment to transform the re-combination-deficient Escherichia coli strain RDP145 (RecA2 RbcB2) (37). The HSV-1 DNA from most colonies consisted exclusively of deleted forms of oriL sequences, whereas the DNA from other colonies contained approximately equal amounts of deleted and undeleted (wild-type) oriL, as deter-mined by Southern blot hybridization. The isolated DNAs from the latter colonies were tested in transient DNA replica-tion assays and shown to be significantly amplified, demon-strating that this 425-bp region of the genome is able to func-tion as an origin of replicafunc-tion. In a second study, Lockshon and Galloway were able to propagate an HSV-2 oriL-contain-ing fragment in E. coli JC9387 (RecBC2SbcB2) (24, 25). In this case, 90% of the large oriL-containing plasmid prepara-tions were full length, while the smaller subclones still retained a strong tendency to delete (24, 25). These investigators local-ized HSV-2 oriL to a 190-bp fragment which was functional in transient replication assays.
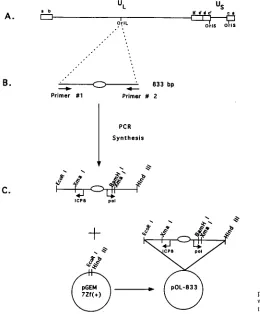
The present report describes the construction of plasmids containing undeleted copies of HSV-1 oriL sequences which remain undeleted upon subsequent subcloning in E. coli SURE cells. The ability of these oriL-containing plasmids to function FIG. 1. Diagram of the HSV-1 genome and nucleotide sequence comparison of oriL and oriS. (A) Beneath the scale of map units is shown a diagram of the HSV-1 genome, indicating the locations of the internal and terminal repeat sequences (a, b, c, b9, a9, and c9), the ULand UScomponents of the genome, the three origins of
replication (oriL and oriS) and the seven genes that encode proteins required for DNA replication, UL5, UL8, UL9, UL29, UL30, UL42, and UL52 (2). (B) Nucleotide sequence comparison of oriS and oriL, including OBP BS-I, BS-II, and BS-III. The vertical dashed line indicates the center of dyad symmetry of the oriL and oriS palindromes (also indicated by the inverted arrowheads). The dashes in the oriS sequence indicate nucleotides that are identical in oriL. The X in the oriS sequence indicates a nucleotide that is not present in oriS.
on November 9, 2019 by guest
as origins of DNA replication was demonstrated in transient replication assays. Although oriS- and oriL-containing plasmids of nearly equal size replicated with similar kinetics, the oriL-containing plasmid replicated somewhat less efficiently than the oriS-containing plasmid. As in the case of oriS flanking se-quences, deletion of sequences flanking oriL reduced replica-tion efficiency significantly. Gel shift analysis demonstrated that the protein-DNA complexes that form with oriL site I and III DNAs are indistinguishable from those that form with the homologous sites in oriS; however, competition analysis indi-cated that the binding affinities of the viral and cellular pro-teins that form these complexes may differ for oriL and oriS.
MATERIALS AND METHODS
Cells and virus.African green monkey kidney (Vero) cells were grown and maintained in Dulbecco’s modified Eagle’s medium (GIBCO Laboratories, Inc., Gaithersburg, Md.) as previously described (38). The wild-type KOS strain of HSV-1 and the oriL2virus, ts17, derived from strain KOS, were grown and assayed as previously described (7). The isolation of ts17 is described elsewhere (30).
DNA isolation.Viral DNA for use in cloning and Southern blot analysis was isolated essentially as described elsewhere (30) except that the viral DNA was further purified by centrifugation in CsCl gradients at 35,000 rpm for 72 h at 258C. Southern blot analysis.Viral DNA restriction fragments were electrophorec-tically separated on 1.2% agarose gels (or 0.8% agarose gels for transient DNA replication assays), transferred to nitrocellulose, and hybridized to32P-labeled
probes as previously described (32). The32P-labeled probes were generated by
nick translation as previously described (32).
Plasmids and cloning.Plasmid pOS-822 containing oriS has been described elsewhere (39). To clone pOL-833, PCR was used to generate an 833-bp frag-ment containing oriL in which additional sequences were added to the 59end of each primer to create new restriction sites to facilitate cloning. The reaction conditions used were essentially as described by the manufacturer of the Gene-Amp PCR reagent kit (Perkin-Elmer Cetus, Norwalk, Conn.). One nanogram of KOS DNA was used for amplification.
The two primers synthesized by the Molecular Biology Core Facility, Dana-Farber Cancer Institute were primer 1 (59-CGGAATTCCGTGGTTGCCGTCT TGGGCTTT-39) and primer 2 (59-CCCAAGCTTGGGGCCGCCGACTTTC CTCCGG-39). Primer 1 contained an EcoRI site, whereas primer 2 contained a HindIII site.
The PCR-generated fragments were digested with EcoRI and HindIII, cloned into pGEM7Zf(1) (Promega, Madison, Wis.), and amplified in E. coli SURE cells (Stratagene, La Jolla, Calif.). Plasmid amplification in E. coli was minimized (12 to 14 h). Amplified plasmids were purified by the standard alkali method (7) and banded twice by CsCl equilibrium centrifugation. pOL-433 was generated by digesting pOL-833 with XmaI and BamHI, isolating the 433-bp oriL-containing fragment, and ligating it to XmaI- and BamHI-digested pGEM7Zf(1). The resulting plasmid was propagated and purified as described above. All plasmids were sequenced by the dideoxynucleotide method as described by the manufac-turer (United States Biochemical Corporation, Cleveland, Ohio).
Replication assay.Vero cells (23106) were plated in 100-mm-diameter dishes
and incubated at 378C overnight. Four hours before transfection, the medium was replaced with fresh medium. Cells were cotransfected with 10mg of test plasmid and 10mg of an internal, nonreplicating transfection standard (pUC19) by the calcium phosphate-N,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid (BES) method (1). At 18 to 24 h posttransfection, cells were infected with HSV-1 strain KOS at a multiplicity of infection of 10 PFU per cell. Except where indicated, cells were harvested and total cellular DNA was isolated 24 h postin-fection as described previously (39). Ten micrograms of DNA was digested with DpnI to restrict unreplicated input DNA and with EcoRI to linearize both test and reference plasmids. The digested DNAs were fractionated on 0.8% agarose gels, transferred to nitrocellulose, and hybridized to32P-labeled, nick-translated
vector sequence (pUC19). The resulting bands were quantitated with a Phos-phorImager (Molecular Dynamics, Sunnyvale, Calif.).
To detect low levels of viral DNA replication in experiments performed to examine the kinetics of replication, the Lipofectin reagent (GibcoBRL, Gaith-ersburg, Md.) method of transfection was used. For these experiments, Vero cells were seeded as described above. At the time of transfection, plasmid DNA (10mg) was diluted in 2 ml of serum free Dulbecco’s modified Eagle’s medium. Lipofectin reagent (final concentration, 8mg/ml) diluted in 2 ml of serum-free medium was mixed with the diluted DNA, and the mixture was held at room temperature for 15 min. Monolayers were rinsed once with serum-free medium, and the DNA mixture was added dropwise to the cells. The monolayers were incubated at 378C for 5 h, at which time the monolayer was rinsed two times with Tris-buffered saline (137 mM NaCl, 5 mM KCl, 25 mM Tris-HCl [pH 7.4]), and fresh medium containing serum was added to the cells. At 18 to 24 h after transfection, the cells were infected with KOS at a multiplicity of infection of 10 PFU per cell, and total DNA was harvested at 3, 6, 9, 12, and 24 h postinfection. Ten micrograms of DNA was digested either with EcoRI and DpnI or with
EcoRI and MboI. EcoRI linearized the test plasmids. DpnI digests unreplicated methylated DNA, whereas MboI digests only replicated unmethylated DNA. Therefore, the newly replicated DNA is cleaved, and the level of input DNA can be calculated following digestion with MboI. The DNAs were analyzed by South-ern blot hybridization as described above.
Oligonucleotides and probes.The oligonucleotide probes used in DNA bind-ing assays (see Fig. 7A) were synthesized by the Molecular Biology Core Facility, Dana-Farber Cancer Institute. Complementary oligonucleotides containing 4-bp overhangs that comprise BamHI or BglII sites were annealed by heating at 708C for 3 min followed by slow cooling to room temperature. The oligonucleotides were gel purified on a 12% polyacrylamide gel as described previously (26). Radiolabeled probes were prepared as described previously (4). Unlabeled dou-ble-stranded oligonucleotides were used as specific competitor fragments in gel shift competition assays. A double-stranded oligonucleotide generated as de-scribed above and containing the consensus binding site for nuclear factor 1 was used as a nonspecific competitor (4).
Gel shift DNA binding assays.Infected (2mg of protein) or uninfected (10mg of protein) cell extracts, prepared as previously described (4), were incubated at 258C for 30 min with 93104
cpm (1 ng) of probe. Competition experiments were performed as described above, premixing the probe and competitor DNAs prior to the addition of cell extracts in DNA binding buffer. Protein-DNA complexes were separated from free probe by electrophoresis at 48C in 6% polyacrylamide gels (37.5:1 acrylamide/bisacrylamide) prepared and run in 0.53 Tris-borate-EDTA buffer.
RESULTS
Cloning of HSV-1 oriL in an undeleted form in bacteria.The functional characterization of oriL requires the availability of a cloned intact copy of oriL. Previous attempts to clone the region of HSV-1 DNA containing oriL into bacteria have rou-tinely resulted in deletion of the large oriL palindrome. In the studies described herein, we used an E. coli strain, designated SURE (Promega), carrying a series of mutations which elimi-nate a number of independent DNA repair pathways. These pathways have been shown to be directly involved in the rear-rangement and deletion of DNA sequences that are prone to the formation of irregular structures such as palindromes. Thus, SURE cells contain a number of mutations, including
uvrC, umuC, sbcC, recJ, and recB. To further minimize
dele-tions, amplification of the oriL-containing fragment in these cells was limited (12 to 14 h) as suggested by Lockshon and Galloway (24).
To clone oriL, PCR was used to generate an 833-bp frag-ment which contains the 144-bp oriL palindrome as well as flanking regulatory and downstream noncoding and coding sequences of the two divergently transcribed genes, ICP8 and DNA polymerase (Fig. 2C). For PCR, we synthesized two oligonucleotide primers which would create new restriction sites on either end of the oriL-containing fragment to facilitate cloning (Fig. 2B and Materials and Methods). The 59 oligonu-cleotide (primer 1) generated a new EcoRI site, while the 39 oligonucleotide (primer 2) generated a new HindIII restriction site. These oligonucleotides were annealed to KOS DNA and amplified by PCR to generate the 833-bp fragment. The re-sulting fragment was digested with EcoRI and HindIII and ligated into vector pGEM7Zf(1) predigested with EcoRI and
HindIII to generate pOL-833 (Fig. 2C). The viral DNA insert
in pOL-833 contained no detectable deletions upon amplifica-tion in SURE cells, as determined by polyacrylamide gel and nucleotide sequence analysis (data not shown).
To confirm the absence of deletions in pOL-833 DNA, the viral DNA insert in this plasmid and the corresponding frag-ments from KOS and ts17 DNA were digested with XmaI and
BamHI and analyzed by Southern blot hybridization (Fig. 3). ts17 DNA was shown previously to contain a 150-bp deletion
in oriL (30). Digestion of KOS DNA with XmaI and BamHI yielded a fragment of the expected size (433 bp) (Fig. 3), whereas the 283-bp ts17 fragment migrated with a greater mobility. The viral DNA fragment derived from pOL-833 mi-grated with the same mobility as the KOS DNA fragment.
on November 9, 2019 by guest
http://jvi.asm.org/
Moreover, no evidence of faster-migrating (deleted) fragments was observed, even after longer exposure of the blot, suggest-ing that the oriL-containsuggest-ing plasmid had sustained no dele-tions upon propagation in SURE cells.
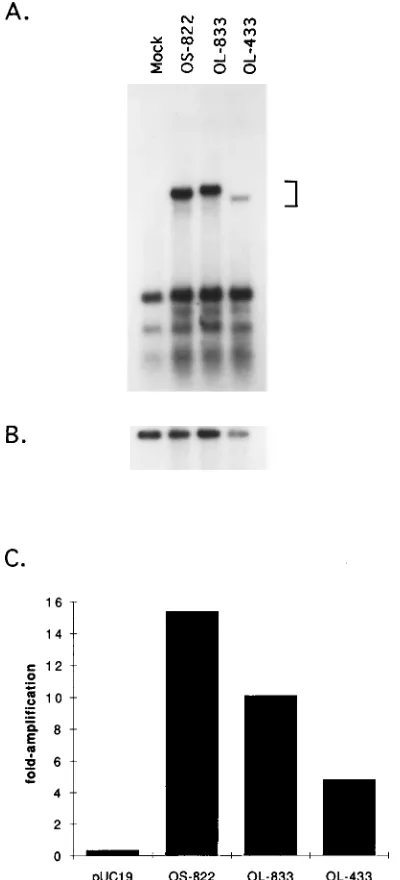
Functional analysis of oriL-containing plasmids.To deter-mine whether the cloned oriL sequences in pOL-833 were functional, transient replication assays were performed (Fig. 4). Vero cells were transfected with either a plasmid containing an 822-bp oriS-containing fragment (39) or a plasmid contain-ing the 833-bp oriL-containcontain-ing fragment. In addition, a 433-bp oriL-containing XmaI-to-BamHI fragment (Fig. 2C and 3B) cloned into the equivalent site in pGEM7Zf(1), pOL-433, was tested for origin function. A nonreplicating reference plasmid (pUC19) was included to control for transfection efficiency and to serve as an internal standard in the calculation of replication efficiencies (Fig. 4B). The replicated DNAs (bracket in Fig. 4A) were quantitated by PhosphorImager scanning, the values were corrected for differences in transfection efficiencies, and the results are presented as fold amplification over input DNA in the graph shown in Fig. 4C.
As shown in Fig. 4A and B, pOS-822, containing 822 bp of oriS sequence, replicated 15-fold more efficiently than the ref-erence plasmid. The replication efficiency determined here is
similar to that reported previously for pOS-822 (39), in which it replicated 16- to 22-fold more efficiently than the reference plasmid. In contrast, pOL-833 containing oriL replicated ap-proximately two-thirds as efficiently as the oriS-containing plasmid but approximately 10-fold more efficiently than the reference plasmid.
Wong and Schaffer (39) have shown that elements in the regulatory region flanking oriS stimulate replication from oriS by at least 80-fold relative to a plasmid containing only 80 nucleotides of oriS, demonstrating a role for flanking se-quences in oriS replication efficiency. On the basis this obser-vation, we compared the replication efficiency of a plasmid containing 433 bp of oriL-containing fragment to that of pOL-833. The oriL clone containing 400 bp less flanking sequence, pOL-433, replicated half as efficiently as pOL-833 but 4.8-fold more efficiently than the reference plasmid. These results sug-gest that as for oriS, elements within oriL flanking sequences may be important for efficient DNA replication.
[image:4.612.58.318.73.387.2]Comparison of the kinetics of DNA replication from oriL and oriS.To determine whether DNA replication from oriL and oriS occurs with similar efficiencies at early as well as late times after infection with HSV-1, transient DNA replication assays were performed as a function of time. Vero cells were transfected by the Lipofectin reagent method, with either the 833-bp oriL-containing plasmid (pOL-833) or the 822-bp oriS-containing plasmid (pOS-822). This method of transfection was chosen in order to increase the transfection efficiency such that lower levels of DNA replication that occur at early times after infection with HSV-1 could be detected. At 18 to 24 h after transfection, cells were superinfected with HSV-1, and total DNA was harvested at the indicated times (Fig. 5). The FIG. 2. Cloning strategy for the 833-bp oriL-containing plasmid pOL-833.
(A) Diagram of the HSV-1 genome as described in the legend to Fig. 1. (B) Diagram of the 833-bp oriL-containing fragment generated by PCR using two oligonucleotide primers. These primers (see Materials and Methods) created new restriction sites at the 59(EcoRI) and 39(HindIII) ends of the oriL-con-taining fragment. The oval represents the 144-bp palindrome. (C) Diagram of the 833-bp PCR-generated oriL-containing fragment. The 833-bp oriL fragment was digested with EcoRI and HindIII and ligated into pGEM7Zf(1) to generate pOL-833. The arrows represent the transcriptional start sites of the divergently transcribed genes encoding ICP8 and DNA polymerase (pol) that flank oriL.
FIG. 3. Southern blot analysis of the viral DNA in oriL-containing plasmid pOL-833. Viral (KOS and ts17) and plasmid (pOL-833) DNAs were digested with XmaI and BamHI, electrophoresed in a 1.2% agarose gel, and transferred to a nitrocellulose filter. The filter was then hybridized to a nick-translated 433-bp XmaI-to-BamHI oriL-containing fragment diagrammed in panel B.
on November 9, 2019 by guest
digested DNAs were analyzed by Southern blot hybridization. The replicated and input DNAs were quantitated by Phosphor-Imager scanning analysis, and the values were corrected for transfection efficiencies; the average values of two separate assays are shown in Fig. 5. The values shown in Fig. 5 were calculated relative to the level of replication that occurred at 3
h postinfection and are presented as fold amplification over the level of replication that occurred at 3 h. The results of these tests demonstrate that the kinetics of replication from plasmids of nearly equal size containing either oriS or oriL were similar, but that the oriS-containing plasmid replicated more efficiently than the oriL-containing plasmid at both early and late times after infection with HSV-1.
Comparison of protein-DNA complex formation with sites I and III of oriL and oriS. (i) Complex formation with infected cell extracts.As shown in Fig. 1B, the nucleotide sequences of oriS and oriL are strikingly similar. On the basis of this high degree of sequence homology, it was anticipated that, like BS-I and BS-III of oriS, OBP would bind the corresponding sites in oriL. Indeed, Olivo et al. reported that oriL-containing frag-ments could be immunoprecipitated by a combination of bac-ulovirus-produced OBP and anti-OBP antibody (29). On the other hand, it is notable that several of the single nucleotide differences between oriL and oriS that do exist occur within a region immediately adjacent to BS-I and BS-III that has been shown to be involved in the binding of a cellular factor(s) (OF-1) to oriS site I, resulting in the formation of complex M (3). Thus, Dabrowski et al. (3) demonstrated by gel shift anal-ysis using mutagenized oriS site I oligonucleotides that 8 of 11 nucleotides in the 59half of the bipartite OF-1 binding site are shared with OBP BS-I, whereas the 39half of the OF-1 binding site is located immediately adjacent to OBP BS-I, as summa-rized in Fig. 6. Given that the function(s) of the protein(s) involved in the formation of complex M (OF-1) is not known and that complexes identical in mobility to complex M are observed in gel shift analysis using both infected and unin-fected extracts, the binding characteristics of OBP and OF-1 to BS-I and BS-III of oriS and the corresponding sequence in oriL may differ.
[image:5.612.79.279.72.512.2]To compare the protein-DNA complexes that form with BS-I and BS-III of oriS with those that form with the corre-sponding sites in oriL, complex formation was examined by gel shift analysis. It was shown previously that incubation of HSV-1-infected cell extracts with a probe containing oriS site I resulted in the formation of two specific protein-DNA com-plexes, designated A and B (4, 10, 23, 35). Both of these complexes were shifted to positions of decreased mobility in FIG. 4. Replication efficiencies of oriL-containing plasmids pOL-833 and
pOL-433 relative to an oriS-containing plasmid, pOS-822. (A) Southern blot analysis of DNA from a transient DNA replication assay. Vero cells were trans-fected with an internal standard plasmid (pUC19) alone (Mock) or the internal standard and either pOS-822, pOL-833, or pOL-433 DNA. Total cellular DNAs were harvested 24 h after infection with KOS. DNA was digested with EcoRI and DpnI, separated on a 0.8% agarose gel, and hybridized to a32P-labeled pUC19
[image:5.612.319.552.73.225.2]probe. The bracket denotes the bands of replicated DNA. The lower bands represent DpnI-digested input DNA. (B) Comparison of levels of unreplicated input vector DNA (pUC19) from the experiment in panel A. Details are as for panel A except that the DNA was digested with EcoRI alone to linearize the plasmid DNA. (C) Replication efficiencies of origin-containing plasmids. The replicated DNAs in the Southern blot in panel A were quantitated by Phospho-rImager scanning, and the results are presented as fold amplification over input DNA.
FIG. 5. Comparison of the kinetics of DNA replication from oriS and oriL. Vero cells were transfected with either pOS-822 or pOL-833. DNAs were har-vested at either 3, 6, 9, 12, or 24 h postinfection. The replicated DNAs were quantitated by PhosphorImager analysis. At all times in two separate experi-ments, oriS replicated more efficiently than did oriL. Replication efficiencies were calculated relative to the level of replication that occurred at 3 h postin-fection. Black bars represent fold amplification from a plasmid containing oriS; white bars represent fold amplification from a plasmid containing oriL.
on November 9, 2019 by guest
http://jvi.asm.org/
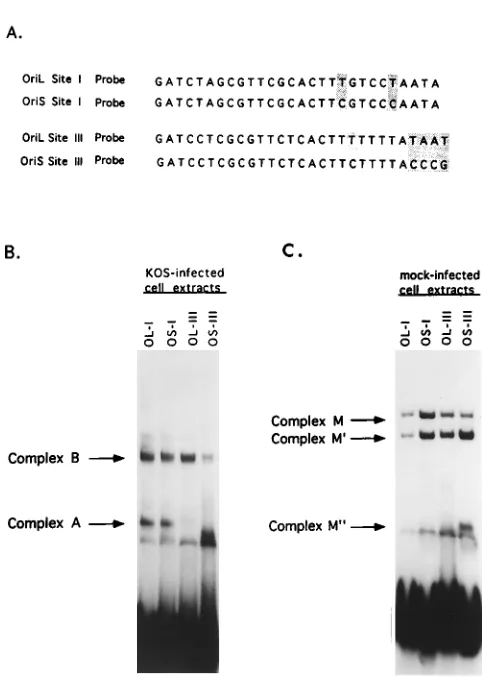
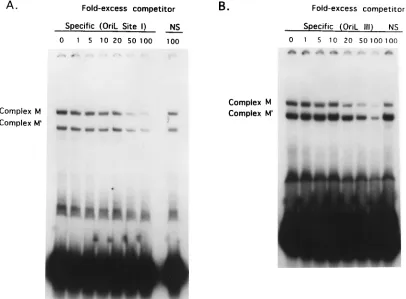
the gel after incubation with OBP-specific antibody, indicating that OBP is a component of both of these complexes (4, 35). When HSV-1-infected cell extracts were incubated with an oriL site I probe, complexes identical to those observed with an oriS site I probe were detected (Fig. 7B). The ability of pro-teins in infected cell extracts to bind an oriL site III probe was also examined (Fig. 7B). Previous studies showed that only one protein-DNA complex was detected when an oriS site III probe was used and that this complex was indistinguishable from complex B which formed with an oriS site I probe (4, 9). When a probe containing oriL site III was used, a single com-plex with a mobility equivalent to that which formed with oriS site III, and to that of complex B which formed with oriS and oriL site I probes, was detected (Fig. 7B). An additional com-plex which exhibited a mobility significantly faster than that of complex B was observed with the oriS site III probe. This complex is also observed when uninfected cell extracts are used as the source of protein and is thought to be the cellular protein-DNA complex previously designated M0(3) (Fig. 7C). To further compare the properties of complexes A and B of oriL and oriS, gel shift assays were performed with infected cell extracts and radiolabeled oriL and oriS site I probes in the presence of anti-OBP antibody (data not shown). In these tests, all four complexes (oriL site I complexes A and B and oriS site I complexes A and B) migrated with equally reduced mobility, demonstrating that OBP is a component of these complexes. It should be noted that while quantitative differ-ences in the levels of cellular complexes that formed with oriS and oriL site I and III DNAs were apparent in Fig. 7, the purpose of these experiments was to examine qualitative dif-ferences in protein-DNA complex formation; thus, further ex-periments are needed to examine the quantitative differences.
[image:6.612.125.488.69.247.2](ii) Complex formation with uninfected cell extracts. Dab-rowski et al. demonstrated that cellular DNA-binding proteins interact with HSV-1 oriS site I, II, and III DNA sequences (3, 4). Two protein-DNA complexes designated M and M9 were observed, and a third complex, M0, was occasionally seen. These investigators further demonstrated that the protein component of complex M, OF-1, binds to a bipartite site. The 59half of this site partially overlaps the OBP binding site, and the 39half lies immediately adjacent to oriS OBP BS-I (Fig. 6).
FIG. 6. Diagram of the binding sites of OBP in complexes A and B and of the cellular factor, OF-1, in complex M within oriS site I. Shown at the top is a nucleotide comparison of DNA sequences containing oriL and oriS site I. The highlighted nucleotides differ between oriL and oriS. The hatched boxes in line 3 represent nucleotides demonstrated by gel shift analysis to be important for the binding of a cellular factor, OF-1, to an oriS site I probe (3). The black boxes in lines 4 and 5 represent nucleotides shown by gel shift analysis to be important for the binding of OBP in complexes A and B (4). The vertical dashed lines indicate the limits of OBP BS-I as defined by DNase I footprint analysis, filter binding assays, and gel shift analysis using mutagenized oriS site I oligonucleotides (4, 9, 10, 11, 23, 25).
FIG. 7. Comparison of protein-DNA complex formation with oriS and oriL sites I and III DNA probes. (A) Nucleotide sequence of the oligonucleotide probes used in the gel shift analyses. The highlighted nucleotides represent nucleotides that are different between oriS and oriL. (B and C) Radiolabeled oriL (OL) or oriS (OS) site I and site III probes, indicated for each lane, were incubated with infected Vero cell extracts (B) or mock-infected Vero cell extracts (C), and resulting complexes were separated on a 6% polyacrylamide gel.
on November 9, 2019 by guest
[image:6.612.313.554.330.669.2]Thus, the sequences of the OF-1 binding site is 59-GTTCGCA CTTCGTCCCAAT-39; the underlined sequence indicates nu-cleotides that partially overlap OBP BS-I. Interestingly, oriS and oriL differ by two nucleotides in the 39 half of the OF-1 binding site (Fig. 1B and 6). Given these minor differences, it was of interest to examine complex formation with oriL site I DNA sequences. As shown in Fig. 7C, identical complexes were observed with all four probes, regardless of whether sites I and III were derived from oriL or oriS. As observed for complex formation with infected cell extracts, a faster-migrat-ing complex (M0) was observed when oriS site III was used as the probe. This complex has also been observed occasionally with oriL site III (data not shown).
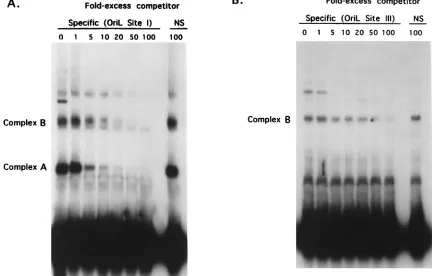
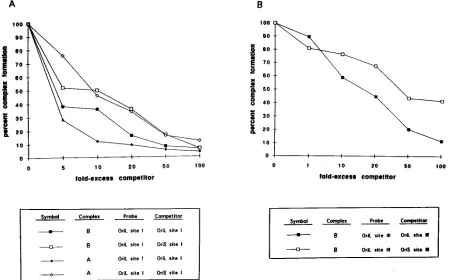
Specificity of infected-cell complex formation at oriL. (i) Competition analysis of oriL site I and III DNAs, using oriL as the competitor.The gel shift analysis shown in Fig. 7B dem-onstrated that, as for oriS, proteins in infected cell extracts form two protein-DNA complexes with the oriL site I probe and one complex with the oriL site III probe. To assess the specificity of complex formation with oriL-derived probes, competition analysis was performed. In these studies, infected cell extracts and radiolabeled oriL site I probe were incubated with increasing concentrations of unlabeled oriL site I-contain-ing sequences as the competitor. As shown in Fig. 8A, the formation of both the A and B complexes decreased progres-sively with increasing concentrations of unlabeled oriL site I-containing DNAs. Complex formation was unaffected follow-ing incubation with a 100-fold excess of nonspecific competitor. Densitometric tracing and data interpolation indicated that the amounts of specific competitor required to reduce complex formation by 50% were similar for complexes A and B, in that
both were reduced to 50% when four- to fivefold excess com-petitor was added.
The specificity of complex B formation with the oriL site III probe was also examined. As described above, radiolabeled oriL site III DNA and infected cell extracts were incubated in the presence of increasing concentrations of unlabeled oriL site III-containing sequences (Fig. 8B). The formation of com-plex B decreased progressively with the addition of increasing amounts of unlabeled oriL site III DNA. The addition of 100-fold excess nonspecific DNA reduced complex B forma-tion only slightly. Densitometric tracing and data interpolaforma-tion demonstrated that complex B formation with the oriL site I probe was reduced to 50% with the addition of 18-fold excess cold competitor.
(ii) Competition analysis of infected-cell complex formation with oriL site I and III DNAs, using oriS as the competitor.
[image:7.612.86.518.78.354.2]Because oriL and oriS differ by two nucleotides in a region important for the binding of a cellular factor(s) (OF-1) to oriS, we attempted to determine if oriS could compete for protein complex formation as efficiently as oriL. For this purpose the oriL site I DNA was radiolabeled and incubated in the pres-ence of infected cell extracts and increasing concentrations of unlabeled oriS site I DNA. The complexes from two or more experiments were analyzed by densitometric tracing and data interpolation, and average densities were compared with re-sults of the competition analysis described above (Fig. 8A), using unlabeled oriL site I DNA as the competitor. The results of these comparisons demonstrated that although oriL com-plexes A and B were reduced to 50% by the addition of 4- to 5-fold excess cold oriL, the reduction in complex A and com-plex B formation to 50% required approximately 10-fold excess FIG. 8. Specificity of infected-cell complex formation with oriL site I and III DNA probes. (A) Infected cell extracts were incubated with radiolabeled oriL site I DNA and increasing concentrations of unlabeled oriL site I-containing DNA or a 100-fold excess of nonspecific (NS) DNA. Protein-DNA complexes were separated on a 6% polyacrylamide gel. (B) Competition analysis was performed as described for panel A except that radiolabeled oriL site III DNA was used as the probe.
on November 9, 2019 by guest
http://jvi.asm.org/
unlabeled oriS as the competitor (Fig. 9A). These results sug-gest that in infected cells, the protein-DNA interactions that occur between oriL site I-containing sequences and the pro-tein(s) in complexes A and B may be stronger than the inter-actions between oriS site I-containing DNA and the protein(s) in complexes A and B.
As noted in Fig. 7A, the oriL site III probe differs from the oriS site III probe by a single nucleotide in the region adjacent to BS-III. Consequently, we wished to determine if the single nucleotide difference in the oriL sequence would alter the ability of oriS to compete for the binding of protein in complex B formation. A competition analysis using radiolabeled oriL site III DNA as the probe and infected cell extracts together with increasing quantities of unlabeled oriS site III DNA as the competitor was performed. The densities of the resulting com-plexes were analyzed as described above and are shown in Fig. 9B. These results were compared with those of tests in which oriL site III DNA was used as the competitor as described above (Fig. 8B) and represent the averages of data from two or more experiments. As observed in competition tests with site I, less unlabeled oriL site III competitor (18-fold) than oriS site III competitor (43-fold) was required to reduce complex for-mation by 50%. These results suggest that the interactions of proteins with oriL site III to form complex B may be stronger than the interactions of proteins with oriS site III.
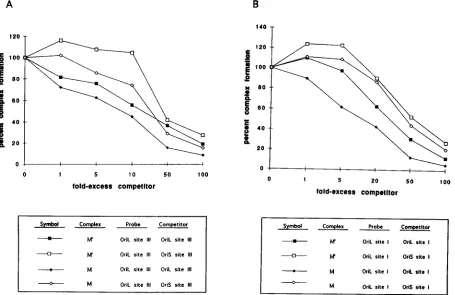
Specificity of cellular complex formation at oriL. (i) Com-petition analysis of oriL site I and III DNAs, using oriL as the competitor. The specificity of the formation of cellular com-plexes with oriL sites I and III was also examined by compe-tition analysis. Shown in Fig. 10 are the results of gel shift
analysis in which the oriL site I (Fig. 10A) or site III (Fig. 10B) probe was incubated with uninfected Vero cell extracts and increasing concentrations of unlabeled oriL site I or site III competitor DNA, respectively. Formation of complexes M and M9 decreased progressively as increasing concentrations of competitor were added. The results of competition analysis for complexes M and M9 with radiolabeled oriL site I and III DNAs were very similar in that 50% reduction in complex M and M9formation occurred in the presence of approximately 10- and 30-fold excesses, respectively, of specific competitor. These findings suggest that the strength of the protein-DNA interactions resulting in the formation of complexes M and M9 are similar.
[image:8.612.82.535.73.353.2](ii) Competition analysis of oriL site I and III DNAs, using oriS as the competitor.Heterologous competition analysis was also performed with radiolabeled oriL site I or III as the probe and increasing concentrations of unlabeled oriS site I and III DNAs as competitors. The results of these tests are shown in Fig. 11. As observed with infected cell extracts, the complexes formed with oriL site I and III probes and uninfected extracts required less specific competitor to reduce complex formation to 50% than with heterologous oriS site I or site III DNA as the competitor. It is notable that less competitor was required to reduce the formation of complex M than complex M9, re-gardless of whether the oriS or oriL sequence was used as the competitor and regardless of whether the probe was site I or site III. These findings are similar to those reported previously in which the formation of complex M was affected more sig-nificantly than formation of complex M9 by the addition of increasing concentrations of homologous competitor DNA (3). FIG. 9. Competition analysis of infected-cell complex formation with oriL site I and III DNA probes, using oriS as a competitor. Gel shift analysis was performed as described in the legend to Fig. 8 except that either oriS site I or III- or oriL site I or III-containing DNA was used as a competitor. (A) The graph represents the interpolated densitometric analysis of competition analysis using radiolabeled oriL site I DNA as a probe and increasing concentrations of either unlabeled oriS site I DNA or unlabeled oriL site I DNA as a competitor. All values are compared with complex formation with no competitor set at 100%. (B) Competition analysis using radiolabeled oriL site III DNA as a probe and increasing concentrations of either unlabeled oriS or oriL site III-containing DNA as a competitor.
on November 9, 2019 by guest
DISCUSSION
Cloning of HSV-1 oriL in an undeleted form.To date, most studies of HSV-1 origin function have focused on oriS because deletions in the oriL palindrome occur after the propagation of oriL-containing plasmids in E. coli. Although both Weller et al. (37) and Lockshon and Galloway (24) were able to clone oriL in an undeleted form in yeast cells and a recombination-defi-cient strain of E. coli, respectively, small fractions of both cloned oriL plasmid populations contained deletions which would be problematic in studies of origin function. In this report, we describe the successful cloning of HSV-1 oriL in an undeleted form, using a strain of E. coli that contains a series of mutations which eliminate a number of independent repair pathways known to be involved in the deletion of irregular structures, including palindromes. As an additional means of reducing the probability of generating deletions, we limited the growth of E. coli, and hence amplification of the cloned oriL containing fragment, to 12 to 14 h, as suggested by Lockshon and Galloway (24). Southern blot and DNA sequence analysis demonstrated that the amplified 833-bp oriL-containing frag-ment had sustained no deletions or point mutations.
The cloned copy of oriL functions as an origin of replication.
To determine whether the 833-bp oriL-containing clone could function as an origin, we used a transient DNA replication system first described by Stow and McMonagle for mapping studies of oriS (33). We compared the replication efficiency of the 833-bp oriL-containing plasmid with that of an 822-bp oriS-containing plasmid. The oriL-containing plasmid consis-tently replicated to only 60 to 70% of the level observed for the oriS plasmid. In a similar study, Lockshon and Galloway (25)
compared the replication efficiencies of an 146-bp oriS-con-taining plasmid and a 190-bp oriL-conoriS-con-taining plasmid. They concluded that oriS and oriL replicated to approximately the same extents. The discrepancy between their results and ours could be due to several factors: (i) the sizes of the origin-containing plasmids used were different in the two studies, (ii) the sensitivities of the assays used to measure replicated DNAs were different, and (iii) different cell types were used in the two studies. The approximately twofold difference in the replica-tion efficiencies of oriS- and oriL-containing plasmids de-scribed herein likely reflect intrinsic differences in the structure and properties of flanking sequences of the two plasmids. Thus, in addition to the fact that oriL is a large and perfect palindrome whereas oriS is not, oriS is located between diver-gently transcribed immediate-early genes whereas oriL lies be-tween divergently transcribed early genes. It is widely recog-nized that the promoter regulatory regions of immediate-early genes are more complex than the promoter regulatory regions of early genes. Consequently, the proteins that bind to the two intergenic regions are clearly different, as discussed in greater detail below. The intergenic region containing oriS is signifi-cantly larger than the region containing oriL such that 536 bp lie between the base of the oriS palindrome and the start site of the ICP4 transcript, and 98 bp lie between the oriS palin-drome and the start site of the ICP22/ICP47 transcripts. In contrast, the start site of the DNA polymerase transcript is only 56 nucleotides, and the start site of the ICP8 transcript is only 92 nucleotides, from the base of the oriL palindrome.
The transcriptional regulatory elements within oriS flanking sequences have been well characterized and consist of a num-FIG. 10. Specificity of cellular protein-DNA complex formation with oriL site I and III DNA probes. (A) Competition analysis was performed as described in the legend to Fig. 8A, using radiolabeled oriL site I, except that mock-infected cell extracts were used. (B) Competition analysis was performed as described for panel A except that radiolabeled oriL site III was used as the probe. NS, nonspecific competitor DNA.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:9.612.102.508.73.372.2]ber of binding sites, including those for the HSV-1 transcrip-tional activator VP16 (TAATGARAT elements), cellular fac-tors SP1, NFIII, and CCAAT box-like-binding proteins (39). Fewer recognized binding sites for regulatory proteins sur-round oriL. These include the recognition sequence for the DNA-binding proteins APREI, LBP-I, and CRE (18). The only elements common to both oriS and oriL flanking se-quences are SP1 binding sites. It is therefore likely that the minor difference in the replication efficiencies of oriL (pOL-833) and oriS (pOS-822) is due, at least in part, to differences in the flanking sequences.
We are currently assessing the role of oriL flanking se-quences in replication efficiency by systematic deletion of these sequences. The results of transient replication assays using a 433-bp and an 833-bp oriL-containing viral DNA fragment provide strong preliminary evidence that oriL flanking se-quences, like those of oriS, enhance replication efficiency. Spe-cifically, the 433-bp oriL clone replicated twofold less effi-ciently than did a clone containing an additional 400 bp of flanking sequence (approximately 200 bp on either side of the oriL palindrome). This observation is in contrast to that of Lockshon and Galloway, who found that a 1,680-bp oriL-con-taining plasmid replicated to approximately the same extent as a 190-bp oriL-containing plasmid (25). This discrepancy is most likely due to differences in the sensitivities of the assays used to measure replicated DNAs.
Comparison of the kinetics of DNA replication from oriS and oriL.The results of initial transient DNA replication as-says demonstrated that 24 h after superinfection with HSV-1,
oriL replicated only 60 to 70% as efficiently as oriS. Because viral DNA replication begins at approximately 3 h postinfec-tion, we next examined whether DNA replication from the two origins occurs with similar efficiencies at early as well as late times after infection with HSV-1. In these studies, the kinetics of replication of plasmids containing the two origins were sim-ilar, but the oriL-containing plasmid replicated two- to four-fold less efficiently than the oriS-containing plasmid at early as well as late times after infection. Whether this is the case in the context of the viral genome remains to be determined. In addition, it is conceivable that while these differences are true for Vero cells, the efficiency of replication may vary for each origin in different cell types, in which specific cellular factors may play a role in initiation and replication.
[image:10.612.81.536.74.369.2]Comparison of protein-DNA complex formation at oriS and oriL sites I and III. The results of gel shift analysis demon-strated that the protein-DNA complexes that formed with oriL and oriS site I and III probes, using infected and mock-infected cell extracts, were indistinguishable. Consequently, we con-clude that the nucleotide differences in the region adjacent to the OBP binding sites in oriL sites I and III (Fig. 1B and 6) have no detectable effect on protein-DNA complex formation in Vero cells. Although complex formation was not detectably different, we did not attempt to determine directly whether the binding affinities of these complexes were the same. Competi-tion analysis suggested that (i) because oriS sites I and III were able to compete with oriL sites I and III for protein-DNA interactions, the proteins involved in the interactions with the two origins are likely the same, and (ii) these proteins may bind FIG. 11. Competition analysis of cellular protein-DNA complex formation with oriL site I and III DNA probes, using oriS site I and III DNAs as competitors. Gel shift analysis was performed as described in the legend to Fig. 10 except that either oriS site I or III- or oriL site I or III-containing DNA was used as a competitor. (A) The graph represents the results of competition analysis using either oriS or oriL site I-containing DNA as a competitor. All values are compared with complex formation with no competitor set at 100%. (B) Graph of the interpolated densitometric analysis of competition assays using oriL site III as a probe and either unlabeled
oriS site III- or oriL site III-containing DNA as a competitor.
on November 9, 2019 by guest
with greater affinity to oriL than oriS. This result was some-what surprising, given that only a single base pair difference distinguishes the oriL and oriS site III probes and two nucle-otide differences distinguish the oriL and oriS site I probes. However, these nucleotide differences occur in a region that has been shown to affect the binding of a cellular factor, OF-1, to the oriS site I probe (4). Therefore, it is possible that the ability of OBP to bind to its cognate binding site is affected by the ability of OF-1 to bind to its adjacent binding site. It is worth noting that although the cellular complexes that bind to oriL and oriS appear to be identical, the identities of the proteins in complexes M and M9 are unknown; it is unclear whether these complexes contain a single protein or multiple proteins. If these complexes contain multiple proteins, gel shift assays are insufficient to determine whether all of the proteins in the oriL and oriS M and M9complexes are identical.
What is the biological significance of two types of origin in the HSV-1 life cycle? Although the locations and nucleotide sequences of oriL and oriS have been known for nearly a decade, and the studies described herein have demonstrated that protein-DNA complexes formed with homologous regions of the two origins are indistinguishable, the biological signifi-cance of the two types of origin remains unclear. Available evidence from studies of mutant viruses lacking oriL (30) or both copies of oriS (22) indicates that none of the three origins is uniquely required for viral DNA replication in cell culture. Rather, it is likely that any one of the three is sufficient in vitro. Why, then, have all three origins been conserved evolutionar-ily? Three plausible explanations include the following: (i) oriL and oriS initiate different types of DNA replication (e.g., theta versus rolling circle) and may, therefore, assemble different types of replication complexes; (ii) the two origins function primarily in different cell types, at different stages of the cell cycle, or at different stages of the viral life cycle (e.g., during productive infection versus reactivation from latency), and (iii) the three origins are functionally redundant, but this redun-dancy confers a survival advantage on the virus. The availabil-ity of an intact, functional source of oriL should prove valuable in investigating these possibilities.
ACKNOWLEDGMENTS
We thank Christine E. Dabrowski for the oriS site I and III oligo-nucleotides and for thoughtful discussions and review of the manu-script.
This work was supported by Public Health Service grant R01AI28537 from the National Institute of Allergy and Infectious Diseases. M.A.H. is the recipient of postdoctoral fellowship F32AI08878 from the National Institutes of Health.
REFERENCES
1. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl (ed.).1988. Current protocols in molecular biology. John Wiley & Sons, New York.
2. Challberg, M. D., and T. J. Kelly. 1989. Animal virus DNA replication. Annu. Rev. Biochem. 58:671–717.
2a.Dabrowski, C. E. Personal communication.
3. Dabrowski, C. E., P. C. Carmillo, and P. A. Schaffer. 1994. Cellular protein interactions with herpes simplex virus type 1 oriS. Mol. Cell. Biol. 14:2545– 2555.
4. Dabrowski, C. E., and P. A. Schaffer. 1991. Herpes simplex virus type 1 origin-specific binding protein: oriS-binding properties and effects of cellular proteins. J. Virol. 65:3140–3150.
5. Deb, S., and S. P. Deb. 1989. Analysis of Ori-S sequence of HSV-1: identi-fication of one functional DNA binding domain. Nucleic Acids Res. 17:2733– 2752.
6. DeLuca, N. A., D. Bzik, V. C. Bond, S. Person, and W. Snipes. 1982. Nucle-otide sequence of herpes simplex virus type 1 (HSV-1) affecting virus entry, cell fusion, and production of glycoprotein gB (VP7). Virology 122:411–423. 7. DeLuca, N. A., and P. A. Schaffer. 1985. Activation of immediate-early, early, and late promoters by temperature-sensitive and wild-type forms of herpes
simplex virus type 1 protein ICP4. Mol. Cell. Biol. 5:1997–2008. 8. Dodson, M. S., and I. R. Lehman. 1993. The herpes simplex virus type 1
origin binding protein. J. Biol. Chem. 268:1213–1219.
9. Elias, P., C. M. Gustafsson, and O. Hammarsten. 1990. The origin binding protein of herpes simplex virus 1 binds cooperatively to viral origin of replication oriS. J. Biol. Chem. 265:17167–17173.
10. Elias, P., C. M. Gustafsson, O. Hammarsten, and N. D. Stow. 1992. Struc-tural elements required for the cooperative binding or the herpes simplex origin binding protein to oriS reside in the N-terminal part of the protein. J. Biol. Chem. 267:17424–17429.
11. Elias, P., and I. R. Lehman. 1988. Interaction of origin binding protein with an origin of replication of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 85:2959–2963.
12. Elias, P., M. E. O’Donnell, E. S. Mocarski, and I. R. Lehman. 1986. A DNA binding protein specific for an origin of replication of herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA 83:6322–62326.
13. Frenkel, N., R. J. Jacob, R. W. Honess, G. S. Hayward, H. Locker, and B. Roizman.1975. Anatomy of herpes simplex virus DNA. III. Characterization of defective DNA molecules and biological properties of virus populations containing them. J. Virol. 16:153–167.
14. Frenkel, N., H. Locker, W. Batterson, G. S. Hayward, and B. Roizman. 1976. Anatomy of herpes simplex virus DNA. VI. Defective DNA originates from the S component. J. Virol. 20:527–531.
15. Frenkel, N., H. Locker, and D. A. Vlazny. 1980. Studies of defective herpes simplex viruses. Ann. N.Y. Acad. Sci. 354:347–370.
16. Friedmann, A., J. Shlomai, and Y. Becker. 1977. Electron microscopy of herpes simplex virus DNA molecules isolated from infected cells by centrif-ugation in CsCl density gradients. J. Gen. Virol. 34:507–522.
17. Gao, M., J. Bouchey, K. Curtin, and D. M. Knipe. 1988. Genetic identifica-tion of a poridentifica-tion of the herpes simplex virus ICP8 protein required for DNA binding. Virology 163:319–329.
18. Ghosh, D. 1990. A relational database of transcription factors. Nucleic Acids Res. 18:1749–1756.
19. Hayward, G. S., R. J. Jacobs, S. C. Wadsworth, and B. Roizman. 1975. Anatomy of herpes simplex virus DNA: evidence for four populations of molecules that differ in the relative orientations of their long short segments. Proc. Natl. Acad. Sci. USA 72:4243–4247.
20. Hazuda, D. J., H. C. Perry, A. M. Naylor, and W. L. McClements. 1991. Characterization of the herpes simplex virus origin binding protein interac-tions with oriS. J. Biol. Chem. 266:24621–24626.
21. Hirsch, I., G. Cabral, M. Patterson, and N. Biswal. 1977. Studies on the intracellular replicating DNA of herpes simplex virus type 1. Virology 81: 48–61.
22. Igarashi, K., R. Fawl, R. J. Roller, and B. Roizman. 1993. Construction and properties of a recombinant herpes simplex virus 1 lacking both S-compo-nent origins of DNA synthesis. J. Virol. 67:2123–2132.
23. Koff, A., and P. Tegtmeyer. 1988. Characterization of major recognition sequences for a herpes simplex virus type 1 origin-binding protein. J. Virol. 62:4096–4103.
24. Lockshon, D., and D. A. Galloway. 1986. Cloning and characterization of oriL2, a large palindromic DNA replication origin of herpes simplex virus type 2. J. Virol. 58:513–521.
25. Lockshon, D., and D. A. Galloway. 1988. Sequence and structural require-ments of a herpes simplex viral DNA replication origin. Mol. Cell. Biol. 8:4018–4017.
26. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
27. Martin, D. W., S. P. Deb, J. S. Klauer, and S. Deb. 1991. Analysis of the herpes simplex virus type 1 oriS sequence: mapping of functional domains. J. Virol. 65:4359–4369.
28. McGeoch, D. J., M. A. Darymple, A. J. Davison, A. Dolan, M. C. Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor.1988. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531–1574.
29. Olivo, P. D., N. J. Nelson, and M. D. Challberg. 1988. Herpes simplex virus DNA replication: the UL9 gene encodes an origin-binding protein. Proc. Natl. Acad. Sci. USA 85:5414–5418.
30. Polvino-Bodnar, M., P. K. Orberg, and P. A. Schaffer. 1987. Herpes simplex virus type 1 oriL is not required for virus replication or for the establishment and reactivation of latent infection in mice. J. Virol. 61:3528–3535. 31. Rabkin, S. D., and B. Hanlon. 1991. Nucleoprotein complex formed between
herpes simplex virus UL9 protein and the origin of DNA replication: inter-and intramolecular interaction. Proc. Natl. Acad. Sci. USA 88:10946–10950. 32. Sandri-Goldin, R. M., A. L. Goldin, L. E. Holland, J. C. Glorioso, and M. Levine.1983. Expression of herpes simplex virus beta and gamma genes integrated in mammalian cells and their induction by an alpha gene product. Mol. Cell. Biol. 3:2028–2044.
33. Stow, N. D., and E. C. McMonagle. 1983. Characterization of the TRS/IRS
origin of DNA replication of herpes simplex virus type 1. Virology 130:427– 438.
34. Vlazny, D. A., and N. Frenkel. 1981. Replication of herpes simplex virus